
Abstract
Serials of Mn doping by substituting Cd sites on Cu2CdSnSe4 are prepared by the melting method and the spark plasma sintering (SPS) technique to form Cu2Cd1−xMnxSnSe4. Our experimental and theoretical studies show that the moderate Mn doping by substituting Cd sites is an effective method to improve the thermoelectric performance of Cu2CdSnSe4. The electrical resistivity is decreased by about a factor of 4 at 723 K after replacing Cd with Mn, but the seebeck coefficient decreases only slightly from 356 to 289 μV/K, resulting in the significant increase of the power factor. Although the thermal conductivity increases with the doping content of Mn, the figure of merit (ZT) is still increased from 0.06 (x = 0) to 0.16 (x = 0.10) at 723 K, by a factor of 2.6. To explore the mechanisms behind the experimental results, we have performed an ab initio study on the Mn doping effect and find that the Fermi level of Cu2CdSnSe4 is shifted downward to the valence band, thus improving the hole concentration and enhancing the electrical conductivity at the low level doping content. Optimizing the synthesis process and scaling Cu2Cd1−xMnxSnSe4 to nanoparticles may further improve the ZT value significantly by improving the electrical conductivity and enhancing the phonon scattering to decrease the thermal conductivity.
Similar content being viewed by others
Introduction
Due to the ability of realizing the conversion between heat and electricity, thermoelectric (TE) materials have been intensively studied for a long time. The efficiencies of power generation and refrigeration in the thermoelectric devices highly depend on the dimensionless figure of merit (ZT), which is defined as ZT = S2T/ρκ, where S, ρ, T and κ are the Seebeck coefficient, the resistivity, the absolute temperature and the thermal conductivity, respectively. Seeking for new type materials with high TE performance and exploring effective methods to improve the TE performance of existing materials via tuning the materials' ρ, S and κ are critical for the TE device applications.
Quaternary chalcogenides, especially, Cu2-II-IV-VI4 compounds have been investigated recently as promising lead free p-type thermoelectric materials for excellent thermoelectric power generation1,2,3,4,5,6,7,8,9. Cu2-II-IV-VI4 are also promising materials for the solar cell applications due to their suitable direct band gap Eg (Eg = 1.44 eV for Cu2ZnSnSe4 and Eg = 0.96 eV for Cu2CdSnSe4) and the high absorption coefficient for wave numbers around 105 cm−1.10 These compounds are one of the members of the large chalcogen based zinc-blende derived structure compounds (such as Ga2Te3, AgGaTe2, AgInSnTe4, Cu3SbSe4, Cu2CdSnSe4, etc) family, which are well known as the low thermal conductivity materials. What's more, the wide band gap property of such quaternary compounds also helps enhance their TE performances, because the conventional TE materials exhibit a narrow band gap in which a bipolar effect may reduce the thermoelectric efficiency8.
In previous studies, improvements of ZT value for these compounds are mainly focused on Cu2ZnSnSe4. Those improvements include enhancing the electrical conductivity through doping/partial substitution and reducing the thermal conductivity by nanocrystalization5,8,9,11,12. Single crystal13, powder X-ray diffraction (XRD)14 and theoretical calculation15 reveal that Cu2CdSnSe4 has a stannite type structure with the space group (S.G.) of I-42m. By contrast, only a few efforts have been done to improve the TE properties for it, such as doping on bulk materials1 or preparing nanocrystalization4,6. The maximum value of ZT observed for bulk materials Cu2.1Cd0.9SnSe4 reached 0.91 at 700 K1. The nanostructured compounds Cu2CdSnSe4, Cu2.15Cd0.85SnSe3.9 using a chemical synthesis route showed peak ZT values 0.65 at 723 K4, 0.71 at 685 K6, respectively. Mn-doping has been used to improve the TE performance of the ternary compound in this family due to the effect of increasing electric conductivity and Seebeck coefficient, or reducing the thermal conductivity. It is observed that the ZT value has been enhanced by over two orders of magnitude with the introduction of Mn into the CuInSe216; The ZT value reaches 1.2 at 573 K for Ag0.9SbMn0.1Te2.0517; Both power factor and ZT value are improved by Mn doping in Cu2MnxSn1−xSe318. It is also reported that the Mn atom can totally replace the Cd atom in Cu2CdSnSe414, but the effect on TE performance is not reported up to now.
In this work, we provide the first report on the effect of Mn doping on the crystal structure and the TE performance for Cu2CdSnSe4. A significant increase of electrical conductivity and power factor has been observed and the ZT value has been improved by a factor of 2.6 at 723 K. To explore the mechanisms behind the experimental results, we have performed an ab initio study on the Mn doping effect and find that the Fermi level of Cu2CdSnSe4 is shifted downward to the valence band, thus improving the hole concentration and enhancing the electrical conductivity at the low level doping content.
Results and Discussion
Structure characterization
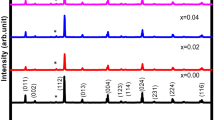
XRD-patterns of the SPS sintered samples are shown in Figure 1(a). The diffraction intensities are shown in the square root manner in order to distinguish the weak peaks. It reveals that all of the samples are Cu2CdSnSe4 with a trace amount of CdSe phases. A white unregular region in SEM image also indicated the existence of CdSe (Figure S1). The CdSe phase may be formed in the melting process, while most of the CdSe materials should be decomposed at the annealing and sintering stage. A trace amount of residual CdSe materials may also exist in the samples due to the high bonding between the Cd and Se atoms in CdSe structure corresponding to the high melt temperature (1540 K). The CdSe phase also has been found from the reported XRD pattern of the polycrystal samples at the melting temperature of 1370 K14. The Rietveld refinement reveals that the Mn atom mainly occupies at the Cd site, when the site occupations of Cd, Sn and Cu are open to Mn atom. The Cu, Cd, Sn atoms occupy at the special Wyckoff positions 4d (0, 0.5, 0.25), 2a (0, 0, 0), 2b (0, 0, 0.25), respectively, the Se atom occupies at the 8i (0.255, 0.255, 0.137) position. The Rietveld refinement shows that the Mn doping slightly changes the atomic position of Se (Table S1), which shortens the bond length of Cu-Se and Cd-Se and slightly enlarges the atomic distance of Sn-Se. Crystal graphic data and reliable factors are listed in Table 1 and the refined patterns are shown in Figure S2. Figure 1(b) shows the plot of lattice parameters as a function of Mn content. We can see that the lattice parameters, such as a, c and V, decrease with the increasing Mn content, due to the smaller ionic radius of Mn2+(0.66 Å) compared with that of Cd2+ (0.78 Å) in the situation of quadridentate19. The dramatic change of lattice parameters reveals the fact that the Mn atoms do incorporate in the crystal structure of Cu2CdSnSe4 at the position of Cd atoms. The value of a, c and c/a for the undoped sample in our work is almost the same as the reported data13,20.

(a) XRD patterns for Cu2Cd1−xMnxSnSe4 (x = 0, 0.05, 0.10 and 0.15). (b) Lattice parameters (a, c and V) of Cu2Cd1−xMnxSnSe4 as a function of Mn content x.
Electrical transport properties
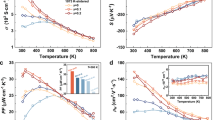
The electrical resistivity data of Cu2Cd1−xMnxSnSe4 samples with x = 0, 0.05, 0.10, 0.15 are plotted in Figure 2(a). Resistivity values of all samples decrease with the increasing temperature from the room temperature to about 600 K. Note that in the temperature range, the resistivity value increases slightly or holds almost the same value for those Mn doped (Cu2Cd1−xMnxSnSe4 with x = 0.05, 0.1, 0.15) samples. The Mn-doping reduces the resistivity of Cu2CdSnSe4 dramatically, which decreases from 1.71 × 10−2 Ω.m for the undoped sample to 5.59 × 10−4 Ω.m for the doped samples of x = 0.05 at the room temperature, nearly by a factor of 30 and from 1.11 × 10−3 Ω.m for the undoped sample to 2.78 × 10−4 Ω.m for the doped samples of x = 0.05 at 723 K, nearly by a factor of 4. However, the higher levels of the Mn doping will not improve the electrical conductivity further. The resistivity of the sample for x = 0.10 is almost the same as the samples for x = 0.05 and the resistivity even increases when the doping amount rises, such as for the sample of x = 0.15.

Electrical transport properties.
(a) Electrical resistivity (ρ) of Cu2Cd1−xMnxSnSe4 as a function of temperature. (b) Seebeck coefficients (S) of Cu2Cd1−xMnxSnSe4 as a function of temperature.
Seebeck coefficients of Cu2Cd1−xMnxSnSe4 as a function of the temperature are shown in Figure 2(b). Positive values of the Seebeck coefficients (S) were found for all samples over the temperature range between 300 K and 723 K. Seebeck coefficients increase almost linearly with the temperature for the undoped sample. However, all doped samples show the Seebeck coefficients increase to the maximum values at about 380 K, then decrease till 480 K and finally follow the increase up to 723 K, which may account for a phase transformation of Cu2Cd1−xMnxSnSe4 induced by the incorporating of Mn. A similar phenomenon was reported in Cu2+xZn1−xGeSe4 system21,22. This phase transformation maybe the second transformation, because there is no obvious endothermic or exothermic effect found by differential scanning calorimetry measurement. At present, the nature of this phase transformation remains unclear and a detailed structural investigation and analysis is necessary. Note that all Seebeck coefficients of Mn-doped samples are lower than the undoped one at the temperature range of 450 to 723 K. The variation tendency of Seebeck coefficients agrees with the variation of resistivity for the Mn-doped samples. In most cases, the lower resistivity, the lower Seebeck coefficient is, because both of Seebeck coefficient and resistivity are related to the inverse proportion with the carrier concentration and mobility. Hall measurements indicate that the carrier concentration dramatically increases when the Mn doping content increases from x = 0 to x = 0.05 and then almost holds the same value as further increasing the Mn content, while the carrier mobility is decreased (Table 1).
Under a given temperature difference, the ability of a material to produce useful electrical power is quantified by its power factor (PF): PF = S2/ρ. As shown in Figure 3, Mn-doping improved the PF from 1.16 × 10−4 W/m/K2 for the undoped sample to 2.88 × 10−4 W/m/K2 for the Mn-doped (x = 0.10) samples at 723 K. Thus, the experiment results show that there is an improvement of PF by about a factor of 2 with and without the Mn-doping, which is mainly attributed to the dramatic decrease of the resistivity in the Mn-doping samples.

Power factors (PF) of Cu2Cd1−xMnxSnSe4 as a function of temperature.
Thermal transport properties
Figure 4(a) displays the temperature dependent thermal conductivity (κ) of all the samples. The thermal conductivity of the undoped sample is 2.9 Wm−1K−1, nearly the same as that obtained by Min-Ling Liu et al. for the undoped samples1. The carrier contribution (κc) was calculated from the electrical resistivity by using the Wiedemann-Franz relation, κc = LT/ρ, with a Lorentz number L = 1.50 × 10−8 W/Ω/K2 (Figure S3). The remaining lattice contribution (κl = κ − κc) is plotted in Figure S4. We can see that values of κc are about two order smaller than that of κl so that the total thermal conductivity κ is depended on the κl, which comes from the lattice contribution.

Thermal transport properties.
(a) Thermal conductivities (κ) of Cu2Cd1−xMnxSnSe4 as a function of temperature. (b) Figure of merit (ZT) of Cu2Cd1−xMnxSnSe4 as a function of temperature.
From the data of Table 1, we can analyze the relationship between the content of Mn-doping and the lattice thermal conductivity. When the Mn content x is increased from 0 to 0.15, values of a- and c- lattices are gradually decreased. The lattice shrinkage in the Mn-doping samples, which proves the atomic distance to be shortened, will enhance the bonding strength to improve the phonon transportation, so as to increase the thermal conductivity. On the other hand, the structure of Cu2CdSnSe4 is aderivation of the cubic zinc blende, so that the lattice distortion can be estimated from the c/a ratio. If the lattice has no distortion, the c/a ratio should be equal to 2. When the Mn content x is increased from 0 to 0.15, the c/a ratio is gradually increased a little bit. It means that the increase of the Mn-doping content reduces the lattice distortion to decrease the phonon scattering, so that the higher thermal conduction can be created. Thus, the increase of the Mn-doping concentration, which makes the lattice shrinkage and the decrease of lattice distortion, respectively, improves the lattice thermal conductivity (κl).
Figure 4(b) shows the temperature dependence of the ZT value. Obviously, ZT values increase with the temperature monotonously due to the decrease of κ and the increase of PF. Even though the increasing thermal conductivity after the substitution of Cd with Mn, which can make the negative contribution to the ZT value, the whole ZT value of the Mn-doped samples still increases from 0.06 (undoped, x = 0) to 0.16 (doped, x = 0.10). Note that the ZT value obtained in our experiments is lower than values reported in previous works for Cu2CdSnSe41. The main reason is the lower electrical conductivity for our samples. For example, our electrical conductivity for the undoped Cu2CdSnSe4 at the room temperature is only 58.5 S.m−1, which is much lower than the value of about 2000 S.m−1 obtained by Min-Ling Liu et al.1. The difference on reported electrical transport properties may be due to the different preparing method resulting in the slight difference in chemical composition, microstructure or trace second phase. The ZT value can also be further improved by careful adjusting the preparing conditions and scaling Cu2CdSnSe4 to nanoparticles to enhance the electrical transport properties and the phonon scattering to decrease the thermal conductivity6, respectively.
Calculation results
In order to identify the effect of Cd atoms partially substituted by the Mn atoms to the thermoelectric performance of Cu2Cd1−xMnxSnSe4, we performed a density functional theory (DFT) study with spin polarization of the density of states (DOS) for our materials. The doping models are shown in Figure 5. The calculated band gap of Cu2CdSnSe4 is 0.8 eV, which is comparable to the experimental value of Eg = 0.96 eV10. For each x value, we have considered at least four substitution structures and found out there is little difference among them. Figure 6 shows the DOS of two spins for Cu2Cd1−xMnxSnSe4 with different substitution degrees. Our calculation clearly revealed a gradual shift of the Fermi level toward the valence band with Cd substituted by Mn and the Fermi level shift reached up to 0.23 eV at x = 0.75. By contrast, the band gap is nearly unchanged when the Cd atoms are substituted by the Mn atoms. We also calculate the Cu doping cases. The total density of states for Cu2+xCd1−xSnSe4 with different substitution degrees are shown in Figure S5, which agree with the DFT calculations conducted by Maria et al.6. The Mn doping effect is similar to Cu doping effect: both shifting the Fermi level to valence band except that the degree of shift for Mn doping is a little weaker than Cu doping.

Configurations of the intrinsic Cu2CdSnSe4 (left panel) cell and two different substitutions of Cu2Cd1−xMnxSnSe4 for x = 0.125 (middle panel) and 0.25 (right panel), respectively.
Arrows denote the substituted ions.

DFT calculation results.
Density of states for Cu2Cd1−xMnxSnSe4 when x = 0 (a), x = 0.50 (b) and x = 0.75 (c), respectively. (d) Fermi level shift (red line) and band gap (blue line) a function of the level of Cd substitution by Mn, respectively. The energy of the valence band maximum of intrinsic Cu2CdSnSe4 is set to 0.
The Fermi level shift downward to the valence band maximum (VBM) shows there is an p-type doping effect when Cd substituted by Mn for Cu2CdSnSe4. From Figure 6, we can see the DOS near the Fermi level is mainly from the up spin for Cu2+xCd1−xSnSe4, which indicates that the doped Mn atoms (3d and 4s orbitals) can contribute to the VBM, because all of the left element atoms show nearly zero magnetic moment according to our calculation results (the magnetic moment of Mn is 4.66 in units of 1/2 μB). This reason accounts for the Fermi level shift with Cd substituted by Mn for Cu2CdSnSe4. The p-type doping effect will increase the hole concentration and contribute to the electrical conductance, as suggested by our experimental results when Cd substituted by Mn at a degree of x = 0.05, 0.1, 0.15. In Figure 3, the electrical conductance first increases with the Mn content increasing from x = 0 to x = 0.05 and then nearly saturates with x = 0.05 to x = 0.1 and finally decreases with the Mn content increasing from x = 0.1 to x = 0.15. When the Mn doping is under low levels (x ≤–– 0.05), the increased electrical conductance is mainly attributed to the increasing intrinsic doping effect caused by the substitution of Cd with Mn. Higher levels of Mn doping not improving the electrical conductivity can be attributed to two reasons: first, the increasing Mn contents could introduce more impurity scattering centers, which will impede the electron transport and decrease the carrier mobility. This reason is also validated in our Hall measurements: the carrier mobility is decreased from 30.4 cm2V−1S−1 to 7.14 cm2V−1S−1 when the Mn doping level is increased from x = 0 to x = 0.15 for Cu2Cd1−xMnxSnSe4 (Table 1); second, it is possible that some of the Cd ions are replaced not by Mn but by Sn ions, which will lead to charge compensation and hinder the hole concentration increasing6.
Besides the electrical conductance, the Seebeck coefficient, the PF and the ZT value all show a nonmonotonic behavior with the increasing Mn doping content. In contrast to the electrical conductance, the Seebeck coefficient first decreases with the Mn content increasing from x = 0 to x = 0.05 and then nearly saturates with x = 0.05 to x = 0.1 and finally increases with the Mn content increasing from x = 0.05 to x = 0.15 (Figure 2(b)). In the first stage with Mn content increasing from x = 0 to x = 0.05, the deceased Seebeck coefficient is mainly attributed to the increased carrier concentration. However, as the Mn content increases, the DOS near the Fermi level also increases due to the Mn contribution to VBM, which will improve the Seebeck coefficient. When the Mn content increases from x = 0.05 to x = 0.15, this effect plus the charge compensation effect caused by the substitution of Cd with Sn and the decreased carrier mobility as mentioned in the last paragraph, would finally surpass the increasing intrinsic doping effect, thus leading to the increased Seebeck coefficient. Because the extent of variation for the electrical conductivity (σ) with the increasing Mn doping content is larger than that for the Seebeck coefficient, according to PF = S2σ, the behavior for the power factor with the increasing Mn doping content is similar to that for the electrical conductivity. At last, the extent of variation for the PF with the increasing Mn doping content is also larger than that for the thermal conductivity (Figure 4(a)), leading to that the behavior for the ZT value with the increasing Mn content is similar to that for the PF (according to ZT = S2σT/κ).
Conclusions
In sumary, we have investigated the Mn doping effect to the thermoelectric performance of Cu2CdSnSe4 experimentally and theoretically. The results of the Rietveld refinement of the X-ray diffraction patterns show that a and c decrease with the increase of the Mn content, revealing that Mn substitutes the Cd position in the structure. The electrical conductivity is increased about four times at 723 K, while the seebeck coefficient decreases slightly from 356 to 289 μV/K, resulting the significant increase of power factor by substituting Cd sites with Mn. Even though the thermal conductivity increases with the doping content of Mn, the ZT values still increase from 0.06 (x = 0) to 0.16 (x = 0.10). Our DFT calculation results prove that the intrinsic doping effect caused by the substitution of Cd with Mn can enhance the electrical conductivity. All our experimental and theoretical results prove that Mn doping by substituting Cd sites is an effective method to improve the thermoelectric performance of Cu2CdSnSe4. According to the experimental and first principles studies, it can be expected that optimizing the synthesis process and scaling Cu2Cd1−xMnxSnSe4 to nanoparticles may further improve the ZT value significantly by improving the electrical conductivity and enhancing the phonon scattering to decrease the thermal conductivity.
Methods
Synthesis
The stoichiometric compounds Cu2Cd1−xMnxSnSe4 (x = 0, 0.05, 0.1, 0.15) are prepared by the melting and the subsequent spark plasma sintering (SPS) method. The starting materials of the elements of Cu, Cd, Mn, Sn and Se with a purity of 99.99% mixture were sealed in the evacuated quartz tube and heated at 1237 K for homogeneity for 172 h in the muffle furnace and then cooled down to 723 K to react for 72 h and then subsequently quenched in the liquid nitrogen. The quenched alloys were powdered in the agate mortar and then ball-milled in a planetary ball millor (QM-4F, Nanjing University, China) by using a hard stainless steel vial and zirconia balls, at 200 rpm for 12 h. The weight ratio of ball to powder was kept at about 20:1 and the mill vial was evacuated and then filled with the purified H2 gas to prevent the powder from oxidation during the milling process. The milled powders were pressed as pills and then sealed in the evacuated quartz tube to react at 723 K for 172 h again and then the pills were ball-milled to powders. The powders were consolidated by SPS at 923 K for 5 min under an axial pressure of 48 MPa with a peak impulse value of 675 A to obtain the high density samples.
Thermoelectric measurements
The bar specimens with a typical dimension of 12.0 mm × 5.0 mm × 5.0 mm were prepared for the electronic property measurements and the disk specimens with 10.0 mm in diameter and 2.0 in thickness for the thermal conductivity measurements. XRD data were collected by a Bruker D8 Advance SS/18 kW diffractometer with the Cu Kα radiation. Accurate lattice parameters were got by the Rietveld refinement method with Topas 3.1 software23. The Seebeck coefficient (S) and the electrical resistivity (ρ) were measured by using ZEM-2 (Ulvac-Riko, Japan) in the helium atmosphere. The thermal conductivity (κ) was calculated by using the equation κ = λCpd, where λ is the thermal diffusivity, Cp is the heat capacity and d is the bulk density of the sample. The thermal diffusivity was measured by a laser flash technique (NETZSCH LFA457) in the argon atmosphere. The bulk density of the sample was calculated by the Rietveld analysis and the realistic density was calculated by the principle of the floating bodies of Archimedes.
Computational details
In order to model the Cd substitution by Mn, a 1 × 2 × 2 supercell based on the unit cell was constructed, allowing us to investigate different substitution degrees of Cu2Cd1−xMnxSnSe4, with x ranging from x = 0 to 0.75 in steps of Δx = 0.125, as shown in Figure 5. All calculations are performed using the plane-wave projector-augmented wave method24,25 as implemented in the Vienna ab initio simulation package26,27. The Perdew-Burke-Ernzerhof (PBE)28 form of generalized gradient approximation (GGA) is chosen as the exchange-correlation potential. Structural properties and electronic properties are calculated by the PBE + U approach29, with a U = 4 eV on Cu 3d and Cd 4d states and U = 6 eV on Mn 3d states. All calculations were performed with spin polarization. To obtain reliable optimized structures, the maximum residual force is less than 0.01 eV/Å and energies are converged to within 5 × 10−6 eV per atom and the k-point mesh is set to 3 × 3 × 5. The k-point mesh is set to 10 × 10 × 5 to calculate electronic properties. An energy cut-off of 400 eV was used in all cases.
References
Liu, M. L., Chen, I. W., Huang, F. Q. & Chen, L. D. Improved thermoelectric properties of Cu-doped quaternary chalcogenides of Cu2CdSnSe4 . Adv. Mater. 21, 3808–3812 (2009).
Sevik, C. & Cagin, T. Assessment of thermoelectric performance of Cu2ZnSnX4, X = S, Se and Te. Appl. Phys. Lett. 95, 112105 (2009).
Shi, X. Y., Huang, F. Q., Liu, M. L. & Chen, L. D. Thermoelectric properties of tetrahedrally bonded wide-gap stannite compounds Cu2ZnSn1-xInxSe4. Appl. Phys. Lett. 94, 122103 (2009).
Fan, F. J. et al. Colloidal synthesis of Cu2CdSnSe4 nanocrystals and hot-pressing to enhance the thermoelectric figure-of-merit. J. Amer. Chem. Soc. 133, 15910–15913 (2011).
Fan, F. J., Wang, Y. X., Liu, X. J., Wu, L. & Yu, S. H. Large-scale colloidal synthesis of non-stoichiometric Cu2ZnSnSe4 nanocrystals for thermoelectric applications. Adv. Mater. 24, 6158–6163 (2012).
Ibanez, M. et al. Composition control and thermoelectric properties of quaternary chalcogenide nanocrystals: the case of stannite Cu2CdSnSe4 . Chem. Mater. 24, 562–570 (2012).
Ibanez, M. et al. Cu2ZnGeSe4 nanocrystals: synthesis and thermoelectric properties. J. Amer. Chem. Soc. 134, 4060–4063 (2012).
Raju, C. et al. Thermoelectric properties of chalcogenide based Cu2+xZnSn1−xSe4 . Aip Advances 3, 032106 (2013).
Zeier, W. G. et al. Phonon scattering through a local anisotropic structural disorder in the thermoelectric solid solution Cu2Zn1−xFexGeSe4 . J. Amer. Chem. Soc. 135, 726–732 (2013).
Matsushita, H., Maeda, T., Katsui, A. & Takizawa, T. Thermal analysis and synthesis from the melts of Cu-based quaternary compounds Cu–III–IV–VI4 and Cu2–II–IV–VI4 (II = Zn,Cd; III = Ga, In; IV = Ge, Sn; VI = Se). J. Cryst. Growth 208, 416–422 (2000).
Chiang, M. H., Fu, Y. S., Guo, T. F., Liu, H. L. & Lin, W. T. Effects of Zn precursors on solvothermal synthesis of Cu2ZnSnSe4 nanocrystals. Mater. Lett. 83, 192–194 (2012).
Fan, F. J. et al. Linearly arranged polytypic CZTSSe nanocrystals. Sci. Rep. 2, 952 (2012).
Olekseyuk, I. D. et al. Single crystal preparation and crystal structure of the Cu2Zn/Cd, Hg/SnSe4 compounds. J. Alloy Compd. 340, 141–145 (2002).
Sachanyuk, V. P., Olekseyuk, I. D. & Parasyuk, O. V. X-ray powder diffraction study of the Cu2Cd1−xMnxSnSe4 alloys. Phys. Status Solidi A 203, 459–465 (2006).
Chen, S. Y. et al. Wurtzite-derived polytypes of kesterite and stannite quaternary chalcogenide semiconductors. Phys. Rev. B 82, 195203 (2010).
Yao, J. et al. Thermoelectric properties of p-type CuInSe2 chalcopyrites enhanced by introduction of manganese. Phys. Rev. B 84, 075203 (2011).
Zhang, H. et al. Synthesis and thermoelectric properties of Mn-doped AgSbTe2 compounds. Chin. Phys. B 21, 106101 (2012).
Lu, X. & Morelli, D. T. Thermoelectric properties of Mn-doped Cu2SnSe3 . J. Electron. Mater. 41, 1554–1558 (2012).
Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751–767 (1976).
Heinrich, C. P., Day, T. W., Zeier, W. G., Snyder, G. J. & Tremel, W. Effect of isovalent substitution on the thermoelectric properties of the Cu2ZnGeSe4−xSx series of solid solutions. J. Amer. Chem. Soc. 136, 442–448 (2013).
Zeier, W. G. et al. Influence of a nano phase segregation on the thermoelectric properties of the p-type doped stannite compound Cu2+xZn1−xGeSe4 . J. Amer. Chem. Soc. 134, 7147–7154 (2012).
Zeier, W. G. et al. Bond strength dependent superionic phase transformation in the solid solution series Cu2ZnGeSe4−xSx . J. Mater. Chem. A 2, 1790–1794 (2014).
Bruker AXS, TOPAS V2.1: General profile and structure analysis software for powder diffraction data. - User's Manual (Bruker AXS, Karlsruhe, Germany, 2003).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943–954 (1991).
Acknowledgements
F.S.L. and J.Q.L. thank the support of the National Natural Science Foundation of China (Nos: 51171117 and 51003060) and Shenzhen Science and Technology Research Grant (No. JCYJ20130329104944356, JCYJ20120613115247045) and Foundation for Distinguished Young Talents in Higher Education of Guangdong, China (No. LYM10119). J.X.Z. and F.P. thank the support of Shenzhen Science and Technology Research Grant (No. ZDSY20130331145131323, CXZZ20120829172325895, JCYJ20120614150338154). We also acknowledge the support of ShenZhen National SuperComputing Ceter.
Author information
Authors and Affiliations
Contributions
F.S.L. and J.Q.L. designed and conduted the experiments. M.J. Huang, L.P.H. and W.Q.A. assisted with the experiments. J.X.Z. and F.P. performed DFT calculation. The data analyses were performed by F.S.L., J.X.Z., F.P. and J.Q.L. L.P.H. and W.Q.A. took part in discussion. This manuscript was written by J.X.Z. and F.S.L. All authors reviewed this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Liu, F., Zheng, J., Huang, M. et al. Enhanced Thermoelectric Performance of Cu2CdSnSe4 by Mn Doping: Experimental and First Principles Studies. Sci Rep 4, 5774 (2014). https://doi.org/10.1038/srep05774
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05774
This article is cited by
-
Improving the Thermoelectric Performance of Tetrahedrally Bonded Quaternary Selenide Cu2CdSnSe4 Using CdSe Precipitates
Journal of Electronic Materials (2019)
-
Heterovalent Substitution to Enrich Electrical Conductivity in Cu2CdSn1-xGaxSe4 Series for High Thermoelectric Performances
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
