
Abstract
The complex architecture of the cortical part of the bovine-femur was examined to develop potential tissue engineering (TE) scaffolds. Weight-change and X-ray diffraction (XRD) results show that significant phase transformation and morphology conversion of the bone occur at 500–750°C and 750–900°C, respectively. Another breakthrough finding was achieved by determining a sintering condition for the nucleation of hydroxyapatite crystal from bovine bone via XRD technique. Scanning electron microscopy results of morphological growth suggests that the concentration of polymer fibrils increases (or decreases, in case of apatite crystals) from the distal to proximal end of the femur. Energy-dispersive analysis of X-ray, Fourier transform infrared, micro-computer tomography and mechanical studies of the actual composition also strongly support our microscopic results and firmly indicate the functionally graded material properties of bovine-femur. Bones sintered at 900 and 1000°C show potential properties for soft and hard TE applications, respectively.
Similar content being viewed by others

Introduction
The beauty of natural bone architecture remains a mystery to scientists, who have attempted to replicate the exact properties of natural bone to produce tissue engineering (TE) scaffolds for bone repair or reconstruction applications1. The development of TE scaffolds from the xenogeneic bone brings significant challenges to researchers because of the complex architecture of real bone, which is a natural composite consisting of inorganic particles and organic fibers1,2,3,4,5,6. In 1926, deJong first identified the structural composition of inorganic bone mineral, which is composed of a solid crystalline phase of calcium and phosphorous-like geological apatite2. The core inorganic constituent of the human bone is hydroxyapatite (HA, Ca5(PO4)3OH) and 7 wt% tri-calcium phosphate (i.e., β-TCP, Ca3(PO4)2)3. The organic part of the bone is mainly composed of polymeric collagen fibrils4,5, which account for almost 38% of the total volume of dry bone6. The overall composition of healthy bone may vary based on several factors, such as species6,7, age7,8,9 and even organ7. Knowing the exact composition of the bone is crucial not only in predicting the mechanical and biological properties of bone, but also in improving the osteogenic influence of xenogeneic bone scaffold design for clinical TE applications10,11,12. In this context, the synthetic HA from different synthetic sources13, which uses various techniques such as solid-state reaction, as reported in our previous study14 and many wet-chemical methods15,16,17, is proven to have one of the best properties that match those of the natural bone mineral14,18. Scientists have attempted to produce synthetic HA scaffolds from various natural sources such as coral19, shell20,21 and bone1,3,22. Recently, HA extracted from bovine bone was proven to offer potential advantages in terms of quantity and quality over materials synthesized from synthetic or other natural sources used as TE scaffolds3,22,23,24. Studies have indicated that an interconnected porous structure is highly desirable for hard tissue TE applications14,25. In this context, the change in bone morphology in the transverse direction (i.e., a highly porous-cancellus bone is encompassed by a densely compact-cortical bone) has significantly contributed to the development of TE scaffolds26,27. However, composition changes along the length of a bone have not yet been investigated. The variation in composition and morphology within a bone, in different directions of a particular organ, could have a significant effect on the load-bearing biomechanical properties of bones. In addition, different properties of scaffold are desired for different types of TE applications. Several fabrication techniques and many synthetic reagents have been used to maintain the exact properties of scaffolds for various TE applications3,20,21,22. However, these techniques are subtle and researchers have yet to produce promising evidence of their use. In addition, the morphology change and growth mechanism of bovine-HA with heat-treatment have yet to be adequately characterized. Therefore, this study is the first attempt at investigating the entire femoral bovine cortical bone (FBCB) to derive the proper heat-treatment conditions suitable for different TE scaffolds. Scaffold density is one of the most important factors in obtaining a desired property for a particular TE application. Osteoconductivity and mechanical properties strongly depend on the combination of porosity and density of the scaffold. In this context, a proper densification mechanism of the heat-treated bioceramic scaffolds derived from abundant bovine bone is potentially useful in designing and developing a desired property of the three-dimensional (3D)-scaffold. Therefore, our study aims to identify the suitable densification mechanisms during sintering of the bovine bone. Furthermore, this study attempts to confirm the reproducibility of the prediction on shape morphology via the XRD technique, as stated in the authors' previous report24.
Methods
Preparation of heat-treated bones
In this study, adult (two to three years old) femoral bovine bones from over five different bodies obtained from the local market were used as starting raw materials. At first, the proximal epiphysis and distal epiphysis were wiped out from the femoral stems. The remained diaphysis part or femoral shaft of a bovine-femur was divided along its length into three equal parts as follows: proximal (i.e., near to the hip joint or the proximal epiphysis), distal (i.e., far from the hip joint or near to the distal epiphysis) and middle (i.e., between the proximal and distal ends) are denoted as P, D and M, respectively. All the muscles, ligaments and cartilages were macroscopically removed from the raw bones after boiling in distilled water for 2.5 h. The spongy bones were then macroscopically scratched out from the cortical bone. Extra fats were removed from the cortical part of each boiled bone via ultrasonication with acetone in a Sono Swiss SW12H ultrasonicator for an optimized time of 5 min. The three different parts of the femoral bovine cortical bones (FBCBs) were excised into different dimensioned specimens for different analyses. The FBCB specimens of proper sizes were dried at 120°C for 12 h in an oven followed by sintering at temperatures 350, 500, 750, 900 and 1000°C for 3 h in ambient conditions by using a Vistec Technology L8-1200 Box furnace. All the heat-treatments were done mainly by two-step sintering method. In step-I, the samples were heated at a heating rate of 5°C/min to an intermediate holding temperature and in step-II, the samples were further heated at a heating rate of 10°C/min to a final temperature. All the samples were furnace-cooled and their heat-treatment conditions are shown in Table 1.
The as-received specimens derived from the distal, middle and proximal parts are to be represented as Das, Mas and Pas, respectively. The distal samples heat-treated at 120, 350, 500, 750, 900 and 1000°C temperatures are to be denoted as D120, D350, D500, D750, D900 and D1000, respectively; and similarly for middle and proximal parts, the heat-treated samples are to be represented as M120, M350, M500, M750, M900 and M1000 and as P120, P350, P500, P750, P900 and P1000, respectively.
Characterizations
The weight-change in all the heat-treated samples after fully completing the heat-treatments was analyzed using a digital weighing machine that maintained up to four decimal digits (±0.0005 g tolerance). The heat-treatment temperatures were determined according to the experience from our previous study24. The weight-change (wt%) measurement was started after completing the final heat-treatment (i.e., step-II) of each sample and was evaluated following the expression Eq. (1). The standard deviation of at least three identical specimens for each part was accounted for error.

where, was is initial weight of the untreated or as-received specimens (i.e., Das, Mas, or Pas) before heat-treatment and wh is weight of the heat-treated specimen.
Phase analysis, crystallinity, crystal size and the relationship between lattice parameters ratio and sintering temperature of the samples were determined via wide angle X-ray diffraction (WAXRD) with Cu-Kα radiation at a wavelength of λ = 1.54056Å by using a PANalytical Empyrean X-ray diffractometer.
The morphology and composition of all the samples were conducted using a Carl Zeiss Auriga dual beam focused ion beam (FIB)–field emission scanning electron microscope (FESEM) equipped with energy dispersive analysis of x-ray (EDAX) detector with secondary electrons. The average value of at least five selected areas in EDAX was employed for each sample. All the samples for the scanning electron microscope (SEM) and EDAX studies were explored on aluminum stubs without any further coating of conducting materials. The subsurface or inner surface, which was already come out during sample preparation by excising, from an outer face parallel to the longitudinal femur bone-axis was scanned under FESEM for each specimen. The SEM images were color-enhanced using Microsoft Office software.
Fourier transform infrared (FTIR) spectroscopy was carried out to determine the functional bonds present in the powder samples by using a Thermo Scientific NICOLET-6700 Fourier transform infrared spectroscope. The diamond (with refractive index: 2.4) attenuated total reflectance (ATR) crystal was used for sampling.
Density of the samples was calculated using Archimedes' principle24,28, which has great advantages over other porosity measuring tests. A minimum of five identical specimens was conducted at 25°C to evaluate the mean and standard deviation in each sintered sample. Open and close porosities were evaluated by this method. The theoretical density (ρth) was considered as 2.2 g/cc for raw bones29,30 and 3.16 g/cc (i.e., theoretical density of HA)14 for the sintered bones to calculate the close porosity of samples.
Micro-computer tomography (microCT) was conducted in a poly(methyl methacrylate) sample holder of size Ø37 mm × 80 mm (diameter × length) by using a Scanco Medical μCT40 analyzer to obtain the bone mineral volume and bone mineral surface area in three different zones of an FBCB, which was heat-treated at 120°C. The each tomographic micrograph of sample was created using multiple X-ray generated images derived from different selected planes with the help of computer in the microCT machine. The resolution, rotation step, rotation angle and scan slices for all the samples were set at 18 μm (pixel size), 0.18°, 360° and 300, respectively. The outer face of the femoral samples was kept perpendicular to the scanning direction.
Three-point (3P) bending was performed using a 5848 Instron Microtester universal testing machine only for the heat-treated bones (at 120°C) to check the role of fibrils and inorganic particles in mechanical properties of femoral bone at three different parts along its length. At least three identical rectangular specimens of size 3 mm × 5 mm × 30 mm (thickness × width × total length) with a fixed span length of 20 mm and crosshead speed of 0.3 mm/min were tested for the D120, M120 and P120 bone samples. Mechanical properties of the other sintered materials could not be able to perform due to showing fragile nature and generation of micro- or macro crack during preparation or machining of samples. Fortunately, authors have able to overcome this difficulty with additional treatment, which will be explored in our next continual study.
Results
In Figure 1, weight-change measurement results indicate almost similar thermal degradation kinetics of the Das and Mas specimens. The weight-change in proximal specimens (i.e., Pas) was noticeably larger and faster than that in distal (Das) and middle (Mas) specimens. A significantly large amount of weight loss (i.e., 10% for Das and Mas and 20% for Pas in weight-change method) was observed at 120°C, which is attributed to the water and moisture content in BCBs. Subsequent drastic and maximum weight loss (i.e., 21% for Das and Mas and 24% for Pas) was observed up to 350°C because most of the organic content, including collagen fibril polymers, was removed from the specimen (see Figure 1). This result was later confirmed via SEM study at 120°C, where more collagen fibrils were prominently revealed at 120°C than at 350°C. Thereafter, a small but noticeable amount of weight loss (i.e., 2 to 3% for all the specimens) was observed up to 500°C, which is attributed to the complete removal of the organic polymers, including collagen fibrils. Later, this finding was confirmed via SEM for specimens at 500°C, where the collagen fibrils left impressions after the complete removal from bone. A trace amount of weight loss (i.e., 1.5 to 2%) was also observed up to 750°C because of the transformation of the HA phase from other phosphates compounds group such as carbonate apatites [Ca10−δZδ(PO4)6−δ(CO3)δ(OH)2, where Z may be sodium (Na), magnesium (Mg) or aluminium (Al)]13,14,17,31. It can also be confirmed by EDAX result where the amount of carbon (C) is found to be significantly decreased at high temperature compared to dried samples (P120 or D120). It is to be noted that the weight change during phase transformation may also vary according to the corresponding phase change since the chemical composition of different calcium phosphates is different. Unlike a structural phase transformation where the chemical compositions are same without any change in weight, for example, transformation of α-TCP (Crystal system: monoclinic, α-Ca3(PO4)2) to β-TCP (Crystal system: rhombohedral, β-Ca3(PO4)2)14,32, in this study at 750°C, the carbonate apatites changed to stoichiometric HA resulting to weight loss accordingly.

A comparison of thermal stability via weight-change measurement in air at two different heating rates for distal (Black open square), middle (Green filled circle) and proximal (Purple open triangle) specimens.
Distal part specimens show maximal and proximal end specimens show least residual weight and there by thermal stability. Maximum degradation of the bone is occurred between 350 to 750°C.
In the temperature range 750–1000°C, the weight loss was negligible and steady because of the very slow decomposition of the remaining inorganic phases, namely, crystalline single-phase pure HA ceramics. This sluggish weight loss also indicates almost complete formation of the HA crystals at 900°C. The transition temperatures for the degradation or decomposition reaction of the FBCBs are nearly at the 120, 350, 500, 750 and 900°C. The residual weights of the FBCBs at 1000°C between proximal and distal ends are 52 and 62 wt%. Conversely, the implication is that the maximum amounts of pure HA, which can be produced from the proximal and distal parts of FBCB by sintering in air, are 52 and 62 wt%, respectively. These values closely match with some reported results22,24. The residual mass of this degradation study also may vary with the raw bone that depends on the initial wet-condition and age of the bone7,8,9.
X-ray diffraction (XRD) presented in Figure 2A (a selected important magnified part is shown in Figure 2B) evaluates the phase transformations or shape morphology and the crystallinity (Xcrys) (Figure 2C) of the FBCBs, following the Eq. (2)29,33. The crystallite size (t) (Figure 2C) was derived from the XRD result using the Debye Scherrer formula34 by neglecting the crystal strain value, i.e., <0.25%24 (see Eq. (S1) in Supplementary Information)

where, Atotal is the total area under the XRD curve and Aamor is the amorphous region only.

(A) XRD patterns of FBCBs from proximal end, after heat-treatments at 750 (Nevi blue), 900 (Green) and 1000 (Red)°C with crystallite dimensions (insets). The crystal shape becomes equiaxed at temperatures 900 and 1000°C as the aspect ratio becomes nearly 1 from the ratio of 0.6891 at 750°C. Areas under the whole curves are computed to evaluate the crystallinity; (B) An enlarged part of a selected important XRD peaks of the samples P750, P900 and P1000; (C) Change in crystallite size (Open square – distal, Open circle – middle and Open triangle – proximal) and crystallinity (Filled square – distal, Filled circle – middle and Filled triangle – proximal) of FBCBs before and after heat-treatments at 120, 350, 500, 750, 900 and 1000°C, as evaluated from XRD patterns. Crystallite size increases with temperature and it is more significant above 500°C, while above 750°C, the change in crystallinity is very slow in all the samples derived from three different sections of bovine-femur; and (D) relationship between c⊥(002)/a⊥(200) ratio and sintering temperature (Purple filled circle) with experimental error (Green bar). The crystal formation of pure HA phase from FBCB is significant only at, or above, 500°C.
The XRD patterns for all bone specimens extracted from the three different femoral parts of femur are almost similar in nature, as the result from a distal part is reported in our previous study24. Therefore, only the essential part of the XRD pattern from the proximal end is depicted in Figure 2A. The XRD patterns of all the specimens from the proximal showed in supplementary Figure S1 depict the phase transformations of FBCBs with heat-treatments. The patterns of the heat-treated bones are also similar to the results reported previously24. The three different crystalline peaks, namely, the 211, 112 and 300 planes at 2θ near 32°, 32.5° and 33°, respectively, closely matched with the peaks those are found in the standard HA possess hexagonal crystal system and the 63/m space group [JCPDS 09-0432]. This result strongly supports the claim that more crystalline pure HA phase formations occur at 900°C than at 750°C because the peaks became more crystalline by significantly reducing the amorphous region at 900°C. When the temperature was increased to 1000°C, Xcrys remained almost unaltered (see Table S1 in Supplementary Information). This result clearly indicates the influence of the rate of grain growth, which is higher than that of the change in crystallinity (see Figure 2C). No significant difference in crystallite size was observed up to 500°C. The average crystallite size abruptly increased, from 27.37 to 66.47 nm, when the temperature was increased from 500 to 750°C, respectively. This sudden increase in crystallite size is a result of the maximum phase transformation of different calcium phosphates or apatite (e.g., carbonate apatite) phases into more ordered or stoichiometric pure HA phase. This result is also strongly supported by a similar phenomenon observed in other study upon influence of thermal treatment on the chemically synthesized HA crystals35. In addition, when the temperature exceeded 750°C, the crystal growth rate became steady (Figure 2C). This indicates that the major stoichiometric phase transformations occur in between 500 and 750°C and mainly morphology conversion takes place in the region 750–900°C. Afterwards, grain growth occurred only at 1000°C. Morphological change or grain growth (e.g., 750–1000°C) may not depend on mass change in the materials. This result is also strongly supported the weight-change experiments22,24 where no significant weight change was observed after 750°C for the all femoral specimens (Figure 1). Here, the crystallinity value became similar in behavior to the crystallite size (see Figure 2C and Table S1 in Supplementary Information). Difference in Xcrys value between the specimens derived from the distal domain and proximal part of a FBCB is not significant nearly at 750°C or above temperature, but below that it is significant (Figure 2C). Therefore, this finding strongly indicates that the upper domain of the bovine femoral bone contains more amorphous phases of polymers compared with the middle and distal parts.
In this investigation, we reproduced the innovative technique24 presented in the XRD study, which can predict the shape morphology of the crystallites (Figure 2D). In brief, the main assumption was that the crystal dimension along a specific axis of its unit cell was perpendicular to its respective plane. In predicting the shape of the HA crystals via XRD analysis, two types of planes, including {200} at 2θ near 22° and {002} at 2θ near 26°, which are perpendicular (⊥) to the a-axis and c-axis, respectively, were selected (Figure 2A). Only three heat-treated specimens (i.e., P750, P900 and P1000) were considered in evaluating of the shape morphology of crystals from FBCB because no other material from the proximal domain of the FBCB prominently showed the {200} peak. The ratio of crystal dimensions, that is, c⊥(002) to a⊥(200) ratio at 750°C, 0.6891, was very close to the ratio obtained from the standard unit cell of HA (i.e., c/a = 0.7309 [JCPDS 09-0432]). This is occurred owing to the lower intensity peak at (002) plane, which makes broader full width at half maxima (θFWHM) value that implies the crystal dimension perpendicular to c-axis. The peak of (002) becomes more intense with increased θFWHM value compare to the peak of (200) at 900 and 1000°C. As a result, both the c⊥(002)/a⊥(200) ratios at 900 and 1000°C, which were 1.0050 and 1.0049, respectively, became almost 1. Such ratios indicate that the high aspect ratio hexagonal HA crystals become equiaxed when the temperature is increased from 750°C to 900°C. When the temperature was further increased to 1000°C, grain growth occurred but the particles remained in an equiaxed shape (see the insets of Figure 2A). This result strongly supports our SEM result, where hexagonal particles at 750°C were converted into equiaxed polycrystalline particles at 900°C. When the temperature was increased to 1000°C, the particles became smoother with a larger grain size. It can also be visualized in the SEM micrographs (Figure 3).

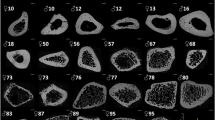
SEM micrographs of as-received and heat-treated cortical bones from distal to proximal parts of a bovine femur.
More polymers present in as-received and low temperature samples and more porosity observed at high temperature proximal part. For A–U, each row consists of same heat-treatment and same colour and three different columns from left to right represent distal, middle and proximal parts of a femur. [A, B, C] as-received: Blue accent. Heat-treated at [D, E, F] 120°C: Olive green; [G, H, I] 350°C: Sepia gray; [J, K, L] 500°C: Red accent; [M, N, O] 750°C: Aqua accent; [P, Q, R] 900°C: Purple; [S, T, U] D1000°C: Orange accent. Yellow arrow indicates inorganic matrix or particles, White arrow indicates collagen fibrils and Green arrow indicates impression left in the sample after removing the polymeric fibrils or fibers. [V] A schematic change in morphology of as-received and heat-treated FBCBs at different temperatures from 120°C to 1000°C – with temperatures, the fibrous polymers gradually decrease and tiny equiaxed inorganic particles convert into hexagonal HA and become large round shaped grains. Three equivalent parts (distal, middle and proximal) of the diaphysis shaft in a bovine femur are also schematically indicated. Growth mechanisms or steps for HA formation and diffusion phenomena of the bovine-apatite are different at different temperatures.
In the above XRD technique, a characteristic curve of c⊥(002)/a⊥(200) ratio (Y) was plotted with the sintering temperatures (X) at 750, 900 and 1000°C (Figure 2D). The average experimental error of the Y-ratio was found to be 0.0038. The curve behavior (Figure 2D) between Y-ratio and X is used to determine the exact sintering temperature for the nucleation of HA crystal from FBCB. From the trend of the experimental curve (see Figure 2D), it can be found that the Y-ratio is going to be substantially differ from the standard value as plotted with dotted line (c/a ratio = 0.7309) in Figure 2D at below 750°C and near at 500°C the difference will be significantly lower than the Y-ratio of standard HA. It has also been noticed that the XRD peak of (200) plane became disappeared at or below 500°C, thereby Y-ratio would turn to be undefined. It indicates that at this temperature, the tiny HA crystals are completely separated out from the polymers or any organic phase. Therefore, it is obvious that the crystal formation of pure HA phase from FBCB becomes significant as close as to 750°C.
The SEM micrographs in Figures 3A–3U depict the proper growth study, including morphology changes at the surface parallel to the bone-length, with sintering temperatures and different regions in longitudinal axis for the FBCB specimens. A same scale bar (0.5 μm) was used for all the SEM images from Figure 3A to Figure 3U. The different colours in different rows represent the different heat-treatment conditions, while three different parts (i.e., Distal, Middle and Proximal) of femur indicate at top of the three columns in the SEM images. In this observation, no significant difference in morphology between the parallel and cross-sectional faces of a same specimen from different parts of a femur in longitudinal axis was found. Here, morphology of cross-sectional face perpendicular to the femoral axis only for D900 specimen was compared with its outer face morphology, is depicted in Figure S2 as supplementary data. The white and yellow colour arrows indicate the fibril polymers and inorganic calcium phosphates or apatites matrix or particles, respectively and the green colour arrow indicates the impressions those were formed after removing the polymeric fibrils or fibers. The as-received bones reveal densely compacted organic molecules, including collagen fibrils that are strongly bonded with the inorganic bone mineral matrix (Figures 3A to 3C). The multi-orientation-polymer fibrils are visible for distal specimen in Figure 3A. The Das resembles a homogeneous composite of polymer and ceramics (Figures 3A) and Mas and Pas show more porous structures with unidirectional polymer fibrils (Figures 3B and 3C).
After drying at 120°C, the fluid contents, including water and part of the organic molecules, decrease. The collagen fibril-surfaces become more cleared, as seen in Figures 3D to 3F. The more nanoparticles are distinctly visible in Figure 3D. The dense structured D120 become more porous in M120 and a larger amount of collagen fibrils with highly porous structure is also seen in P120 specimen. The collagen fibrils are also merely triple helix protein bundles (see inset of Figure 3F)26. This result strongly indicates that the proximal end of the femoral bone has higher amounts of polymer fibrils compared with the distal and middle parts. The submicron size (i.e., 0.5 to 2 μm in diameter) collagen polymer fibrils could provide extra toughness to the bone. More unidirectional polymer frills are revealed for the samples of middle or proximal part in Figures 3E and 3F. After sintering at 350°C, most of the collagen polymer molecules form aggregates (see Figure 3G) and are degraded or fused (Figures 3H and 3I). Unidirectional polymer frills are also visible for the samples of middle or proximal part in Figures 3H and 3I. A very few polymer fibril bundles still remain in the P350 specimen since it contains largest amount of organic polymer parts (Figure 3I). The morphologies of the sintered bovine bones changes significantly at or above 500°C temperatures (Figures 3J to 3U) in comparison to the sample up to 350°C treat samples (Figures 3A to 3I). After that at 500°C, all the collagen fibrils just become disappeared and in few places the impressions of the fully decomposed collagen fibrils are also visible. At 500°C, almost all the polymer fibrils are fully oxidized and in few places, the impressions of the polymer fibrils have also been observed after burning out from the inorganic matrix, as indicated by green coloured arrow in the SEM micrographs (Figures 3J to 3L). The inorganic calcium phosphates comprised of smaller-sized (i.e., <30 nm) particles are fully exposed in the SEM images in Figures 3J to 3L because most of the organic molecules and polymers are removed at 500°C. The inorganic particles form highest dense morphology in D500 sample (Figure 3J) and the inorganic tiny particles also aggregate into a fibrous shape and they are clearly viewed in the proximal bone specimen (see inset of Figure 3L). This result proves that the bone is a natural nanocomposite22,23,24. Figures 3M to 3O clearly show that the hexagonal HA particles, including single crystals, with a wide range of dimensions (i.e., 80 nm to 400 nm in length and 60 nm to 250 nm in width) are nucleated at, or just below, 750°C. The particle size of the P500 and P750 specimens is smaller in dimensions compared with the respective middle or distal specimens. This indicates that the initial inorganic particles present in the proximal end of the FBCB are smaller in size compared with those in the middle or distal ends. At 900°C, most hexagonal crystals and particles become equiaxed polycrystalline, with sizes from 280 to 600 nm (avg. ~390 nm), having uniform porosity (Figures 3P to 3R). Pore size is higher at the proximal end (i.e., 0.3–1.0 μm, avg. ~0.5 μm) than in the middle (i.e., 0.5–1.5 μm, avg. ~0.7 μm) or distal ends (i.e., 0.8–2.2 μm, avg. ~0.9 μm). A clear grain growth was observed when the specimens were sintered at a temperature of 1000°C. Furthermore, the higher number of polymer fibrils creates more intra-particle pores in the heat-treated specimens at the proximal end of femur, as clearly displayed in the micrographs in Figures 3S to 3U. At 1000°C, the overall porosity of the samples decreases and the density abruptly increases according to grain growth. Similar morphological properties of the SEM result were also found in our XRD study, which was illustrated in Figure 2B. In this context, it is to be noted that the dense HA had also shown better osteoblast-like cell growth compare to the less dense needle like nanostructures36. A general change in morphology of FBCB at different heat-treatment temperatures are schematically illustrated in Figure 3V. It shows that the collagen fibers are gradually decreased from the natural bone composite, made of organic fibers-inorganic particles, with heat-treatment temperatures up to 350°C. After that at 500°C, all the collagen fibrils just become disappeared by fully decomposition and their leaving impressions can be visible. At 750°C, the inorganic phosphate particles begin to form hexagonal HA particles. At 900°C, the hexagonal HA particles become equiaxed and further increase of temperature at 1000°C, the equiaxed particles possess grain growth. Therefore, the schematic representation of the micrographs depicted the actual steps in the phase transformation or morphology of pure HA crystals from an FBCB based on temperature. The schematic diagrams are also used to understand the proper growth mechanism or step of HA formation with sintering temperatures (Figure 3V). Therefore, the SEM result also confirms that the major phase transformation occurs in the region 500 to 750°C through the lattice diffusion and morphology conversion taking place in the region 750 to 900°C via surface diffusion. When the temperature is further elevated to above 900°C, the grain growth occurs through grain boundary diffusion. In this context, the nucleation and growth mechanism can be different for different synthesis technique and different source materials24,35. This result was later confirmed by density measurement. The interconnected and highly uniform porous morphology of the D900 specimen observed via SEM may be a promising candidate for the ideal hard TE scaffolds because the submicron sized, uniformly distributed porous structure has potential for use in soft bone TE scaffolds3,22,25.
Essential elements, including carbon (C), oxygen (O), nitrogen (N), calcium (Ca) and phosphorous (P), present in the FBCB were detected via EDAX (Figure 4). The C (~40 At%) and N (~12 At%) atoms present in the as-received and dried (120°C) specimens come from the amide (–CONH2) or amine (–NH2) group of organic biomolecules33,37. The element, N disappears after the complete removal of amino acids of collagen fibrils from the bone at high temperatures, but a small amount of C-atoms still remains (~4 At%) because of the presence of carbonates in the bones (i.e., D900 and P900), as depicted in the inset of Figure 4. The carbon content is higher in the proximal end (P120) than in the distal end (D120) (see inset of Figure 4). Other trace elements (not more than 5 At%), such as sodium (Na), magnesium (Mg) and aluminum (Al), are also detected via EDAX. In this context, one other vital element, hydrogen (H), which must be present in each specimen, cannot be detected in this analysis method because of the limited ability of the analyzer in detecting the lighter atomic weight elements. The carbon content in the materials rapidly decreases when the temperature is increased to 500°C. Afterwards, the rate of decrement becomes very slow and almost steady. The trend in the C-content vs. temperature plot behaved as opposite to the oxygen or Ca/P molar ratio (see Figure 4) since the maximum carbon atoms were removed at 500°C by burning organic polymers. The oxygen concentration and Ca/P molar ratio rapidly increase to 350°C and thereafter, the rate of increment becomes very slow up to 750°C. The Ca/P ratio slightly increases when the temperature increases from 750 to 1000°C. This increase is found due to both the removal of other phosphates, excluding calcium phosphates and the formation of more stoichiometric HA or calcium-apatite. The Ca/P ratios of D120 and P120 specimens are almost the same (i.e., 1.20), which implies that both contains the same main calcium-phosphate compound. The Ca/P ratio (~1.62) for the samples sintered at 900°C is close to the stoichiometric ratio of the pure HA phase (i.e., 1.667). This result proves the formation of a maximum amount of pure phase HA at 900°C, which was predicted by the TGA test. At 1000°C, the Ca/P ratio exceeds the stoichiometric value without producing significant changes in the C or O concentrations. This excess ratio is attributed to the movement of the phosphate group from pure HA to other apatites at elevated temperature.

Trends in atomic percentages of carbon (Grey filled pentagonal) and oxygen (Open circle) and Ca/P molar ratio (Half-filled square) from EDAX data for the proximal end specimens of FBCB change with heat-treatment temperature.
Inset: a comparison of EDAX data between the dried (D120 – red and P120 – black) and high-temperature sintering (D900 – green and P900 – blue) materials; Ca, P, O, N and C elements present in bovine bone significantly change with heat-treatment conditions due to change in phases; N is completely eliminated at high temperature (e.g., 900°C).
The Fourier transform infrared (FTIR) result in Figure 5 reveals all the desired functional bonds present in the bone, which are responsible for organic molecules, including collagen proteins and non-collagen proteins and inorganic phosphates, including HA. The overall molecules are same in a whole femur since same functional groups have been found in both the distal and proximal parts. The FTIR result confirms that the carboxylic acid (a small peak near wavenumber 1740 cm−1) and adsorbed water (a broad adsorption peak in the region from 3000 to 3300 cm−1) of the bovine bone gradually decrease with sintering temperature because the hydroxyl (–OH) and carboxyl (>C = O) groups are reduced with temperature (Figure 5). The hydrated OH from adsorbed water and the acidic OH from proteins or organic biomolecules present in the bone completely disappear when the temperature exceeds 500°C. The C = O bond of the –COOH group near 1740 cm−1 and three N-H bonds of amine (–NH2) from glycine molecule and amide (–CONH2) groups from proline or hydroxyproline molecule near 1640, 1550 and 1240 cm−1 are attributed to the amino acids (i.e., consisting mainly of amine, amide and carboxylic acid groups) of collagen33,37. The C-H stretching bonds near 2920 cm−1 (asymmetric stretching) and 2820 cm−1 (symmetric stretching) are attributed to the –CH2 group in the polymer chain. All the peaks due to the amino acid groups and polymer chains disappear after heating above 350°C. The two peaks of the amino acids, namely, N-H and C = O and both the C-H stretching peaks are more prominent in the P120 specimen than in the D120 specimen (see Inset-A in Figure 5). This implies that the proximal part of the FBCB contains the maximum amount of organic protein molecules, which were clearly revealed as collagen fibrils in the SEM images (see Figure 3F). A small peak near 3300 to 3500 cm−1, which is attributed to the molecular hydroxyl (–OH) group, becomes sharp after sintering at temperatures above 500°C. The increase in molecular OH indicates the formation of HA crystals at 750°C. This molecular OH peak also becomes more sharp and stable at 900 and 1000°C, as revealed in Figure 5. All the materials show small peaks near 1450, 1950 and 2170 cm−1, which are attributed to the carbonate (CO32−) group38. This indicates that a small amount of carbonate compound is always present before and after heat-treatment of the bone. Similar observation has also been found in our EDAX result. In this context, the peak bands near 1380 to 1500 cm−1 correspond to the asymmetric stretching vibrations of CO32−, are also superimposed with the absorptions from –CH2 wagging. This peak band in the distal part (see Inset-B in Figure 5) also gradually decreases with temperature. In the proximal part, it first decreases in the drying sample (P120) because of the removal of adsorbed environmental carbonaceous substance. In this context, to illustrate a precise comparison between the proximal and distal parts, the important peaks only for as-received and dried at 120°C samples are also depicted in Inset-A of Figure 5. The peak then increases up to 500°C during the removal of retained fats and excess polymer content. The latter result clearly implies that the polymer content at the proximal end is higher than at the distal end. Once the temperature exceeds 500°C, this peak, near 1380 to 1500 cm−1, at the proximal end behaves similar to that at the distal end because the polymer had no effect above 500°C in both specimens. The peaks near 1020 and 960 cm−1, which are attributed to the PO43− asymmetric and symmetric stretching, respectively, change quite anomalously because of the combined effect of the formation and decomposition of several apatites and phosphate compounds at a particular heat-treatment condition. The peak near 870 cm−1, which is attributed to P-OH and superimpose of CO32− bending mode, also behaves similar to the other CO32− peak as explained before.

FTIR spectra of FBCBs at the proximal end before (Black – as-received) and after heat-treatments at 120°C (Red), 350°C (Blue), 500°C (Dark cyan), 750°C (Magenta), 900°C (Dark yellow) and 1000°C (Navy blue).
Inset-A: A precise comparison between the two selected specimens from proximal and distal parts – as-received (Black firm and Gray dot lines, respectively) and heat-treated at 120°C (Red firm and Green dot lines, respectively) samples at same absorbance scale; both asymmetric and symmetric C-H stretching peaks are still much higher with the heat-treatment at 120°C for P120 than that of D120. Inset-B: A comparison of an enlarged view of all the specimens from distal part at same intensity scale for two important regions (1850 to 1150 cm−1 and 3650 to 2650 cm−1).
Porosity is an important function of mechanical properties of ceramic materials14,39. In this context, Archimedes' principle have been exploited widely and precisely in several explores to measure the density and porosity28,40,41. Figure 6 depicts the changes in the trends in bulk density (i.e., mass per unit volume of a specimen), open porosity (i.e., ability to hold air or fluid by open voids present in a specimen) and close porosity (i.e., empty space without connecting via any open path or channel present inside a specimen) with the temperature at which the FBCBs were heat-treated (see Table S3 in Supplementary Information). Bulk density (ρ, g/cc), open porosity (Popen, %) and close porosity (Pclose, %) were measured using Archimedes' principle, following Eqs. (3)24, (4)24 and (5), respectively.



where, w1 is the mass of the specimen in air, w2 is the mass of the specimen in distilled water, w3 is the mass of the wet specimen after removal from water and  is water density at 25°C.
is water density at 25°C.

Trends in density, open porosity and close porosity changes with heat-treatment temperature for cortical bones from the distal (Black filled square), middle (Blue open circle) and proximal (Red open triangle) parts of a bovine femur.
Density of scaffolds derived from the distal part of bovine-femur is normally higher compared to proximal or conversely porosity is higher for the scaffolds at proximal domain.
The diffusion mechanism of the growth of apatite particle was identified in this study. The bulk density of the as-received (~1.98, 1.70 and 1.66 g/cc for Das, Mas and Pas, respectively) and of the 120°C-dried bone (~2.02, 1.91 and 1.80 g/cc for D120, M120 and P120, respectively) specimens is higher than that of sintered bones (see Table S3). The density of the other heat-treated materials depends on the initial density of the bone, which can vary according to different factors6,7,8,9, as mentioned in the introduction section. Our initial bone-density exceeds the density range, 1.24 to 1.71 g/cc, of the bovine bone limb reported elsewhere42. After sintering, the bone density initially decreases as a result of the removal of organic polymers that function as binding agents in the bone. Eventually, these natural organic binders create porosity in the material, as confirmed by the porosity value. Further increasing of temperature to 750°C, a densification is observed because of the combination of larger apatite particles and newly formed HA nanoparticles via lattice diffusion43. Thereafter, at 900°C, the density (~1.35, 1.33 and 1.30 g/cc are for D900, M900 and P900, respectively) again decreases via surface diffusion, which results in a more uniform porous microstructure. Prior grain growth occurs, which increases bulk density by significantly reducing porosity (i.e., both open and close porosities) through grain boundary diffusion when the temperature is further increased to 1000°C. These findings are clearly presented in the SEM micrographs (Figures 3P to 3R).
In microCT test (see Figure 7), the FBCBs heat-treated at 120°C show that both porosity (Por, %) and surface-to-volume ratio, that is, bone mineral surface (BS)/bone mineral volume (BV), of the bone mineral are highest in the P120 (i.e., 15.50% and 2.7480 mm−1) and lowest in the D120 (i.e., 3.15% and 0.9594 mm−1) specimen. The microCT data are also illustrated in Table S4 of Supplementary Information. In contrast, bone has a wide range of pore sizes. A change in porosity of the D120, M120 and P120 samples from distal to proximal end was observed in microCT, where minimum pore size may be same or larger than the used resolution (18 μm) during study. This result together with SEM result depicts the gradient of porosity in the longitudinal direction. But the large size micropores were not revealed in SEM micrographs due to using of higher magnification to show the fibril structures present in specimens heat-treated at low temperature (120°C) more clearly. On the other hand, this microCT result could be obtained since a relatively larger amount of organic polymers was removed by heat-treatment at the proximal part than in the distal part. The flexural modulus of the P120 specimen decreases compared with the M120 or D120 specimen as the elastic part of the stress-strain curves becomes stiffer from the proximal to the distal direction (see Figure 7). Here flexural strength and flexural extension gradually increase from the distal (i.e., 23 ± 5 MPa and 4.5 ± 1%) to the proximal (i.e., 51 ± 3 MPa and 9.3 ± 2%) bone (see Figure 7) because the collagen fibrils play a major role during loading at the proximal bone. High modulus value and unlike flexural strain-strain behaviour of the D120 are attributed to the more number of nanoparticles (see SEM Figure 3D) and multi-orientation-polymer fibrils (see SEM Figure 3A), respectively present at the distal part compared to other femoral parts. On other hand, the polymer frills are more unidirectional (see SEM Figures 3E, 3F, 3H and 3I) in middle or proximal part that is responsible for their improved flexural strength and toughness. Since more inorganic particles are present in the distal end, they produce greater pinning effect on the polymer fibrils during loading and the behavior of the stress-strain curve shows as ups and down in nature more distinctly compared to middle or proximal samples. Therefore, this result obviously suggests that the properties of femoral bone along the longitudinal axis are similar to those of functionally graded materials (FGMs).

3-Point bending property of the bone specimens (Black open star – D120, Red open circle – M120 and Blue open triangle – P120) heat-treated at 120°C, with corresponding microCT image, bone mineral volume and porosity.
Porosity and elongation increase from distal to proximal direction of the bovine-femur while strength reduces.
Discussion
The entire FBCB, from the distal to the proximal end, has not yet been systematically studied. Morphology in transverse or radial direction within a cortical part has not been found much difference26,27. Therefore in this study, we investigated only the cortical bone of the bovine-femur in axial direction. This investigation is the first successful attempt at revealing the complex architecture of the cortical bone in bovine-femur in order to design a potential tissue engineering scaffold. An obvious morphological change between D and P parts has been observed. In this context, the junction or gradient section, i.e., in between the two parts like D and M or M and P, could not focus owing to the lack of different sample quantities derived from the exactly edge sections of D, M or P parts for different tests. Despite unveiling the junction properties, we can conclude that the gradation in axial direction of FBCB may be there since the D, M and P parts have shown different gradual change in morphology like FGM. From the weight-change study, we conclude that the bovine-femur is a functionally graded type composite material because it thermally degrades from 38 to 48 wt%, depending on its location on the femoral axis. In the weight-change study, the proximal part of the femoral bone exhibits higher amounts of weight loss compared with the middle or distal ends. This study also showed that the proximal part of the bone contains higher amounts of adsorbed water and more organic molecules. The high polymer fibril content in the proximal part of the FBCB generally withstands more flexural loads at the hip joint compared with the distal end. This investigation demonstrated a reproducibility of the prediction on the shape morphology of the bovine-HA crystallites by a novel technique via XRD, as reported in our previous study24. The relationship between c⊥(002)/a⊥(200) ratio and sintering temperature clearly indicates that the crystal formation of pure HA from FBCB becomes lower below 750°C and the obtention of pure HA crystal becomes insignificant at more lower temperature since the ratio goes down substantially compared to the standard HA. In addition, the XRD peak of (200) plane becomes insignificant at or below 500°C and Y-ratio is to found to be undefined. Thus, nucleation of pure stoichiometric HA crystals may not occur only at or below 500°C. The XRD result also indicates that a major phase transformation and morphology conversion of the bone occur at 500 to 750°C and 750 to 900°C, respectively.
The XRD and SEM results show that the HA crystal also starts to grow as a hexagonal particle at 750°C and becomes converted into equiaxed polycrystalline at 900°C. Increasing the temperature to 1000°C causes the particles to become smoother with larger-sized grains having a dense structure via grain growth. Our SEM, EDAX and FTIR results confirm that both bone morphology and compositions of femoral bone not only change with temperature, but also according to its position along the bone-axis. For example, polymer content at the proximal part is higher than the distal part as evaluated in the present study. The submicron-sized (i.e., 0.5 to 2 μm in diameter) collagen polymer fibrils (see Figure 3F) provide extra toughness to the proximal part of the bone. The proximal or middle part samples showed improved flexural strength and toughness due to the more unidirectional polymer fibrils. The high modulus value in distal samples indicates the presence of large amount of nanoparticles and dissimilar flexural strain-strain behaviour was attributed to the presence multi-orientation-polymer fibrils. Porosity and elongation increase from distal to proximal direction of the bovine-femur while strength reduces. It indicates a relatively larger amount of organic polymers, which was present in the proximal part compared to the distal part, is removed by heat-treatment. The microCT and 3-point bending test results also suggest that the property of femoral bone along the longitudinal axis is similar to the FGMs. Therefore, for the first time, we declare that the maximum amount of collagen polymer fibrils, which are mainly responsible for resisting the flexural and the impact loads at the hip joint, is present at the proximal end of the bovine-femur. The maximum body load exerted on the hip joint is easily endured by the top part of the femur without any fracture in normal conditions. From our typical observation we suggest that bone scaffolds heat-treated at 350 and 500°C are not suitable for tissue engineering scaffold applications due to their fragile in nature. Profuse amount of free carbon present in the blackish scaffolds sintered at 350 and 500°C may responsible for fragile nature in the scaffolds. On the other hand, above 750°C, the sintered cortical bone scaffolds, white in colour, from different longitudinal parts of a bovine-femur can be used according to the desired morphology in different suitable tissue engineering applications. In this context, most of the long specimens, especially more than 20 mm, contain micro- or macro- cracks, which may promote to get anomalous results in the white coloured scaffolds sintered at or above 750°C. Therefore, in the current study, we present the mechanical properties of the samples heat-treated only at 120°C.
The close porosity, which cannot be evaluated via SEM of a whole scaffold, was measured via density analysis. The density measurement confirms the probable diffusion phenomenon, which is an effect of heat-treatment, associated with the phase transformation mechanism. The growth mechanisms for the apatite particles were via lattice diffusion (i.e., from 500 to 750°C – densification and abrupt increase in particle size), surface diffusion (i.e., from 750 to 900°C – decrease in density and change in particle shape with little change in size) and grain boundary diffusion (i.e., from 900 to 1000°C – densification and grain growth). A proper densification mechanism of the abundant animal-bone can apparently help to design the desired property of a TE scaffold. In this context, the cell growth property is not only depends on the porosity but also on the morphology of the grain or particles13,36. Therefore, within the used sintered conditions, a temperature of 900°C exhibits interconnected, uniformly porous morphology that is perfect for soft tissue or soft bone and a temperature of 1000°C provides a dense morphology, which is a potential for 3D-scaffold design in hard tissue repair or reconstruction applications13,36. Considering the combination of close porosity as well as bulk density values and microstructures of all the specimens, the scaffolds heat-treated above 500°C temperatures can be designed according to its intended applications based on the recent developments24,25,44.
References
Hunter, G. K. & Goldberg, H. A. Nucleation of hydroxyapatite by bone sialoprotein. Proc. Natl. Acad. Sci. USA 90, 8562–8565 (1993).
Rey, C., Combes, C., Drouet, C. & Glimcher, M. J. Bone mineral: update on chemical composition and structure. Osteoporosis Int. 20, 1013–1021 (2009).
Lin, F.-H., Liao, C.-J., Chen, K.-S. & Sun, J.-S. Preparation of a biphasic porous bioceramic by heating bovine cancellous bone with Na4P2O7.10H2O addition. Biomaterials 20, 475–485 (1999).
Chen, J. et al. In vitro mineralization of collagen in demineralized fish bone. Macromol. Chem. Phys. 206, 43–51 (2005).
Wang, J. et al. Site-specific in vivo calcification and osteogenesis stimulated by bone sialoprotein. Calcif. Tissue Int. 79, 179–189 (2006).
Biltz, R. M. & Pellegrino, E. D. The chemical anatomy of bone I. A comparative study of bone composition in sixteen vertebrates. J. Bone Joint Surg. Am. 51A, 456–566 (1969).
Kramer, B. & Shear, M. J. Composition of bone. IV. Primary calcification. J. Biol. Chem. 79, 147–160 (1928).
Termine, J. D. & Posner, A. S. Infrared analysis of rat bone: age dependency of amorphous and crystalline mineral fractions. Science 153, 1523–1525 (1967).
Legeros, R., Balmain, N. & Bonel, G. Age-related changes in mineral of rat and bovine cortical bone. Calcif. Tissue Int. 41, 137–144 (1987).
Salama, R. Xenogeneic bone grafting in humans. Clin. Orthop. Rel. Res. 174, 113–121 (1983).
Taschieri, S., Del Fabbro, M., Testori, T. & Weinstein, R. Efficacy of xenogeneic bone grafting with guided tissue regeneration in the management of bone defects after surgical endodontics. J. Oral Maxil. Surg. 65, 1121–11721 (2007).
Kim, S. W., Seo, D. S. & Lee, J. K. Fabrication of xenogeneic bone-derived hydroxyapatite thin film by aerosol deposition method. Appl. Surf. Sci. 255, 388–390 (2008).
Pramanik, S. Syntheses and characterizations of nano-hydroxyapatite, functional polyetheretherketone, carbon nanofibers and their nanocomposites for biomedical applications: high strength and biocompatible. Ph.D. Thesis, Materials Science Programme, I. I. T. Kanpur, India June 2011, 392 (2011).
Pramanik, S., Agarwal, A. K., Rai, K. N. & Garg, A. Development of high strength hydroxyapatite by solid-state-sintering process. Ceram. Int. 33, 419–426 (2007).
Suchanek, W. & Yoshimura, M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 13, 94–117 (1998).
Kar, K. K. & Pramanik, S. Hydroxyapatite-poly(etheretherketone) nanocomposites and method of manufacturing same. Pat. US 8652373 B2, (2014 February 18).
Pramanik, S. & Kar, K. K. Nanohydroxyapatite synthesized from calcium oxide and its characterization. Int. J. Adv. Manuf. Technol. 66, 1181–1189 (2013).
Zambonin, G. & Grano, M. Biomaterials in orthopedic surgery: effects of different hydroxyapatites and demineralized bone matrix on proliferation rate and bone matrix synthesis by hymen osteoblast. Biomaterials 16, 397–402 (1995).
Roy, D. M. & Linnehan, S. K. Hydroxyapatite formed from coral skeletal carbonate by hydrothermal exchange. Nature 247, 220–222 (1974).
Mohammad, N. F., Zahid, M. A., Awang, S. A., Zakaria, Z. & Abdullah, A. A. Synthesis and characterization of bioceramic from malaysian cockle shell. 2010 IEEE Symposium: Industrial Electronics & Applications (ISIEA), Penang, Malaysia: IEEE Malaysia. (10.1109/ISIEA.2010.5679432)(2010 October 3–5).
Sanosh, K. P., Chu, M.-C., Balakrishnan, A., Kim, T. N. & Cho, S.-J. Utilization of biowaste eggshells to synthesize nanocrystalline hydroxyapatite powders. Mater. Lett. 63, 2100–2102 (2009).
Ooi, C. Y., Hamdi, M. & Ramesh, S. Properties of hydroxyapatite produced by annealing of bovine bone. Ceram. Int. 33, 1171–1177 (2007).
Boskey, A. L. & Posner, A. S. Extraction of a calcium-phosphate complex from bone. Calcif. Tissue Res. 19, 273–283 (1976).
Pramanik, S., Hanif, A. S. M., Pingguan-Murphy, B. & Abu Osman, N. A. Morphological change of heat treated bovine bone: a comparative study. Materials 6, 65–75 (2013).
Pramanik, S., Pingguan-Murphy, B. & Abu Osman, N. A. Progress of key strategies in development of electrospun scaffolds: bone tissue. Sci. Technol. Adv. Mater. 13, 043002 (2012).
Rho, J.-Y., Kuhn-Spearing, L. & Zioupos, P. Mechanical properties and the hierarchical structure of bone. Med. Eng. Phys. 20, 92–102 (1998).
Sansalone, V. et al. Anatomical distribution of the degree of mineralization of bone tissue in human femoral neck: Impact on biomechanical properties. Bone 50, 876–884 (2012).
Moradi, A., Pramanik, S., Ataollahi, F., Kamarul, T. & Pingguan-Murphy, B. Archimedes revisited: computer assisted microvolumetric modification of liquid displacement method for porosity measurement of highly porous light materials. Anal. Methods 6, 4396–4401 (2014).
Bonar, L. C., Rouffousse, A. H., Sabine, W. K., Grynpas, M. D. & Glimcher, M. J. X-ray diffraction studies of the crystallinity of bone mineral in newly synthesized and density fractionated bone. Calcif. Tissue Int. 35, 202–209 (1983).
Aerssens, J., Boonen, S., Lowet, G. & Dequeker, J. Interspecies differences in bone composition, density and quality: potential implications for in vivo bone research. Endocrinology 139, 663–670 (1998).
Murphy, W. L. & Mooney, D. J. Bioinspired Growth of Crystalline Carbonate Apatite on Biodegradable Polymer Substrata. J. Am. Chem. Soc. 124, 1910–1917 (2002).
Viswanath, B., Raghavan, R., Gurao, N. P., Ramamurty, U. & Ravishankar, N. Mechanical properties of tricalcium phosphate single crystals grown by molten salt synthesis. Acta Biomater. 4, 1448–1454 (2008).
Pramanik, S. & Kar, K. K. Functionalized Poly(ether ether ketone): improved mechanical property and acellular bioactivity. J. Appl. Polym. Sci. 123, 1100–1111 (2012).
Weibel, A., Bouchet, R., Boulch, F. & Knauth, P. The big problem of small particles: A comparison of methods for determination of particle size in nanocrystalline anatase powders. Chem. Mat. 17, 2378–2385 (2005).
Zanotto, A., Saladino, M. L., Martino, D. C. & Caponetti, E. Influence of Temperature on Calcium Hydroxyapatite Nanopowders. Adv. Nanoparticles 1, 21–28 (2012).
Okada, S., Ito, H., Nagai, A., Komotori, J. & Imai, H. Adhesion of osteoblast-like cells on nanostructured hydroxyapatite. Acta Biomater. 6, 591–597 (2010).
Leaver, A. G. & Shuttleworth, C. A. Fractionation of the acid-soluble nitrogen of bone and dentine. Arch. Oral Biol. 12, 947–958 (1967).
Rey, C., Collins, B., Goehl, T., Dickson, I. R. & Glimcher, M. J. The carbonate environment in bone mineral. A resolution enhanced Fourier transform infrared spectroscopy study. Calcif. Tissue Int. 45, 157–164 (1989).
Lian, C., Zhuge, Y. & Beecham, S. The relationship between porosity and strength for porous concrete. Constr. Build. Mater. 25, 4294–4298 (2011).
Yan, W. et al. Effect of particle size on microstructure and strength of porous spinel ceramics prepared by pore-forming in situ technique. Bull. Mat. Sci. 34, 1109–1112 (2011).
Li, S. & Li, N. Influences of composition of starting powders and sintering temperature on the pore size distribution of porous corundum-mullite ceramics. Sci. Sinter. 37, 173–180 (2005).
Fursey, G. A. J. A note on the density of bovine limb bones. J. Anim. Prod. 21, 195–198 (1975).
Gupta, T. K. & Coble, R. L. Sintering of ZnO: I, Densification and Grain Growth. J. Am. Ceram. Soc. 51, 521–525 (1968).
Mastrogiacomo, M. et al. Role of scaffold internal structure on in vivo bone formation in macroporous calcium phosphate bioceramics. Biomaterials 27, 3230–3237 (2006).
Acknowledgements
This study was supported by UM/MOHE/HIR grant (Project number: D000010-16001). Authors also thank to Hi-Tech Instruments Sdn Bhd for providing the facility of microCT test.
Author information
Authors and Affiliations
Contributions
S.P. performed the experiments and wrote the manuscript. B.P.-M., J.C. and N.A.A.O. provided all the facilities for characterizations. All authors interpreted the data and reviewed the entire manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Pramanik, S., Pingguan-Murphy, B., Cho, J. et al. Design and Development of Potential Tissue Engineering Scaffolds from Structurally Different Longitudinal Parts of a Bovine-Femur. Sci Rep 4, 5843 (2014). https://doi.org/10.1038/srep05843
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05843
This article is cited by
-
Synthesis, fabrication and characterization of CaMgFexTiyO12-based electro-ceramics sensor
Journal of Materials Science: Materials in Electronics (2024)
-
Enhancement of H13 Tool Steel Performance for Die-Casting Process Components via Heat Treatment
JOM (2023)
-
Beta-tricalcium phosphate granules improve osteogenesis in vitro and establish innovative osteo-regenerators for bone tissue engineering in vivo
Scientific Reports (2016)
-
In Vitro Study of Surface Modified Poly(ethylene glycol)-Impregnated Sintered Bovine Bone Scaffolds on Human Fibroblast Cells
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
