
Abstract
Native collagen is arranged in bundles of aligned fibrils to withstand in vivo mechanical loads. Reproducing such a process under in vitro conditions has not met with major success. Our approach has been to induce nanolinks, during the self-assembly process, leading to delayed rather than inhibited fibrillogenesis. For this, a designed synthesis of nanoparticles - using starch as a template and a reflux process, which would provide a highly anisotropic (star shaped) nanoparticle, with large surface area was adopted. Anisotropy associated decrease in Morin temperature and superparamagnetic behavior was observed. Polysaccharide on the nanoparticle surface provided aqueous stability and low cytotoxicity. Starch coated nanoparticles was utilized to build polysaccharide - collagen crosslinks, which supplemented natural crosslinks in collagen, without disturbing the conformation of collagen. The resulting fibrillar lamellae showed a striking resemblance to native lamellae, but had a melting and denaturation temperature higher than native collagen. The biocompatibility and superparamagnetism of the nanoparticles also come handy in the development of stable collagen constructs for various biomedical applications, including that of MRI contrast agents.
Similar content being viewed by others

Introduction
Structural proteins, well distributed in mammals determine the functions of the body and thereby are considered as the most advanced biopolymers1. The translation of these biopolymers into products, though a complicated process, can be exploited for biomedical applications. For instance, engineering biomaterials which closely emulate the extracellular matrix (ECM), which plays a vital role in storing, releasing and activating a wide range of biological processes has been considered as pivotal in successful tissue regeneration2,3. Collagen is the principal structural element of ECM and uses weak interactions to self-assemble, the hydrogen bonding interaction being the most popular4. The biochemical interaction of collagen molecule, a well-studied topic, brings about crosslinks between the side chain aminoacids of collagen molecule, leading to stiffness of collagen fibres. A simple but very effective crosslinking of collagen molecule prevents the 300 nm long tropocollagen molecules from sliding past each other under stress5. Isolation of collagen from ECM leads to dissociation of the natural crosslinks, which in turn results in biomaterials formed lacking strength and eventually degrading. Research in the area of exogeneous crosslinking has gained importance as the molecular structure of the crosslinked collagen has high mechanical firmness and collagenase resistance6 A survey of the literature indicates that exogeneous crosslinking methods such as a ferrocene group at the N-terminal of the peptide strand7, a cationic residue at N-terminus and an aromatic residue at the C-terminus to generate interactions between the termini of collagen triple helices8, introduction of covalent bonds through cyanogen bromide activation9, leading to enhanced stability have been explored. Though all these methods provide stability to the collagen and thus the resultant biomaterial, the possible side effects from the use of these reagents like cyanogen bromide has not been studied. One of the approaches to overcome the cytotoxic effects is the use of alternative chemicals or physical methods. Use of natural products, such as those with therapeutic applications for enhancing collagen crosslinks is a new direction. A common condensed tannin molecule such as epigallocatechin gallate provided for structural stability, along with high resistance to bacterial collagenase and MMP-110. Hydrogels based on polysaccharides, either individually or in combination, such as alginate11, chitosan or its derivatives like N,O-(carboxymethyl)chitosan12, N-carboxyethyl chitosan, oxidized dextran13, could also be used for crosslinking to collagen, directly or through zero length or bifunctional crosslinkers such as EDC-NHS, PEG and its derivatives etc14,15. Our coworkers, have employed small molecules and amino acids to understand their role in bringing about inter and intramolecular crosslinks in collagen through a host of physicochemical and molecular modeling techniques16,17,18,19,20,21,22. Fibril stabilization being an enthalpy driven process with water molecules playing an important role in the fibril structure formation23, exposing collagen to high temperatures under vacuum (dehydrothermal treatment), removes water mediated H-bonds and promotes condensation reactions, either through esterification or amide formation, leading to improved mechanical properties24,25.
The advent of nanotechnology and the ability to functionalize nanoparticles has enhanced the ability to employ them to crosslink collagen. Gold nanorods modified with polyelectrolyte multilayers or carrying different polymer chemistries have been reported to influence the self-assembly process of collagen26. The ability of chromium(III) oxide nanoparticles functionalized with a polystyrene-block-polyacrylic acid to provide thermomechanical stability to collagen has been reported by us recently27.
It has been reported that the interaction of nanoparticles with protein can induce cooperative effects leading to promotion or inhibition of self-assembly and nanotoxic consequences28. It thus becomes imperative to understand the molecular mechanism of nanoparticle-collagen interactions for further advances in this area. A significant amount of research leading to nanoparticle stabilized collagen has dependent on the thiol or EDC linkages.
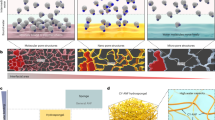
During the 50's, the ability of polysaccharides to stabilize collagen was reported. The involvement of hydroxyl groups in the polysaccharide attached to collagen surface in the process of fibril stabilization was put forth29. However, in later years, the involvement of acidic polysaccharides was discounted30. While direct involvement of starch to bring about fibril stability is scarce, there are reports on the ability of oxidized starch, such as the dialdehydic starch to stabilize collagen, It is known that polycarboxylic acids crosslink to starch31. Combining this information, our goal was to develop polysaccharide (starch) functionalized iron oxide nanoparticles, which combines the bioactive features of starch and the anisotropy and superparamagnetism of the nanoparticle, to achieve synergy between the desired physical properties and biological compatibility in collagen (Figure 1).

1. Rat tail tendons were teased out from Male albino rats. 2. Collagen solution is prepared by established procedures. 3. A reaction between starch capped iron oxide nanoparticles synthesized by a reflux process (48 h), wherein rhombohedral shaped nanoparticles with a fluorescent emission occurring at λ540 nm and having a superparamagnetic character is ensured. 4. The resulting collagen-nanoparticle construct has a higher denaturation and melting temperature than native collagen owing to the fluorescent crosslinks established between tropocollagen units and the starch capped nanoparticles. At appropriate collagen to nanoparticle ratio, these crosslinks form without affecting the natural self assembly process. 5. Such constructs can be employed as T1 contrast agents and is expected to have applications in tissue engineering and magnetically targeted drug delivery/imaging (scheme not drawn to scale). Photographs taken by M.N.
The choice of α-Fe2O3 nanoparticles for such a study was due to its unique electric, optical, catalytic and magnetic properties which have been extended to several applications in biological sciences32. For this work, a greater degree of control over size, size distribution and morphology was desired so as to gain a better control over the understanding of the nanoparticle mediated changes to collagen. Synthesis methodologies ranging from wet chemical co-precipitation to sol-gel synthesis and hydrothermal treatment have been investigated to achieve greater control over morphological properties33,34. For application studies, synthesized nanoparticles need cap to avoid agglomeration. In recent years, our group as well as other has been involved in developing green alternatives to surface active agents, such as the use of polysaccharides to prevent aggregation of nanoparticles35,36. In this work, a reflux synthesis approach, starting with the preparation of a Fe-polysaccharide complex and leading to a polysaccharide stabilized nanoparticles was developed, with a goal to achieve morphological control over the nanoparticles as per Figure 2 presented.

Interaction of iron(III) with starch and formation of α-Fe2O3 nanoparticles by reflux method.
Experimental
Materials
All chemicals employed in this work were procured from M/s. Aldrich Chemicals, USA and used as such without further purification. Milli-Q deionized water was used throughout the study.
Synthesis of α-Fe2O3 nanoparticles
A solution containing FeCl3, acetic acid and starch was prepared such that the effective concentration of Fe3+, acetic acid and starch in the solution was 0.02, 1 and 0.2 mM respectively. The solution was refluxed in a 250 ml round bottom flask, so as to thermally accelerate the reaction between Fe3+ and polysaccharide and subsequently convert Fe3+ to Fe2O3. Care was taken to ensure no loss of vapors occurred, by maintaining adequate cooling of the reflux condenser. Three different experiments, with reflux time of 24, 32 and 48 h were performed. The solution, after adequate cooling was centrifuged to obtain starch coated nanoparticles. The synthesis procedures were repeated thrice to check consistency of synthesized nanoparticles.
Analysis of corona effect (nanoparticle – polysaccharide interaction) on cell viability
The effect of nanoparticle-polysaccharide interaction (corona effect) on cell viability was analyzed as described earlier37,38. Nanoparticles at various concentrations ranging from 10–200 μM were incubated with 1.5 ml DMEM medium containing 10% FCS for 24 hrs at 37°C under sterile condition. The nanoparticles were then collected by centrifugation and re-suspended in fresh 1.5 mL DMEM containing 10% FCS. The medium contacting nanoparticles was treated to NIH/3T3 cells (10,000 cells per well) and maintained in culture in a CO2 incubator at 95%air/5%CO2 atmosphere at 37°C. The MTT assay was performed as described above and the viability was analyzed and compared with that of untreated controls.
Preparation of collagen solution
Collagen solutions were prepared from freshly dissected 6-month old male albino rat tails as per established protocols39. The tails were sourced from rats maintained by other researchers as a part of their control experiments and no animals were separately sacrificed for these measurements. This is with the approval of the institutional (CLRI) ethical committee constituted under the chairmanship of the head biological sciences. All experiments reported in this work were in accordance with relevant guidelines and regulations.
Purity of the collagen solution was confirmed by SDS polyacrylamide gel electrophoresis. Hydroxyproline content was estimated for indirect estimation of collagen concentration in solution40.
To a stock solution of collagen in 5 mM acetic acid (3 mg/mL), adequate volumes of a stock solution of nanoparticles in 5 mM acetic acid (10 mg/mL) were added such that the weight ratio of collagen to nanoparticles was 1:0.02; 1:0.04; 1:0.06; 1:0.08 and 1:0.1 respectively. The solutions marked as 1, 2, 3, 4 and 5 were mixed well and incubated at 4°C overnight.
Collagen fibril formation was initiated by mixing collagen with phosphate buffer (0.2 M) and sodium chloride (2 M) in an ice bath. The pH of the solution was adjusted to 7.4 with sodium hydroxide (1.25 N). The reconstituted solution of collagen in the absence and presence of the nanoparticles were poured in polythene trays and incubated at 37 ± 1°C and air dried. The films were extensively washed with phosphate buffered saline to remove adhering salts.
Characterization of the nanoparticles
Transmission electron microscopic (TEM) images and selected area electron diffraction (SAED) pattern were obtained on a JEOL 3010 field emission electron microscope operating at an accelerating voltage of 300 kV. For this, the iron oxide nanoparticles were dispersed in water by an ultrasonic treatment, 10 μL of the same placed on a carbon film supported copper grid, excess solution wicked away using a filter paper and dried under natural conditions. Diffraction pattern was obtained using a Rigaku, Miniflex(II) desktop X-ray diffractometer (Operating conditions: Cu-Kα (λ − 1.5406Å), 30 kV, 15 mA, scan speed of 4°C/min, step size 0.05°). Zetasizer 3000 HSA (Malvern Instruments, UK) was employed to obtain the particle size, size distribution and zeta potential of the nanoparticle dispersion in water, using concentrations and methods previously standardized by us41. The size and zeta potential measurement were performed in triplicate for independent batches of synthesized nanoparticles. CONTIN analysis was performed to determine the intensity average diameter and size distribution. The polydispersity index, a dimensionless number extrapolated from the autocorrelation function and ranging from 0.01 to 1 was employed to further confirm the monodispersity of the nanoparticles.
Measurement of magnetic resonance relaxivities
MR relaxivities of iron oxide nanoparticles synthesized at 24 h and 48 h reflux were measured using a clinical 1.5 T MR scanner (MAGNETOM Avento Tim System, M/s. Siemens, Germany) equipped with a head coil. For this, phantoms of different concentration of iron oxide (0–0.45 mM) were prepared in deionized water and used. For T2 relaxometry calculations, a modified T2 relaxometry spin echo sequence with TE varying from 15–120 ms with Repetition Time (TR) of 2000 ms were run at three different planes of the phantoms and the pixel intensity with respect to concentration extracted. From the pixel intensity output, the transverse relaxation for each concentration was calculated by employing a linear fit program.
For T1 measurements, an inversion-recovery sequence was used with 7 non equidistant different time delays of 50, 100, 300, 700, 1200, 2000 and 3000 ms between inversion and the first 90° excitation pulse. Time of Echo (TE) and Time of Repetition (TR) are chosen as 15 and 4000 ms respectively. From the MR images corresponding to these inversion times, signal intensities for all the T1s were obtained. T1 relaxation time of each sample was calculated applying these data to the intensity function of the MR signal42,43.
Nanoparticle – collagen interaction studies
Contact angle of nanoparticles were determined by drop shape method44. Kinetic studies were carried out with UV-1800 Shimadzu spectrophotometer. Fibril formation was measured by monitoring turbidometric increase in absorbance at λ313nm immediately after mixing the solution of nanoparticle and collagen in appropriate quantities. The fibril formation rate, represented by t1/2, (half the value of the plateau initiation in the kinetic curve) was determined for both native collagen and nanoparticle-collagen composites. Denaturation temperature (Td) of the samples was measured using Ubbelohde viscometer. For this the solution was incubated at predetermined temperatures of 20–50°C, till equilibrium and the efflux temperature (t) was recorded. The reduced viscosity was calculated from the efflux time of the sample and acetic acid with concentration.
CD-spectra were recorded in the wavelength range of 190–300 nm for every 0.2 nm with a bandwidth of 1 nm at a scan speed of 100 nm/min, under nitrogen atmosphere on a JASCO J-815 CD spectrometer carrying a Peltier temperature controller. To understand temperature induced conformation changes, the spectra were measured with a step wise increase of 0.5°C, with 2 min equilibrium time between each step. For these measurements, the samples were incubated at 4°C for 24 h before recording the spectra. The measure of triple helical content (Rpn), defined as the ratio of positive to negative peak height was also calculated.
For the determination of mechanical properties, the reconstituted collagen films, in the absence and presence of the nanoparticles were placed on an Instron Universal Testing Machine operating at a crosshead speed of 5 mm min−1 and stretched till rupture of film. The thickness of the film was determined using a thickness gauge. From the load employed for rupture, the tensile strength, elongation at break and Young modulus of the samples were measured. The measurements were carried out in triplicate.
High resolution scanning electron microscopic analysis of the reconstituted film in the presence of the nanoparticles were performed using a Quanta 200 FEG SEM. For this, the samples were rinsed with methanol and then sputter-coated with gold to avoid possible contamination. Morphology of the reconstituted films were observed.
Results and Discussion
Morphological features of the nanoparticles
The nanoparticles had rhombohedral geometry (Figure 1), with aspect ratio of 1.26 (major axis = 99 ± 6 nm; minor axis = 79 ± 10 nm), 1.22 (major axis = 93 ± 7 nm; minor axis 76 ± 8 nm) and 1.32 (major axis = 89 ± 7 nm; minor axis = 67 ± 9 nm) respectively for 24, 32 and 48 h of refluxing time. While the presence of starch influences the geometrically alignment of the nanoparticle phases to a rhombohedra, increasing reflux time enhanced the aspect ratio. A decrease in size of nanoparticles (both major and minor axis) was also observed at higher reflux duration. No further change in size was observed beyond 48 h of reflux. Chemical modifications such acid hydrolysis adopted in this work results in starch nanocrystals with reactive surface hydroxyl groups to react with Fe3+45. Acid hydrolysis also impairs the three dimensional network in starch, leading to more percolation of Fe3+ into the matrix and thus a change in the aspect ratio. The crystallite size (Dc) of the nanoparticles was calculated from powder XRD data, using the Debye-Scherrer equation ( , where B is the Scherrer constant, 0.9, λ the wavelength of the Cu-Kα radiation employed, 0.1542 nm, β the full width and half maximum of the plane (110) and θ the Bragg angle). In all three cases, the crystalline size was found to be 38 ± 5 nm. An increase in size of the particle as against crystal size could be attributed to the possible joining of crystal lattices into particles46.
, where B is the Scherrer constant, 0.9, λ the wavelength of the Cu-Kα radiation employed, 0.1542 nm, β the full width and half maximum of the plane (110) and θ the Bragg angle). In all three cases, the crystalline size was found to be 38 ± 5 nm. An increase in size of the particle as against crystal size could be attributed to the possible joining of crystal lattices into particles46.
HRTEM image (Figure 3) shows a typical crystalline domain with inter planar spacing of about 2.69 and 2.47 Å which are comparable with literature values of 2.700 and 2.519 Å, corresponding to the (014) and (110) planes of the hexagonal phases of α - Fe2O3 rhombohedra crystal respectively47. SAED pattern confirmed the formation of α-Fe2O3 phase, the ring being indexed according to the rhombohedra α-Fe2O3 structure (72-469 JCPDS-ICDD).

(a). TEM image of the α-Fe2O3 nanoparticles obtained after 24 h of reflux; (b). after 32 h of reflux; (c). 48 h of reflux; (d). HRTEM image of the nanoparticles after 48 h of reflux, with SAED pattern as inset (e). powder-X-ray diffractogram for the nanoparticles at various durations of reflux; (f). particle size distribution plot (intensity average diameter – CONTIN analysis) as obtained from DLS measurements; (g). fluorescence emission spectra for the nanoparticles; (h). room temperature hysteresis loop for the nanoparticles obtained after various durations of reflux, as obtained by VSM.
Particle size distribution (Figure 3) as measured by dynamic light scattering (DLS), showed an intensity average diameter of 100 ± 5 nm, 93 ± 4 nm and 90 ± 3 nm and polydispersity index value of 0.32, 0.28 and 0.27. Low polydispersity index value is a clear demonstration of monodisperse character of the synthesized nanoparticles47. Usefulness of zeta potential measurement to describe in vivo interactions between implants and cells has gained major relevance48. Zeta potential of the synthesized nanoparticles was found to be 43 ± 2 mV. Compared to previous experiments where the calcination reactions were performed, the nanoparticles from reflux synthesis were highly stable, probably due to a higher level of capping provided by starch. It has been reported that nanoparticles with a positive zeta potential adsorbed more protein than negatively charged nanoparticles49.
Effect of nanoparticle corona on cell viability
When nanoparticles interact with starch and other components in the serum, the direct interaction of the nanoparticle with the cell is replaced with that of nanoparticle corona with the cell37,38. The cellular morphology of the NIH/3T3 cells did not undergo any major change when treated with nanoparticle corona in the concentration range investigated. Cell viability greater than 95% was also observed (Figure 4) for the nanoparticle concentration range investigated. It is known that nanoparticles possessing a high surface charge and large particle size are more efficiently phagocytized50. Our observations on the cell viability in the presence of polysaccharide coated nanoparticles is in tune with earlier observations, wherein chitosan coated nanoparticles with a positive zeta potential has been reported to lack toxicity51.

(a). Morphology of NIH/3T3 cells treated with different concentration (50–200 μM) of α-Fe2O3 nanoparticles; (b). histogram depicting the changes in absorbance (MTT assay) with concentration of nanoparticles.
Magnetic resonance relaxivities
The longitudinal relaxivity (r1) and transverse relaxivity (r2) of the 48 h reflux sample was determined and compared with that of 24 h reflux sample (Figure 5). It is clear from the Figure 3 that 48 h reflux sample had better positive contrast enhancement than the 24 h reflux sample. 48 h reflux sample had an r1 of 3.022 mm−1 s−1, more than seven times that of 24 h reflux sample, of 0.412 mm−1 s−1, as shown in Figure (linear fit). Also the ratio between transverse and longitudinal relaxivity (r2/r1) was found to be low for 48 h reflux sample (5.65174), compared to that of 24 h reflux sample (12.10437). This increase in the r1 value coupled with reduction in r2/r1, is specific and important property for use of a material as T1 contrast agent.

Contrasting effect (a) T1 and (b) T2 relaxation time of iron oxide nanoparticles synthesized at 24 h reflux and 48 h reflux in MRI.
Inset shows the linear fit plot employed for the calculation of r1 and r2.
Superparamagnetic materials such as iron, cobalt or manganese based nanosystems were often used as T2 contrast materials for liver or lymph node MRI52,53,54. But here, we have developed iron based nanosystem as potential T1 contrast agent which is a more preferred way of imaging by clinicians. Moreover, this material shows enhanced positive contrast than recently reported studies42,55. Kim et al56 in a recent report have developed iron oxide nanoparticle as a potential T1 contrast material, with r2/r1 ratio comparable with our current study. But here, we have adopted a greener route synthesis to reduce toxic effects and makes it more suitable for clinical application, compared to the organic one adopted by Kim et al56. Most widely used Gadolinium based T1 MR contrast materials have a tendency to cause osmotic nephropathy, which leads to chronic renal failure57,58. Our results also indicate that the aspect ratio of the nanoparticle is an important parameter, which can be used to tune the properties of nanoparticles for specific applications. A 48 h reflux sample thus has the ability to serve as a T1 contrast agent.
Nanoparticle – collagen interaction: conformation changes
Contact angle of rhombohedra shaped α-Fe2O3 nanoparticles with collagen, in the concentration range employed for 5 was 31.45°, revealing a hydrophilic interaction between the two. It has been reported earlier that modified collagen surfaces with contact angle of 32° showed remarkable human dermal fibroblast and myoblast attachment and proliferation59.
Reconstituting collagen from solution into fibres carrying the signature biological and physic-chemical properties, yet possessing advanced features such as thermal, physical and enzymatic stability than native collagen is a challenge in basic science. For instance, collagen fibres electrospun out of fluoroalcohols were found to denature collagen60. Such materials have been reported to have potential applications in areas such as tissue engineering. Being an optically active protein with a polyproline II like helical conformation, CD spectral analysis provides direct information on the conformational stability of collagen. Native collagen has a negative minima absorption band at 190 nm and a weak positive maxima absorption band at 210–230 nm, with a crossover from negative absorbance to positive absorbance at around 213 nm19. CD spectral analysis is a very useful tool to understand protein-ligand interactions and protein denaturation. Additives, more so metal complexes have been reported to influence collagen stability in a concentration dependent manner61. In this work, the starch capped nanoparticle concentration dependent changes to the CD spectra of collagen was evaluated (Figure 6). Any change in the CD spectra of protein upon addition of a ligand is proportional to amount of protein perturbed62. A complete denaturation of the protein is reported to result in complete disappearance of the positive absorbance band and red shift of negative absorbance band63. With increase in concentration of nanoparticles, the positive peak at 220 nm diminished, though marginally. However, no red shift of the negative absorbance band was noticeable up to a weight ratio of 1:0.08. At 1: 0.1 ratio, the negative peak at 197 nm showed a tendency to red shift. Rpn value (which refers to the ratio of positive maximum to negative minimum), which is an indicative of triple-helix formation64 was also evaluated for native as well as collagen-nanoparticle composites with increasing concentration of nanoparticles. The Rpn value was found to remain more or less constant (ranging from 0.115 to 0.110, with the value being 0.115 for native collagen), indicating that the nanoparticles were not involved in bringing about changes to collagen conformation.

Circular dichroic spectra of native collagen and collagen treated with various quantities of rhombohedra shaped α-Fe2O3 nanoparticles (0.02–0.1 wt%).
Nanoparticle – collagen interaction: thermal stability of collagen
Changes in the CD ellipticity at 220 nm for native collagen with increasing temperature gave a very sharp thermal transition, with a melting temperature, Tm of 38°C. Collagen – nanoparticle composite (5) also showed a similar transition at 39.5°C, indicating an increase of 1.5°C in melting temperature upon treatment with starch capped Fe2O3 nanoparticles. A temperature dependent change in the hydrodynamic radius of collagen as well as collagen – nanoparticle composite (5) was also observed, indicating an unfolding process, wherein the collagen molecules underwent a transition from native trimmers to monomers65.
Nanoparticle – collagen interaction: fibril assembly
Fundamental to mechanical integrity of collagen is its crosslinks. Healthy development and injury repair is progressively dependent on enzymatic rather than non-enzymatic crosslinks66. In vitro collagen self-assembly is a sigmoidal process, with diameter of the fibrils increasing with pH. It has been reported that at pH 7.1, there is an electrostatic interaction promoted unusual alignment of collagen molecules in fibrils67. To replicate the function of native tissues that can be used for tissue engineering or regenerative medicine, in vitro fibrillogenesis is the key. While natural molecules such as proteoglycans are known to modulate collagen fibrillogenesis68, sugars and polyols inhibited fibrillogenesis. Rate of fibre formation of collagen, in the presence and absence of starch capped nanoparticles was measured through turbidity kinetic curves27. The formation of collagen – iron composites have been confirmed from scanning electron microscopic images (Figure 7), where iron oxide nanoparticles have been found effectively coated all over the fibres. Inter fibrillar crosslinks mediated by the functionalized nanoparticles have been observed in the micrographs. Collagen-nanoparticle composite (5) showed delayed fibril formation, with a t1/2 of about 133.33 s, as against 127.06 s for native collagen (Figure 7). It is reported that collagen fibrillogenesis requires formation of hydrogen bonded water clusters, which bridge recognition sites on the opposing helices. When the additive or the partner to collagen in the composite competes with water for crucial hydrogen bonds, then water bridges are disrupted69. In our case, the specific stereochemistry of starch hydroxyls, which defines its preferred hydrogen-bonding pattern, was not conducive for a complete inhibition of the fibrillogenesis process. Binding of starch-capped iron oxide nanoparticles to collagen is akin to the binding of proteoglycans to collagen, i.e. fibrillogenesis is modulated but not completely inhibited. Interestingly, earlier reports had suggested that soluble starch did not affect the collagen assembly70, as against our observation on a retarded process. It can therefore be indirectly concluded that the presence of iron oxide nanoparticles could have influenced the stereochemistry of the starch hydroxyls, thereby retarding the rate of fibrillogenesis. Collagen fibrillogenesis being a time dependent process, a lag period in fibrillogenesis is comparable to the enhanced denaturation temperature71. This was confirmed through viscometric analysis, wherein an increase in denaturation temperature by 3.1°C, from that of native collagen was observed for the composite (5) (data not shown).

(a) Fibril formation of collagen in the absence (self-assembly) of starch coated nanoparticles and with intervention of nanoparticles (b) Mechanical properties of the fibres without and after treatment with functionalized nanoparticles (Tensile strength, elongation at break and Young's modulus have been expressed in N mm−2, % and N m−2 respectively).
The SEM image of the collagen – nanoparticle composite film is depicted in Figure 8. It can be clearly seen that the functionalized nanoparticles clearly coat over the fibres and are involved in bringing about multipoint crosslinking, which could lead to an enhanced mechanical properties. The mechanical properties of the collagen – nanoparticle composite film have been compared against that of the native collagen in Figure 7. The enhancement in the tensile strength, Young's Modulus and elongation at break for the collagen-nanoparticle composite could be attributed to the changes in the spatial arrangement of fibre bundles and interweaving of the fibres. The functionalized iron oxide nanoparticles carrying surface hydroxyl groups are able to bring about interfibrillar crosslinking thus providing resistance to deformation.

Scanning electron microscopic image of collagen fibrils formed in the presence of starch coated nanoparticles.
It is known that crosslinking methods such as glutaraldehyde and EDC/NHS, which are frequently employed in biomedical application, are endowed with disadvantages such as aldehyde cytotoxicity and lower degree of crosslinking72. Our results clearly indicate that the collagen scaffolds are ideal for biomedical applications.
Starch capped anisotropic iron oxide as a fluorescent nanoparticle
The emission spectrum (Figure 3) of the synthesized rhombohedra shaped α-Fe2O3 nanoparticles (reflux duration of 24 h) when dispersed in water showed fluorescence, with λem = 540 nm (λex = 420 nm). As the refluxing time was increased, a corresponding red shift (540–560 nm) in the fluorescent emission was also observed. The observed fluorescence can probably be attributed to recombination of electron hole pair between d band and sp conduction band in the α-Fe2O3 nanoparticles73. We perhaps report such utility for α-Fe2O3 nanoparticles for fluorescent imaging for the first time. Earlier works on fluorescent magnetic nanoparticles are related to the coupling of fluoroprobes to magnetic nanoparticles74, rather than to obtain fluorescent behavior from the α-Fe2O3 nanoparticle itself.
Starch capped iron oxide nanoparticles for targeted delivery
The use of superparamagnetic nanoparticles and a magnetic field gradient to exert a force on the particles have been in wide use for biomedical applications75. Nanoparticles obtained after 24 and 32 h of refluxing time, indicated weak ferromagnetic behavior (Figure 3), with remnant magnetization, Mr values of 0.31 and 0.15 emu/g and coercivity values of 3657 and 1123 Oe, respectively for 24 and 32 h of reflux. However, the nanoparticles synthesized at 48 h of reflux had negligible Mr and coercivity values of 0.004 emu/g and 16 Oe, indicating a shift towards superparamagnetic behavior. Such a change is attributable to the presence of starch capping over the nanoparticle surface, as well as shape anisotropy of α-Fe2O3 nanoparticles. Interestingly, from the temperature dependent zero field cooled and field cooled magnetization measurements (Figure 9), a decrease in Morin transition temperature (TM = 240 K, extrapolated to H = 0), compared to a value of 263 K for bulk α-Fe2O3 was observed for nanoparticles synthesized at 48 h of reflux. Such a decrease in TM can be attributed to the crystalline anisotropy, lattice strain and crystal defects generated by the rhombohedral shaped nanoparticles.

Temperature dependent field cooled (FC) and zero field cooled (ZFC) magnetization for α-Fe2O3 nanoparticles after (a) 24; (b) 32; and (c) 48 h refluxing time.
It is interesting to understand how starch modulates the shape of the synthesized nanoparticles. Based on the information available from literature a plausible mechanism for such a growth has been proposed (Figure 2). Ferric chloride on interaction with starch forms ferric hydroxide. Under reflux conditions, a change from hydroxide to oxyhydroxide is observed, which further condense to α-Fe2O3 nuclei as suggested in literature76. Reflux synthesis in aqueous solution aids process of seed formation and oriented growth by generating numerous hot spots that nucleate crystal growth and generate massive seeds throughout the bulk solution. More seeds created under homogenous conditions will lead to faster crystal growth and the final products will have narrower size distribution and higher yields. Once the α-Fe2O3 seed start to grow under reflux conditions, the available source of Fe3+ becomes promptly depleted. Such reactions will be determined by a non-equilibrium kinetic process77. Adsorption behavior of anions on the oxides and hydroxides can be explained by surface complexation. Unlike other iron oxides, the extent of adsorption on α-Fe2O3 is face specific because of its intrinsic crystal structure. α-Fe2O3 crystallizes in the rhombohedra corundum structure, where the hexagonal unit cell contains six formula units (30 atoms). The oxygen anions form a hexagonal close packed sublattice with exclusively octahedrally coordinated Fe3+ species located in two third of the octahedral sites78. Morphology control, in this instance, has been derived from the specific adsorption of the groups (polysaccharides) on the crystal planes parallel to the c axis, which restrains the crystal growth perpendicular to the c axis. We have been able to demonstrate a fine control over morphology, crystalline character (Figure 3) and size of α-Fe2O3 through a change in the refluxing time.
Starch molecules provide necessary platform for the nuclei to adsorb. Aggregation of the α-Fe2O3 nuclei, so as to reduce their surface energy, will be controlled by the starch platform. The shape and size of the synthesized rhombohedra shaped α-Fe2O3 nanoparticles are determined by the preferential adsorption of starch over the crystalline phases of α-Fe2O3 nuclei during the initial nucleation stage and the subsequent growth stage through the delicate balance between kinetic growth and thermodynamic growth regimes. Subsequent to the growth, starch molecules will provide the necessary capping and colloidal stability to the synthesized rhombohedra shaped α-Fe2O3 nanoparticles79.
Conclusion
The present study describes the development of a simple collagen – nanoparticle construct for potential applications in tissue engineering and imaging by crosslinking collagen to starch capped α-Fe2O3 nanoparticles. The starch capped nanoparticles were found to be non toxic to fibroblast cells and thus used for developing collagen constructs. The enhancement in thermal stability of collagen without changes to its conformational stability was systematically explored. The results showed that the rhombohedral shaped nanoparticles with a starch cap provided for a 1.5°C increase in Tm and 3.1°C increase in Td of collagen. Anisotropy of nanoparticles and presence of a dead layer of starch on surface, conferred superparamagnetic behavior, with a temperature field dependent decrease in Morin transition temperature to 240 K. This observation has implications in the development of magnetically targeted collagen constructs. The starch capped anisotropic nanoparticles also possessed fluorescent properties (λex − 420 nm; λem − 540 nm), thus enhancing their applications as fluorescent probes as well. The tunable relaxivity values of the nanoparticles enhance their value through their ability to function as T1 contrast agents in MRI. The observations made in this study are in variation to already existing reports on starch-collagen interactions, wherein starch was found not to influence thermal stability of collagen, indicating the presence of the capped nanoparticle was responsible for the enhanced properties. This is the first such report wherein a magnetically targetable collagen construct is able to carry both fluorescent and magnetic resonance probe. Compared to known collagen composites, the functionalized nanoparticle – collagen construct developed in this study had higher mechanical properties, associated with increased level of nanoparticle-mediated collagen – collagen crosslinks. For instance, the elongation at break, i.e. the resistance to change for the composite developed in this study was greater than 19% as against 3% reported for poly (lactide-ε-caprolactone)/collagen/nano-hydroxyapatite composite80. Compared to previous reports81 the nanoparticles developed in this study had a lower cytotoxicity due to the presence of the polysaccharide corona, leading to the possibility of offering higher doses of collagen-nanoparticle composites for imaging purposes. The polysaccharide corona also provides for a higher r1 at low r2/r1, leading to its function as a better T1 contrast agent.
In conclusion, the collagen – functionalized iron oxide scaffold developed in this study is a superior material owing to its ability to provide higher cell viability due to presence of polysaccharide corona, better super paramagnetic character, higher mechanical strength and ability to serve both as fluorescent and MRI probe. These features are ideal for the use of scaffold in tissue engineering and biomedicine, such as bio-implants and imaging. These results may offer a new promising approach to tissue engineering.
References
Baer, E., Cassidy, J. J. & Hiltner, A. Hierarchical structure of collagen composite systems - lessons from biology. ACS Sym. Ser. 489, 2–23, 10.1021/bk-1992-0489.ch001 (1992).
Krishnamoorthy, N., Yacoub, M. H. & Yaliraki, S. N. A computational modeling approach for enhancing self-assembly and biofunctionalisation of collagen biomimetic peptides. Biomaterials 32, 7275–7285, 10.1016/j.biomaterials.2011.06.074 (2011).
Shi, J., Votruba, A. R., Farokhzad, O. C. & Langer, R. Nanotechnology in Drug Delivery and Tissue Engineering: From Discovery to Applications. Nano Lett. 10, 3223–3230, 10.1021/nl102184c (2010).
Sun, T. & Qing, G. Biomimetic Smart Interface Materials for Biological Applications. Adv. Mater. 23, H57–H77, 10.1002/adma.201004326 (2011).
Bailey, A. J., Light, N. D. & Atkins, E. D. Chemical cross-linking restrictions on models for the molecular organization of the collagen fibre. Nature 288, 408–410, 10.1038/288408a0 (1980).
Friess, W. Collagen - biomaterial for drug delivery. Eur. J. Pharm. Biopharm. 45, 113–136, 10.1016/s0939-6411(98)00017-4 (1998).
Dey, S. K. & Kraatz, H.-B. Ferrocene-Assisted Stabilization of Collagen Mimetic Triple Helices: Solid-Phase Synthesis and Structure. Bioconjugate Chem. 17, 84–89, 10.1021/bc050268l (2005).
Chen, C.-C., Hsu, W., Kao, T.-C. & Horng, J.-C. Self-Assembly of Short Collagen-Related Peptides into Fibrils via Cationic Interactions. Biochemistry 50, 2381–2383, 10.1021/bi1018573 (2011).
Tabata, Y., Lonikar, S. V., Horii, F. & Ikada, Y. Immobilization of collagen onto polymer surfaces having hydroxyl groups. Biomaterials 7, 234–238, 10.1016/0142-9612(86)90110-9 (1986).
Goo, H. C., Hwang, Y.-S., Choi, Y. R., Cho, H. N. & Suh, H. Development of collagenase-resistant collagen and its interaction with adult human dermal fibroblasts. Biomaterials 24, 5099–5113, 10.1016/S0142-9612(03)00431-9 (2003).
Lee, M., Lo, A. C., Cheung, P. T., Wong, D. & Chan, B. P. Drug carrier systems based on collagen-alginate composite structures for improving the performance of GDNF-secreting HEK293 cells. Biomaterials 30, 1214–1221, 10.1016/j.biomaterials.2008.11.017 (2009).
Chen, R.-N., Wang, G.-M., Chen, C.-H., Ho, H.-O. & Sheu, M.-T. Development of N,O-(Carboxymethyl)chitosan/Collagen Matrixes as a Wound Dressing. Biomacromolecules 7, 1058–1064, 10.1021/bm050754b (2006).
Weng, L., Romanov, A., Rooney, J. & Chen, W. Non-cytotoxic, in situ gelable hydrogels composed of N-carboxyethyl chitosan and oxidized dextran. Biomaterials 29, 3905–3913, 10.1016/j.biomaterials.2008.06.025 (2008).
Shazly, T. M., Artzi, N., Boehning, F. & Edelman, E. R. Viscoelastic adhesive mechanics of aldehyde-mediated soft tissue sealants. Biomaterials 29, 4584–4591, 10.1016/j.biomaterials.2008.08.032 (2008).
Rafat, M. et al. PEG-stabilized carbodiimide crosslinked collagen-chitosan hydrogels for corneal tissue engineering. Biomaterials 29, 3960–3972, 10.1016/j.biomaterials.2008.06.017 (2008).
Usha, R., Maheshwari, R., Dhathathreyan, A. & Ramasami, T. Structural influence of mono and polyhydric alcohols on the stabilization of collagen. Coll. Surf. B 48, 101–105, 10.1016/j.colsurfb.2006.01.015 (2006).
Usha, R., Rajaram, A. & Ramasami, T. Stability of collagen in the presence of 3,4-dihydroxyphenylalanine (DOPA). J. Photochem. Photobiol. B 97, 34–39, 10.1016/j.jphotobiol.2009.07.009 (2009).
Usha, R., Raman, S. S., Subramanian, V. & Ramasami, T. Role of polyols (erythritol, xylitol and sorbitol) on the structural stabilization of collagen. Chem. Phys.Lett. 430, 391–396, 10.1016/j.cplett.2006.09.023 (2006).
Usha, R. & Ramasami, T. Influence of hydrogen bond, hydrophobic and electrovalent salt linkages on the transition temperature, enthalpy and activation energy in rat tail tendon (RTT) collagen fibre. Thermochim. Acta 338, 17–25, 10.1016/S0040-6031(99)00223-3 (1999).
Usha, R. & Ramasami, T. Effect of crosslinking agents (basic chromium sulfate and formaldehyde) on the thermal and thermomechanical stability of rat tail tendon collagen fibre. Thermochim. Acta 356, 59–66, 10.1016/S0040-6031(00)00518-9 (2000).
Usha, R. & Ramasami, T. Structure and conformation of intramolecularly cross-linked collagen. Coll. Surf. B. 41, 21–24, 10.1016/j.colsurfb.2004.11.001 (2005).
Usha, R., Sreeram, K. J. & Rajaram, A. Stabilization of collagen with EDC/NHS in the presence of l-lysine: A comprehensive study. Coll. Surf. B. 90, 83–90, 10.1016/j.colsurfb.2011.10.002 (2012).
Tiktopulo, E. I. & Kajava, A. V. Denaturation of Type I Collagen Fibrils Is an Endothermic Process Accompanied by a Noticeable Change in the Partial Heat Capacity Biochemistry. 37, 8147–8152, 10.1021/bi980360n (1998).
Yannas, I. V., Burke, J. F., Gordon, P. L., Huang, C. & Rubenstein, R. H. Design of an artificial skin. II. Control of chemical composition. J. Biomed. Mater. Res. 14, 107–132, 10.1002/jbm.820140203 (1980).
Gorham, S. D. et al. Effect of chemical modifications on the susceptibility of collagen to proteolysis. 2. Dehydrothermal cross-linking. Int. J. Biol. Macromol. 14, 129–138, 10.1016/s0141-8130(05)80002-9 (1992).
Wilson, C. G., Sisco, P. N., Gadala-Maria, F. A., Murphy, C. J. & Goldsmith, E. C. Polyelectrolyte-coated gold nanorods and their interactions with type I collagen. Biomaterials 30, 5639–5648, 10.1016/j.biomaterials.2009.07.011 (2009).
Sangeetha, S., Ramamoorthy, U., Sreeram, K. J. & Nair, B. U. Enhancing collagen stability through nanostructures containing chromium(III) oxide. Coll. Surf. B 100, 36–41 (2012).
Shemetov, A. A., Nabiev, I. & Sukhanova, A. Molecular Interaction of Proteins and Peptides with Nanoparticles. ACS Nano 6, 4585–4602, 10.1021/nn300415x (2012).
Holt, P. F. & Went, C. W. Studies on the nature of silicosis: a suggested mechanism of fibrogenesis. Brit. J. Ind. Med. 17, 25–30, 10.1136/oem.17.1.25. (1960).
Katzman, R. L. & Jeanloz, R. W. Are acidic polysaccharides involved in collagen fibril formation or stabilization? Biochim. Biophys. Acta 229, 516–521, 10.1016/0005-2795(71)90213-3 (1971).
Reddy, N. & Yang, Y. Citric acid cross-linking of starch films. Food Chem. 118, 702–711, 10.1016/j.foodchem.2009.05.050 (2010).
Meng, Y., Chen, D. & Jiao, X. Fabrication and Characterization of Mesoporous Co3O4 Core/Mesoporous Silica Shell Nanocomposites. J.Phys.Chem.C 110, 15212–15217, 10.1021/jp0626465 (2006).
Cölfen, H. & Antonietti, M. Mesocrystals: Inorganic Superstructures Made by Highly Parallel Crystallization and Controlled Alignment. Angew. Chem. 44, 5576–5591, 10.1002/anie.200500496 (2005).
Rechtin, M. D. & Averbach, B. L. Short-Range Magnetic Order in CoO. Phys. Rev. B. 5, 2693–2704, 10.1103/PhysRevB.5.2693 (1972).
Sreeram, K. J., Nidhin, M. & Nair, B. U. Microwave assisted template synthesis of silver nanoparticles. Bull. Mater. Sci. 31, 937–942, 10.1007/s12034-008-0149-3 (2008).
White, M. A., Johnson, J. A., Koberstein, J. T. & Turro, N. J. Toward the Syntheses of Universal Ligands for Metal Oxide Surfaces:Controlling Surface Functionality through Click Chemistry. J. Am. Chem. Soc. 128, 11356–11357, 10.1021/ja064041s (2006).
Mahmoudi, M. et al. A new approach for the in vitro identification of the cytotoxicity of superparamagnetic iron oxide nanoparticles. Coll. Surf. B. 75, 300–309, 10.1016/j.colsurfb.2009.08.044 (2010).
Walczyk, D., Bombelli, F. B., Monopoli, M. P., Lynch, I. & Dawson, K. A. What the Cell “Sees” in Bionanoscience. J. Am. Chem. Soc. 132, 5761–5768, 10.1021/ja910675v (2010).
Rajan, N., Habermehl, J., Cote, M.-F., Doillon, C. J. & Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nature Protocols 1, 2753–2758, 10.1038/nprot.2006.430 (2006).
Woessner, J. F., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch. Biochem. Biophys. 93, 440–447, 10.1016/0003-9861(61)90291-0 (1961).
Sreeram, K. J., Nidhin, M. & Unni Nair, B. Formation of necklace-shaped haematite nanoconstructs through polyethylene glycol sacrificial template technique. J. Expt. Nanosci., 1–13, 10.1080/17458080.2010.538085 (2011).
Wan, J., Jiang, X., Li, H. & Chen, K. Facile synthesis of zinc ferrite nanoparticles as non-lanthanide T1 MRI contrast agents. J. Mater. Chem. 22, 13500–13505, 10.1039/c2jm30684k (2012).
Nidhin, M. et al. Flower shaped assembly of cobalt ferrite nanoparticles: application as T2 contrast agent in MRI. RSC Adv. 3, 6906–6912, 10.1039/c3ra23232h (2013).
Muthuselvi, L. & Dhathathreyan, A. Contact angle hysteresis of liquid drops as means to measure adhesive energy of zein on solid substrates. Pramana 66, 563–574, 10.1007/bf02704499 (2006).
Lin, N., Huang, J., Chang, P. R., Anderson, D. P. & Yu, J. Preparation, Modification and Application of Starch Nanocrystals in Nanomaterials: A Review. J. Nanomat. 2011, 10.1155/2011/573687 (2011).
Kim, F., Connor, S., Song, H., Kuykendall, T. & Yang, P. Platonic Gold Nanocrystals. Angew. Chem. 43, 3673–3677, 10.1002/anie.200454216 (2004).
Park, T.-J. & Wong, S. S. As-Prepared Single-Crystalline Hematite Rhombohedra and Subsequent Conversion into Monodisperse Aggregates of Magnetic Nanocomposites of Iron and Magnetite. Chem. Mater. 18, 5289–5295, 10.1021/cm061503s (2006).
Kim, Y.-W., Kim, J.-J., Kim, Y. H. & Rho, J.-Y. Effects of organic matrix proteins on the interfacial structure at the bone-biocompatible nacre interface in vitro. Biomaterials 23, 2089–2096, 10.1016/S0142-9612(01)00340-4 (2002).
Patil, S., Sandberg, A., Heckert, E., Self, W. & Seal, S. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials 28, 4600–4607, 10.1016/j.biomaterials.2007.07.029 (2007).
He, C., Hu, Y., Yin, L., Tang, C. & Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31, 3657–3666, 10.1016/j.biomaterials.2010.01.065 (2010).
Yin, L. et al. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 30, 5691–5700, 10.1016/j.biomaterials.2009.06.055 (2009).
Saraswathy, A. et al. Synthesis and characterization of dextran stabilized superparamagnetic iron oxide nanoparticles for in vivo MR imaging of liver fibrosis. Carbohyd. Polym. 101, 760–768, 10.1016/j.carbpol.2013.10.015 (2014).
Saraswathy, A. et al. Citrate coated iron oxide nanoparticles with enhanced relaxivity for in vivo magnetic resonance imaging of liver fibrosis. Colloids Surf. B. 117, 216–224, 10.1016/j.colsurfb.2014.02.034 (2014).
Wagner, M. et al. Coronary MR Angiography Using Citrate-Coated Very Small Superparamagnetic Iron Oxide Particles as Blood-Pool Contrast Agent: Initial Experience in Humans. J. Magn. Reson. Im. 34, 816–823, 10.1002/Jmri.22683 (2011).
Li, Z. et al. Ultrasmall Manganese Ferrite Nanoparticles as Positive Contrast Agent for Magnetic Resonance Imaging. Adv Health Mater 2, 958–964, 10.1002/adhm.201200340 (2013).
Kim, B. H. et al. Large-scale synthesis of uniform and extremely small-sized iron oxide nanoparticles for high-resolution T1 magnetic resonance imaging contrast agents. J Am Chem Soc 133, 12624–12631, 10.1021/ja203340u (2011).
Marckmann, P. et al. Nephrogenic systemic fibrosis: Suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol 17, 2359–2362, 10.1681/Asn.2006060601 (2006).
Yerram, P., Saab, G., Karuparthi, P. R., Hayden, M. R. & Khanna, R. Nephrogenic systemic fibrosis: A mysterious disease in patients with renal failure-role of gadolinium-based contrast media in causation and the beneficial effect of intravenous sodium thiosulfate. Clin J Am Soc Nephro 2, 258–263, 10.2215/Cjn.03250906 (2007).
Cheng, Z. & Teoh, S.-H. Surface modification of ultra thin poly (caprolactone) films using acrylic acid and collagen. Biomaterials 25, 1991–2001, 10.1016/j.biomaterials.2003.08.038 (2004).
Zeugolis, D. I. et al. Electro-spinning of pure collagen nano-fibres: Just an expensive way to make gelatin? Biomaterials 29, 2293–2305, 10.1016/j.biomaterials.2008.02.009 (2008).
Gayatri, R., Sharma, A. K., Rajaram, R. & Ramasami, T. Chromium(III)-induced structural changes and self-assembly of collagen. Biochem. Biophys. Res. Commun. 283, 229–235, 10.1006/bbrc.2001.4713 (2001).
Greenfield, N. J. Applications of circular dichroism in protein and peptide analysis. TrAC, Trends Anal. Chem. 18, 236–244, 10.1016/s0165-9936(98)00112-5 (1999).
Kwak, J., Jefferson, E. A., Bhumralkar, M. & Goodman, M. Triple helical stabilities of guest-host collagen mimetic structures. Bioorg. Med. Chem. 7, 153–160, 10.1016/s0968-0896(98)00230-2 (1999).
Feng, Y. B., Melacini, G., Taulane, J. P. & Goodman, M. Acetyl-terminated and template-assembled collagen-based polypeptides composed of Gly-Pro-Hyp sequences .2. Synthesis and conformational analysis by circular dichroism, ultraviolet absorbance and optical rotation. J. Am. Chem. Soc. 118, 10351–10358, 10.1021/ja961260c (1996).
Mohs, A. et al. Mechanism of stabilization of a bacterial collagen triple helix in the absence of hydroxyproline. J.BIol.Chem. 282, 29757–29765, 10.1074/jbc.M703991200 (2007).
Fessel, G., Gerber, C. & Snedeker, J. G. Potential of collagen cross-linking therapies to mediate tendon mechanical properties. J. Shoulder Elbow Surg. 21, 209–217, 10.1016/j.jse.2011.10.002 (2012).
Li, Y. P., Asadi, A., Monroe, M. R. & Douglas, E. P. pH effects on collagen fibrillogenesis in vitro: Electrostatic interactions and phosphate binding. Mater. Sci. Eng. C-Biomimetic Supramol. Syst. 29, 1643–1649, 10.1016/j.msec.2009.01.001 (2009).
Paderi, J. E. & Panitch, A. Design of a synthetic collagen-binding peptidoglycan that modulates collagen fibrillogenesis. Biomacromolecules 9, 2562–2566, 10.1021/bm8006852 (2008).
Kuznetsova, N., Chi, S. L. & Leikin, S. Sugars and Polyols Inhibit Fibrillogenesis of Type I Collagen by Disrupting Hydrogen-Bonded Water Bridges between the Helices. Biochemistry 37, 11888–11895, 10.1021/bi980089+ (1998).
Nomura, Y., Ishii, Y. & Takahashi, K. Control of Collagen Molecular Assembly with Anionic Polysaccharides. Biosci. Biotechnol. Biochem. 73, 926–929, 10.1271/bbb.80576 (2009).
Tiktopulo, E. I. & Kajava, A. V. Denaturation of type I collagen fibrils is an endothermic process accompanied by a noticeable change in the partial heat capacity. Biochemistry 37, 8147–8152, 10.1021/bi980360n (1998).
Zhong, S. P. & Yung, L. Y. L. Enhanced biological stability of collagen with incorporation of PAMAM dendrimer. J. Biomed. Mater. Res. Part A 91A, 114–122, 10.1002/jbm.a.32188 (2009).
Fei, H. et al. Luminescence of coated a-Fe2O3 nanoparticles. J Lumin. 66–67, 345–348, 10.1016/0022-2313(95)00167-0 (1995).
Jayapaul, J. et al. FMN-coated fluorescent iron oxide nanoparticles for RCP-mediated targeting and labeling of metabolically active cancer and endothelial cells. Biomaterials 32, 5863–5871, 10.1016/j.biomaterials.2011.04.056 (2011).
Sensenig, R., Sapir, Y., MacDonald, C., Cohen, S. & Polyak, B. Magnetic nanoparticle-based approaches to locally target therapy and enhance tissue regeneration in vivo. Nanomedicine 7, 1425–1442, 10.2217/nnm.12.109 (2012).
Jia, B., Gao, L. & Sun, J. Synthesis of Single Crystalline Hematite Polyhedral Nanorods Via a Facile Hydrothermal Process. J. Am. Ceram. Soc. 90, 1315–1318, 10.1111/j.1551-2916.2007.01523.x (2007).
BarrÃ3n, V. & Torrent, J. Surface Hydroxyl Configuration of Various Crystal Faces of Hematite and Goethite. J. Coll. Interface Sci. 177, 407–410, 10.1006/jcis.1996.0051 (1996).
Xi, G. et al. Nucleation and dissolution and recrystallization: A New Growth Mechanism for t-Selenium Nanotubes. Cryst. Growth Des. 6, 577–582, 10.1021/cg050444c (2006).
Jia, C.-J. et al. Single-Crystalline Iron Oxide Nanotubes. Angew. Chem. 44, 4328–4333, 10.1002/anie.200463038 (2005).
Xu, H., Su, J., Sun, J. & Ren, T. Preparation and characterization of new nano-composite scaffolds loaded with vascular stents. Int. J. Mol. Sci. 13, 3366–3381, 10.3390/ijms13033366 (2012).
Mandal, A. et al. Collagen based magnetic nanobiocomposite as MRI contrast agent and for targeted delivery in cancer therapy. Biochim. Biophys. Acta 1830, 4628–4633, 10.1016/j.bbagen.2013.05.018 (2013).
Acknowledgements
The authors acknowledge the financial support from CSIR, New Delhi under the suprainstitutional project S&T Revolution in Leather with a Green Touch (STRAIT) CSIR-CLRI Communication No. 1018. MN and SN thank the CSIR for the senior research fellowship. Authors also thank Dr. A. Bharathi, Head Low Temperature Studies Section, Condensed Matter Physics Division, Materials Science group, IGCAR, Kalpakkam India, for providing the magnetization measurements. KJS thanks Dr Usha Ramamoorthy, Principal Scientist CSIR-CLRI for her contributions to this branch of science and her support in his initiation into this research field.
Author information
Authors and Affiliations
Contributions
K.J.S. wrote the main manuscript text. M.N., M.V., S.S. and S.S.N. prepared the figures 1–7. M.S.K. carried out the cytotoxic measurements, R.S.J. and S.S.N. performed the MRI studies. B.U.N. reviewed the work. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Nidhin, M., Vedhanayagam, M., Sangeetha, S. et al. Fluorescent nanonetworks: A novel bioalley for collagen scaffolds and Tissue Engineering. Sci Rep 4, 5968 (2014). https://doi.org/10.1038/srep05968
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05968
This article is cited by
-
Near infrared-emitting multimodal nanosystem for in vitro magnetic hyperthermia of hepatocellular carcinoma and dual imaging of in vivo liver fibrosis
Scientific Reports (2023)
-
Asialoglycoprotein receptor targeted optical and magnetic resonance imaging and therapy of liver fibrosis using pullulan stabilized multi-functional iron oxide nanoprobe
Scientific Reports (2021)
-
Catechin tuned magnetism of Gd-doped orthovanadate through morphology as T1-T2 MRI contrast agents
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
