
Abstract
Global threats of ssDNA geminivirus and ss(-)RNA tospovirus on crops necessitate the development of transgenic resistance. Here, we constructed a two-T DNA vector carrying a hairpin of the intergenic region (IGR) of Ageratum yellow vein virus (AYVV), residing in an intron inserted in an untranslatable nucleocapsid protein (NP) fragment of Melon yellow spot virus (MYSV). Transgenic tobacco lines highly resistant to AYVV and MYSV were generated. Accumulation of 24-nt siRNA, higher methylation levels on the IGR promoters of the transgene and suppression of IGR promoter activity of invading AYVV indicate that AYVV resistance is mediated by transcriptional gene silencing. Lack of NP transcript and accumulation of corresponding siRNAs indicate that MYSV resistance is mediated through post-transcriptional gene silencing. Marker-free progenies with concurrent resistance to both AYVV and MYSV, stably inherited as dominant nuclear traits, were obtained. Hence, we provide a novel way for concurrent control of noxious DNA and RNA viruses with less biosafety concerns.
Similar content being viewed by others
Introduction
Global threats of crop diseases caused by ssDNA geminiviruses, such as beet curly top, cassava mosaic, cotton leaf curl, maize streak and tomato leaf curl viruses, have led to tremendous economic losses1. Thrips-borne ssRNA tospoviruses also cause serious damages to many economically important crops worldwide2,3.
Whitefly-borne viruses of the genus Begomovirus, the largest genus of the family Geminiviridae4, contain one or two genomic components of circular ssDNA with a bidirectional promoter located in the intergenic region (IGR) for leftward and rightward transcription. The IGRs of geminiviuses have a common region (CR) of about 230 bp that contains motifs required for the control of replication and gene expression5. These include a stem-loop structure containing the highly conserved nonanucleotide TAATATTAC that functions in the initiation of rolling circle replication and the conserved reiterated sequences located upstream of the stem-loop required for specific recognition and binding by replication protein (Rep) during replication6.
The ssRNA genome of a tospovirus is tripartite, with a negative sense L (large) RNA and two ambisense M (medium) and S (small) RNAs. The L RNA codes for the replicase L protein7. The M RNA codes for the movement protein NSm8 and the precursor to the membrane glycoproteins GN and GC embedded in membrane envelope for vector transmission9. The S RNA codes for the nucleocapsid protein (NP) for viral RNA encapsidation and a nonstructural (NSs) protein that forms filamentous inclusion bodies10 and functions as RNA silencing suppressor11.
The different genetic structures and molecular mechanisms of geminiviruses and tospoviruses necessitate applications of transcriptional gene silencing (TGS)- and post-transcriptional gene silencing (PTGS)-, respectively, for their control. Both TGS and PTGS depend on small interfering RNAs (siRNA) or microRNAs (miRNA) that are produced from double-stranded RNA (dsRNA) precursors. TGS occurs in nuclei via RNA-directed DNA methylation (RdDM) at CG, CHG and CHH sequence contexts (where H = A, T or G), whereas PTGS operates in the cytoplasm through mRNA cleavage or inhibition of translation12,13,14.
PTGS-mediated transgenic resistance has been well applied for the control of plant RNA viruses15, including tospoviruses, by siRNA targeting NP gene16,17,18, NSs gene19, NSm gene20 and by artificial micro RNA targeting the conserved regions of L gene21. However, in the case of ssDNA geminiviruses, most attempts using PTGS approach targeting different genes of the viral genome, such as coding regions for replication-associated protein22, movement protein23, coat protein24, or transcriptional activator protein25, have only resulted in short delay in symptom development or reduced disease severity. However, transgenic tomato plants harboring 81 nt of the non-coding IGR plus 426 nt of the 5′ end of the Rep gene of Tomato yellow leaf curl virus (TYLCV) was found to confer resistance against TYLCV26. Hence, TGS-based transgenic strategies are considered more effective than PTGS for controlling geminiviruses.
TGS can be triggered by ectopic expression of specific RNA sequence to induce DNA methylation at the targeted promoter region27. In plant nuclei, the 24-nt siRNAs are processed from dsRNA by Dicer-like 3 (DCL3) and predominantly loaded into Argonaute 4 (AGO4)28,29 to guide RdDM pathway. Thus, a construct generating a hairpin RNA sequence (int-hpRNA) targeting a specific promoter, residing in an intron sequence, is able to be processed to trigger specific RdDM on the targeted promoter sequence for transcription suppression in transgenic tobacco plants30. Similar transgene construct is expected to trigger specific RdDM on geminivirus promoter and incapacitate the virus.
The transgenic plants carrying a marker gene of selectable antibiotic- or herbicide-resistant genes likely to cause potential risks to ecology and also are concerns for food safety. Thus, the selection marker genes are encouraged to be eliminated and appropriate technologies to remove them have been developed31,32. Agrobacterium-mediated transformation using a two-T-DNA binary vector carrying a transgene and selection marker gene in two different T-DNAs with their own sets of left and right T-DNA border sequences causes insertion of the marker gene and the transgene in different loci of host genome. The transgene and selection marker genes tend to segregate in the progeny after selfing and individual transgenic plants only for the transgene can be selected33. Such two-T-DNA approach has been used elsewhere for generation of several marker-free transgenic plants, including the major crops of soybean, tomato, wheat maize and rice32. Similar approach was used in the present study for generation of marker-free transgenic model plant Nicotiana benthamiana concurrently resistant to geminivirus and tospovirus. Presently, the same approach is being extended to the real crop tomato.
Ageratum yellow vein virus (AYVV) is a monopartite begomovirus, widely distributed in Southeast Asia34. The AYVV DNA A component can systemically infect the weed host of Ageratum conyzoides L., French bean and tomato and induces severe leaf curl symptoms in these hosts35. However, no transgenic resistance has been reported for AYVV so far. Similarly, Watermelon silver mottle virus (WSMoV)36 and Melon yellow sport virus (MYSV)37 are two tospoviurses threatening the cultivation of cucurbits in Taiwan, Japan and Southeast Asian countries3. Transgenic resistance in watermelon carrying a single chimeric construct containing the partial NP gene of WSMoV has been reported38, but transgenic resistance to MYSV has not been reported.
In the transgene constructed in this study, a hairpin construct of AYVV IGR was placed in an intron of Arabidopsis to mediate RdDM of IGR of AYVV infecting transgenic plants. This int-hpIGR construct was inserted into untranslatable MYSV nucleocapsid protein (NP) coding sequence. Following splicing of int-hpIGR region from transgenic transcript as an intron, the NP sequence region was released as an exon to induce PTGS against MYSV. After selfing of selected transgenic lines, marker-free transgenic plants conferring concurrent resistance to both AYVV and MYSV underlying TGS and PTGS mechanisms, respectively, were generated. Thus, our approach provides a valuable way for generating marker-free transgenic resistance for control of a ssDNA virus and a ssRNA virus at the same time, also eases the biosafety concerns for the selection marker.
Results
Generation of the construct MY-int-hpIGR-NP and the fidelity of splicing
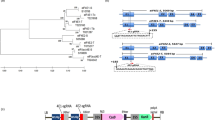
A two-T-DNA binary vector with the nptII selection marker and the target gene containing their own T-DNA border sequences was constructed for generation of marker-free transgenic plants. To induce RdDM, the primary construct int-hpIGR containing a hairpin structure of IGR of AYVV was first constructed (Fig. 1a). The IGR (284 bp) flanked by 54 bp corresponding to N-terminal 18 amino acids of C1 gene coding for viral replication protein that initiates rolling circle replication and 56 bp corresponding to N-terminal 19 amino acids of V2 gene coding for movement protein of AYVV was linked with the inverted repeat of the same by a 96 bp (nt 142–257 At3947160) spacer. This int-hpIGR construct was further inserted into an untranslatable NP sequence (840 bp) of MYSV (Fig. 1a), which acted as exonic sequences after splicing, to form the construct MY-int-hpIGR-NP that was intended to induce transgenic resistance against MYSV based on PTGS mechanism after splicing. In order to compare the transgenic resistance to AYVV conferred by this primary construct with that conferred by a regular PTGS approach, the constructs of IGR, hpIGR and untranslatable MYSV NP were generated in a conventional way without an inserted intron (Fig. 1a). All these constructs were further constructed in the expression cassette of the two-T-DNA binary vector, in which the kanamycin selection marker and the transgene have their own T-DNA border sequences (see Supplementary Fig. 1), to form pk2T-MY-int-hpIGR-NP, pk2T-hpIGR, pk2T-IGR and pk2T-MYSV-NP (Fig. 1a), which were then separately transferred into Agrobacterium tumefaciens strain ABI. Individual constructs were used to transform tobacco (Nicotiana benthamiana Domin) plants via agroinfiltration and the corresponding transgenic lines were regenerated.

Construction of different transgenes in pK2T binary vector and analysis of transcript splicing in transgenic tobacco plants.
(a) Physical map of individual constructs. LB: T-DNA left border; 2X35S-P: Cauliflower mosaic virus (CaMV) double 35S promoter; ★★: stop codons; MY-: 5′ part of MYSV-NP coding sequence; AT-In-: 5′ part of the intron of gene At3947160 of Arabidopsis thaliana; IGR: fragment of the intergenic region (IGR) flanked by 54 bp of C1 gene at the right (shaded in dark gray) and 56 bp of V2 gene at the left (shaded in light gray) of Ageratum yellow vein virus (AYVV); spacer: a 96 bp fragment of the middle part of the At3947160 intron; Inverted IGR: the fragment of the inverted IGR repeat of AYVV; -tron: 3′ part of the At3947160 intron; -SV-NP: 3′ part of the MYSV-NP coding sequence; 35S-T: CaMV 35S terminator; RB: T-DNA right border; nos-P: nopaline synthase gene promoter; nptII: neomycin phosphotransferase gene; nos-T: nos terminator. pK2T-MY-intron -NP: A positive control for confirmation of the action of the splicing process. (b) RT-PCR analysis for the splicing of transcripts from individual constructs, using primers targeting regions flanking the intron sequence. RNAs extracted at 3 dpi from tobacco leaves agroinfiltrated with individual constructs of pK2T-MYSV-NP, pK2T-MY-intron -NP and pK2T-MY-int-hpIGR-NP were analyzed by RT-PCR to examine the splicing of the intron. Total DNAs extracted from the leaf tissues agroinfiltrated with individual constructs were analyzed by PCR as un-spliced controls.
In order to examine whether the hairpin construct was properly processed, total RNAs were extracted from tobacco leaves at 3 days after agroinfiltration with individual constructs. RT-PCR using specific primers of MYSV NP gene, targeting regions flanking the intron sequence, revealed a fragment of 0.8 kb similar to the control pK2T-MYSV-NP which contains no intron, from the tissues agroinfiltrated with pk2T-MY-intron -NP (an untranslatable MYSV NP with the intron as a positive control) or pk2T-MY-int-hpIGR-NP. For un-spliced controls, total DNAs extracted from leaf tissues agroinfiltrated with individual constructs of pk2T-MY-intron -NP and pk2T-MY-int-hpIGR-NP (Fig. 1a) were amplified by PCR using the same primers; products of 1.5 kb and 2.2 kb, respectively, were noticed (Fig. 1b).
When the RT-PCR amplified 0.8 kb fragment was sequenced, the result indicated that the intron was spliced at the correct sites of pk2T-MY-int-hpIGR-NP to form the untranslatable MYSV NP transcript in tobacco plants (see Supplementary Fig. 2).
Greenhouse evaluation of resistance to AYVV
IGR, hpIGR and MY-int-hpIGR-NP transgenic tobacco lines were obtained and evaluated by challenge inoculation with the infectious construct pAYVV by agroinfection under greenhouse conditions. The results are summarized in Table 1. All AYVV-inoculated non-transgenic plants developed leaf curl symptom 10 days post-agroinfection (dpa). The delay-type resistant lines were defined as more than 30% individuals developed delay in symptom development as compared to that of the non-transgenic plants. At 20 dpa, 28% of the MY-int-hpIGR-NP lines showed resistance, while only 4% of the hpIGR lines and none (0%) of the IGR lines showed resistance (Table 1). Among the MY-int-hpIGR-NP lines, two lines showed symptoms at 26 dpa (16 days delay) and one line, MY-int-hpIGR-NP-6, showed symptoms at 31 dpa (21 days delay) (Table 1). One hpIGR line, hpIGR-3, showed symptoms at 21 dpa (11 days delay), while the best IGR line, IGR-2, showed symptoms at 16 dpa (only 6 days delay).
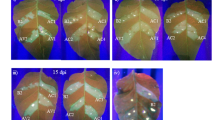
AYVV infection on all transgenic plants with symptoms was confirmed by PCR with AYVV C4 gene-specific primers during the test period of 38 days following inoculation. At 27 dpa, various degrees of leaf curl symptom were observed on leaves of AYVV-inoculated transgenic lines: plants of MY-int-hpIGR-NP-6 line were symptomless in contrast to line hpIGR-3 that showed mild leaf curl symptom and line IGR-2 that showed severe leaf curl symptom (Fig. 2a). Hence, our results indicated that the construct MY-int-hpIGR-NP confers a better degree of transgenic resistance to AYVV than those conferred by the constructs of hpIGR and IGR.

Greenhouse evaluation of transgenic tobacco lines against Ageratum yellow vein virus (AYVV) and Melon yellow spot virus (MYSV).
(a) Transgenic lines showed significant delay in symptom development as compared to non-transgenic (NT) plants when challenged with AYVV. Mock, a NT plant inoculated with buffer. MY-int-hpIGR-NP-6, hpIGR-3 and IGR-2 indicate transgenic lines transformed with the constructs pK2T-MY-int-hpIGR-NP, pK2T-hpIGR and pK2T-IGR, respectively. All the inoculated NT plants developed leaf curl symptom 10 days post-agroinfection (dpa). The hpIGR-3, IGR-2 and NT plants showed leaf curl symptom at 27 dpa, while the plants of MY-int-hpIGR-NP-6 line was symptomless. (b) Northern blot analysis for the detection of IGR siRNAs in MY-int-IGR-NP, hpIGR and IGR lines. Ribosomal RNA (5S) and tRNA were used as loading controls. Different resistant (R) lines showed different days (the number in parentheses) of delay in symptom development were used for siRNA analysis. S: susceptible line. (c) Evaluation of transgenic MY-int-hpIGR-NP and MYSV-NP lines by mechanical inoculation with MYSV. All NT plants developed leaf yellow spot and mosaic symptoms at 8 dpi. The photographs were taken at 20 dpi. HR: highly resistant, all inoculated plants did not show symptoms and were ELISA negative to MYSV NP. MR: moderately resistant, symptom development delayed up to 10 days. S: susceptible. (d) Northern blot analysis for the detection of MYSV-NP transcript and siRNAs in MY-int-hpIGR-NP and MYSV-NP lines before inoculation. (e) Southern blotting analyses of MY-int-hpIGR-NP transgenic lines. Susceptibility (S) and resistance (R) were analyzed comparing to the negative control of a NT plant. The putative insert numbers of the transgene are indicated.
Accumulation of 24-nt siRNA was correlated to higher degrees of AYVV transgenic resistance
In the selected MY-int-hpIGR-NP-6, hpIGR-3 and IGR-2 transgenic lines, which respectively showed 21, 11 and 6 days delay in development of leaf curl symptom, the accumulation of siRNA was detected by northern blotting using an IGR probe (Fig. 2b, left panel). However, the accumulation of siRNAs was not noticed for the susceptible lines MY-int-hpIGR-NP-2, hpIGR-7 and IGR-11 (Fig. 2b, left panel). The autoradiograph of a larger gel with longer electrophoresis revealed accumulation of 24-nt siRNA as the dominant siRNA for line MY-int-hpIGR-NP-6, 24-nt and 21-nt siRNAs for line hpIGR-3 and 21-nt siRNA for line IGR-2 (Fig. 2b, right panel). Thus, here our results indicated that the higher degrees of resistance to AYVV in MY-int-hpIGR-NP-6 and hpIGR-7 lines are correlated to the accumulation of 24-nt siRNA.
Methylation levels of IGR region of the transgene are correlated to levels of transgenic resistance to AYVV
Genomic DNAs of resistant or susceptible lines carrying transgene MY-int-hpIGR-NP or hpIGR were analyzed by bisulfite sequencing for examining the status of methylation on the transgene. When pMY-int-hpIGR-NP or phpIGR was mixed with genomic DNA of healthy plant and used as controls, no methylation was observed in PCR products. In contrast, the amplified 247-bp (IGR-247) fragment, which reflects the IGR containing the leftward promoter region and 54 bp of the 5′-C1 coding sequence, from the MY-int-hpIGR-NP resistant lines (lines 6 and12) reached an average methylation rate of 92 ± 2%, while that of the MY-int-hpIGR-NP susceptible lines (lines 2 and 19) showed only an average methylation rate of 40 ± 2%, from three independent repeats. In susceptible lines, average methylation rate at asymmetric CHH (where H = A, T or G) sites was low as 25 ± 1%, while in resistant lines the average methylation rate of CHH was 91 ± 2%. Methylation did not spread into intron sequence flanking the IGR sequence, as reflected by the results that all cytosines within the intron/spacer were not methylated (Fig. 3a).

Methylation of the transgene analyzed by bisulfite sequencing and fluorescence assay of rightward promoter activity of an auto-replicating vector of Ageratum yellow vein virus (AYVV) after agroinfiltration on transgenic tobacco lines.
(a) Physical map of the MY-int-hpIGR-NP transgene, in which the regions analyzed by bisulfite sequencing are indicated. The 247 bp (IGR-247) and the 350 bp (IGR-350) fragments were analyzed by the primer pairs BisP1/BisM1and BisP2/BisM2, respectively. Circles indicate sites of methylation identified from resistant MY-int-hpIGR-NP lines (6 and12) and squares indicate sites of methylation identified from the susceptible MY-int-hpIGR-NP lines (2 and19). Open circles (for resistant lines) and squares (for susceptible lines) represent 0–25% methylation of cytosines, half-filled symbols 26–50%, three-quarters-filled symbols 51–75% and full-filled symbols 76–100%. Sequencing data were averaged from three repeats. TAATATT↓AC: the conserved replication origin, TATA box: an essential motif of predicted promoter. Translation start sites of C1and V2 ORFs are also boxed. (b) Analysis of the transcriptional activity of an AYVV-derived auto-replicating GFP expression vector pAY-ST (described in Supplemental Experimental Procedure). AYVV shutter vector carrying GFP driven by the IGR rightward promoter was used to infect MY-int-hpIGR-NP, hpIGR and non-transgenic (NT) lines by agroinfiltration and GFP expression was examined at 1.5 and 2.5 days. (c) Relative fluorescence intensity of GFP in transgenic MY-int-hpIGR and hpIGR using pAY-ST vector at 1.5 days post-agroinfiltration. The ANOVA analysis was conducted by measuring four-time repeats for each sample. The significant differences in fluorescence intensity are indicated (P < 0.05).
When the amplified 350 bp (IGR-350) fragment, which reflects the whole IGR containing the leftward and the rightward promoter regions, 54 bp of 5′-C1 coding region and the 9 bp of the 5′-V2 coding region, was analyzed, the methylation of the IGR sequence from resistant lines reached an average rate of 93 ± 1%, while the susceptible lines showed an average rate of 37 ± 2%. In susceptible lines, methylation rate at asymmetric CHH sites was only 22 ± 3%, while in resistant lines the average methylation rate of CHH was 91 ± 1%.
We also analyzed the methylation status in IGR-247 and IGR-350 fragment of hpIGR lines. The hpIGR transgene in hpIGR-3 resistant line was methylated up to 80–100%, whereas hpIGR-7 susceptible lines showed lower methylation rates of 35–40%, especially at asymmetric CHH sites was only 20–30%. Thus, our results demonstrated that RdDM derived from the construct MY-int-hpIGR-NP specifically targets at the IGR sequence of the transgene and the resulted higher levels of methylation are correlated to higher levels of the transgenic resistance to AYVV.
Transcriptional activity of IGR promoter of auto-replicating AYVV is suppressed in transgenic lines
In order to examine whether the siRNA induced RdDM suppresses IGR promoter to reduce the activity of an invading begomovirus, the auto-replicating AYVV vector pAY-ST (see Supplementary Fig. 3) with GFP driven by AYVV rightward promoter was constructed and introduced into transgenic MY-int-hpIGR-NP and hpIGR lines by agroinfiltration. A defective variant pAY-ST-d lacking C2 and C3 genes was also constructed. Another vector of pAYIGR-GFP, in which only one IGR was present, was used as non-auto-replicating control (see Supplementary Fig. 3). Following agroinfiltration, both pAY-ST and pAY-ST-d were auto-replicating in vivo, as evidenced by the products of 2116 and 1667 bp, respectively, which were amplified from a circular replicating form of dsDNA, with the primers P-GFP-625 and M-GFP-82 targeting at the 3′ and 5′ portions of the GFP-ORF in opposite orientations (see Supplementary Fig. 3). In pAYIGR-GFP (non-auto-replicating) and pAY-ST-d (auto-replicating) infiltrated plants, the GFP expression was not detected (see Supplementary Fig. 3). Therefore, our results indicated that GFP expressed by the rightward promoter of pAY-ST was mainly resulted from the rolling-circle DNA replication and not from the construct inserted in host genome.
The PCR-amplified IGR-350 of MY-int-hpIGR-NP construct covers the complete rightward and leftward promoter sequences of IGR (Fig. 3a). At 1.5 days after agroinfilitration, the GFP expression in the infiltrated areas of resistant lines MY-int-hpIGR-NP-6 and 12 and hpIGR-3 were significantly reduced as compared to that of susceptible lines MY-int-hpIGR-NP-2 and 19 and non-transgenic plants (Fig. 3b). However, at the 2.5 days after agroinfiltration, the GFP expression of the resistant lines increased up to levels similar to that of the pAY-ST control in non-transgenic plants (Fig. 3b). From four repeated experiments, relative fluorescence intensity of the resistant lines MY-int-hpIGR-NP-6, MY-int-hpIGR-12 and hpIGR-3 were more than 2–3-fold lower than that of the susceptible lines MY-int-hpIGR-NP-2 and 19 and non-transgenic plants at 1.5 days post-infiltration (Fig. 3c). Thus, here we demonstrate that in the lines resistant to AYVV, the capability to drive GFP by the rightward promoter of the auto-replicating AYVV was reduced, as compared to the full promoter activity in the susceptible lines and the non-transgenic control.
Evaluation of resistance to MYSV
Transgenic tobacco lines transformed with individual constructs were evaluated by challenge inoculation with MYSV under greenhouse conditions. The results are summarized in Table 2. All the infected non-transgenic plants, hpIGR and IGR lines developed symptom of yellow spots and mosaic on upper leaves 8 days post-inoculation (dpi), followed by wilting at 20 dpi (Fig. 2c). Two MYSV-NP lines showed 10 days delay in symptom development and line MYSV-NP-5 did not show any symptoms up to 38 dpi. Three AYVV-resistant MY-int-hpIGR-NP lines showed 10 days delay in symptom development and among them, line MY-int-hpIGR-NP-6 was found to be resistant to MYSV infection at 38 dpi (Table 2 and Fig. 2c). MYSV accumulation was not detected in lines MYSV-NP-5 and MY-int-hpIGR-NP-6 by indirect ELISA during 5 to 38 dpi, indicating that these two lines are completely resistant to MYSV.
Transgenic resistance to MYSV is mediated by PTGS
The results of northern blotting analyses for detection of the transgene transcript and siRNA in MYSV-NP and MY-int-hpIGR-NP lines before inoculation are shown in Fig. 2d. The two completely resistant lines, MYSV-NP-5 and MY-int-hpIGR-NP-6 showed siRNA signal, but accumulation of transgene transcript was not detected (Fig. 2d). In contrast, accumulation of transgene transcript in the moderately resistant lines (MYSV-NP-7 and MY-int-hpIGR-NP-12) and susceptible lines (MYSV-NP-8, MY-int-hpIGR-NP-2 and 19) was noticed, but siRNA signal was not detected. In these MY-int-hpIGR-NP lines, transcripts of 2.2 kb and 0.8 kb, corresponding to the unspliced and spliced forms of the untranslatable NP gene transcript, respectively, were noticed. Thus, here our results indicated that the untranslatable NP construct of MY-int-hpIGR-NP-6 line confers complete resistance to MYSV through PTGS mechanism, as evidenced by the silencing of the NP transcript and the accumulation of the transgenic siRNA.
Southern blotting analysis
When the MYSV NP probe was used in Southern blotting analysis, the results showed that the transgene MY-int-hpIGR-NP was integrated into the genome of the line MY-int-hpIGR-NP-6 at a single locus, while in those of the lines MY-int-hpIGR-NP-12, 2 and 19 at eight, five and twelve loci, respectively (Fig. 2e).
Segregation analysis
A total of 90 T1 seedlings obtained from the selfing of the AYVV-resistant MY-int-hpIGR-NP-6 were evaluated by agroinfection with AYVV. As shown in Table 3, 64 of the seedlings were PCR-positive for the transgene MY-int-hpIGR-NP, while the other 26 seedlings were negative, indicating a 3:1 segregation. Ten days after inoculation, 55 resistant plants were symptomless and all of them were PCR-positive for the transgene MY-int-hpIGR-NP, whereas all the other plants displayed severe symptoms (Table 3). A total of 23 resistant plants (26%) displayed symptoms during 41–50 dpa. The longer delay in symptom development was apparently due to the homozygotic combination of the single-insert transgene in the T1 population.
Another set of 110 T1 seedlings from the selfing of the line MY-int-hpIGR-NP-6 were analyzed by PCR with MY-int-hpIGR-NP primers and their transgenic resistance was evaluated by challenge inoculation with MYSV. As shown in Table 3, 79 of the seedlings were PCR-positive for the transgene, while the other 31 plants were negative, also indicating a 3:1 segregation. When the PCR-positive seedlings were inoculated with MYSV, none of the 79 plants showed symptoms and all were ELISA negative, when tested with MYSV NP antiserum at 50 dpi. All the other 31 plants without the transgene displayed severe symptoms within 8 days after inoculation.
Thus, the segregation analyses of T1 seedlings indicated that the single-inserted transgene in line MY-int-hpIGR-NP-6 is nuclearly inherited as a single dominant trait, conferring concurrent resistance to both AYVV and MYSV.
Double-virus resistant transgenic plants without nptII selection marker
Individual plants PCR-positive for the transgene MY-int-hpIGR-NP, but PCR-negative for nptII selection marker, were selected from T1 progeny of the resistant line MY-int-hpIGR-NP-6 (see Supplementary Fig. 4) to obtain T1 progeny of marker-free transgenic plants. When a total of 50 marker-free T2 plants, generated from the selfing of a selected T1 individual MY-int-hpIGR-NP-6-4, were tested by PCR with MY-int-hpIGR-NP specific primers, all of them showed the presence of the transgene, indicating that the T1 individual MY-int-hpIGR-NP-6-4 and its T2 progeny carry the homozygotic transgene.
When these T2 homozygous plants were agroinfected with AYVV, 15 plants showed 10-30 days delay in symptom development and the other 35 plants (70%) developed leaf curl symptom during 41–50 dpa. In comparison, all hemizygote of T0 plants showed symptoms within 31 dpa. AYVV infection in all the plants with symptoms was confirmed by PCR detection. When another set of 40 T2 homozygotic plants were mechanically challenged with MYSV, all of them showed complete resistance to MYSV at 50 dpi, as reflected by lack of symptom and MYSV ELISA negativity. The results indicated that the homozygotic T2 plants inherited complete resistance to MYSV from the T0 plants of the line MY-int-hpIGR-NP-6 (Table 3).
Taken all together, our results demonstrate that nptII selection marker gene was completely removed in individuals of T2 progeny of the resistant line MY-int-hpIGR-NP-6, in which the transgene is inherited as a single dominant nuclear trait conferring double resistance to both AYVV and MYSV. Furthermore, the homozygotic MY-int-hpIGR-NP-6 T1 and T2 plants confer higher degrees of resistance to AYVV than the hemizygotic T0 and T1 plants, as reflected in longer delay for symptom development.
Discussion
Due to the trends of global warming, rampancy of geminivirus vector whitefly and tospovirus vector thrips have promptly expanded from tropical areas into template zones, creating serious threats on many economically important crops4,39. In this study, we designed a novel binary vector pK2T carrying the transgene MY-int-hpIGR-NP and the selection marker nptII, each with its own T-DNA border sequences and used for Agrobacterium-mediated transformation of N. benthamiana plants. MY-int-hpIGR-NP transgenic tobacco lines showed delay in symptom development up to three weeks after agroinfection with AYVV. Accumulation of IGR 24-nt siRNA, higher methylation levels of IGR region of the transgene and suppression of IGR promoter activity on an auto-replicating AYVV in resistant lines indicate that the resistance to the DNA virus is resulted from siRNA-directed TGS. When challenged with MYSV, AYVV-resistant lines were also highly resistant to MYSV. Lack of expressed MYSV NP transcript and accumulation of corresponding siRNAs indicated that the resistance to the RNA virus is resulted from PTGS. Marker-free transgenic tobacco plants concurrently resistant to both AYVV and MYSV were obtained, stably inherited as dominant nuclear traits. Thus, our novel approach provides a valuable way for concurrent control of a ssDNA and a (-)ssRNA virus, also eases the biosafety concerns for the selection marker.
The IGR-specific 24-nt siRNAs are considered the products of DCL3 processing30 of the spliced int-hpIGR in the nucleus. Accumulation of AYVV IGR-probe-detectable 24-nt siRNA as the dominant species of siRNA in the MY-int-hpIGR-NP-6 transgenic line suggests the nuclear retention of the spliced int-hpIGR as previously reported30,40. Corroboratively, our results of bisulfite sequencing revealed de novo cis-methylation of the IGR sequence of the host genome-incorporated MY-int-hpIGR-NP transgene. The cis-methylation of IGR sequence was much higher in the AYVV-resistant MY-int-hpIGR-NP lines, including both leftward and rightward promoter regions, than that in the susceptible lines. Thus, higher degrees of AYVV-resistance of the MY-int-hpIGR-NP lines are positively correlated to the accumulation levels of 24-nt siRNAs, which efficiently triggers RdDM by de novo DNA methylation machinery on the corresponding sequences41,42. This phenomenon of RdDM is similar to that by a hairpin RNA construct residing in an intron of target promoter to trigger specific RdDM targeting the corresponding promoter region30. The MY-int-hpIGR-NP-6 resistant line also accumulated low amount of 21-nt siRNAs (Fig. 2b, right panel); thus the operation of PTGS in this line cannot be ruled out. Although the IGR's flanking with short C1 (54 bp) and V2 (56 bp) coding regions may confer a limited resistance mediated by PTGS, we believe that the transgenic resistance to AYVV is mainly conferred by 24-nt siRNAs that trigger RdDM.
RNA-mediated chromatin-based silencing in plants is controlled by several collaborative proteins mediating de novo cytosine methylation at symmetric CG/CHG and asymmetric CHH sites (where H = A, T or G)12. CHH methylation is mostly conducted by DRM2 and requires frequent methylation signals provided by the de novo methylation pathway43. Thus, methylated CHH is an indication for the ongoing de novo RdDM44,45,46. Our data showed that the AYVV-resistant lines has, but susceptible lines lack, this competent CHH methylation and de novo RdDM signal.
Following agroinfiltration, both pAY-ST and pAY-ST-d were auto-replicating in vivo, as evidenced by the PCR-amplified products from the replication of circular single-stranded DNA (see Supplementary Fig. 3). GFP expression from pAY-ST is dependent upon C2 and C3, as reflected by the absence of GFP expression from pAY-ST-d. The results shown in Supplementary Fig. 3 indicate that the GFP expression is mainly from auto-replicating pAY-ST, not from the linear form of inserted pAY-ST in the trasnsformed cells, because the GFP expression in plant tissues infiltrated with the pAY-ST-d and non-autoreplicating control pAYIGR-GFP was not detected. GFP expression from the leftward IGR promoter could not be assessed, because the leftward IGR promoter drives C1 gene that is indispensable for replication.
In pAY-ST-infiltrated plants, the GFP expression was observable only in the infiltrated leaf tissues. In Cotton leaf curl Kokhran virus, V2 protein was shown to interact with coat protein (CP, the V1 protein) and a role of the complex in virus movement was suggested47. Hence, the lack of systemic spreading of pAY-ST is apparently attributed to the replacement of indispensable V2 and V1 coding regions by the GFP ORF. Our results suggest that the IGR-specific 24-nt siRNAs detected in the AYVV-resistant MY-int-hpIGR-NP transgenic line trigger trans-methylation of the IGR rightward promoter of pAY-ST and suppress the GFP transcription, as recorded by the significant reduction in GFP expression 1.5 days after agroinfiltration (Fig. 3b). However, the GFP expression in these lines was enhanced 2.5 days after agroinfiltration, probably because the fast replication of the auto-replicating recombinant virus derived from pAY-ST increased copy numbers beyond the suppression capacity resulted from IGR-specific 24-nt siRNA.
The presently used in planta auto-replicating AYVV-based shuttle vector pAY-ST may be fast replicating similar to the Bean yellow dwarf virus mild strain (BeYDV-m)-based pRIC vectors that can amplify from the input level of 106 to 109 within 3 days post-inoculation48. Moreover, since pAY-ST was delivered by agroinfiltration, its initial copy number might be higher than that of the agro-delivered infectious clone pAYVV. Reflecting this disparity, AYVV-resistant lines inoculated with pAYVV were able to display three weeks delay in symptom development. However, in these evaluated plants, since AYVV was fast-proliferating from its cDNA clone pAYVV under the control of 35 S promoter, this level of resistance may predict far better performance against whitefly-borne AYVV under natural conditions.
In plants, RNAs that are inducers of PTGS can also mediate sequence-specific DNA methylation27,46,49. We also observed that the double stranded RNA from the transgene hpIGR designed for inducing PTGS also triggered RdDM and methylated transgene sequences. Since the 24 nt siRNA was also detected for hpIGR transgenic line (Fig. 2b, right panel), we believe that the higher degrees resistance to AYVV of hpIGR lines than the IGR lines, in terms of longer delay in symptom development, is due to the combination of PTGS and RdDM.
Though our present attempt did not evidence complete elimination of AYVV, the transgenic plants did show effective delay-type resistance, much longer than the PTGS controls that only exhibited short delay. In the segregation analysis, 26% and 70% of the T1 plants of MY-int-hpIGR-NP-6 and T2 plants of MY-int-hpIGR-NP-6-4, respectively, did not show symptoms at 40 dpi (Table 3). The higher resistance levels of the progeny, as reflected by longer delay in symptom development, is apparently due to the effect of homozygotism of the transgene in T1 (25%) and T2 (100%) plants resulted from the selfing of the parental plants carrying single insert of the tansgene (Fig. 2e). This is similar to the higher ratio of resistance of T2 plants than T1 plants of TYLCV CP transgenic lines50. The longer delay-type resistance of homozygotic MY-int-hpIGR-6 plants implies the effectiveness of MY-int-hpIGR-NP construct in economically important short-term crops.
In MY-int-hpIGR-NP-6 line, the untranslatable segments of MYSV NP sequence flanking the AYVV IGR-carrying intron of the transgene were precisely processed as exons. However, by northern blotting analysis, the mature untranslatable MYSV NP transcript was not detectable, while the corresponding 21-nt siRNA was detected. Thus, in MY-int-hpIGR-NP-6 line, following splicing from the primary transcript of MY-int-hpIGR-NP, the mature untranslatable MYSV transcript triggers PTGS and confers MYSV resistance similar to the resistance conferred by the MYSV NP transgenic plants. Our results show that the complete resistance to MYSV of the T0 plants of this line is inherited to T1 and T2 progenies carrying the transgene (Table 3). However, since the hemizygotic T0 and T1 plants already showed complete resistance, the effect of homozygotism of the transgene could not be assessed.
The present outcome opens a scope for intronically targeting other sequences of geminivirus, including the highly conserved Rep (replication) gene essential for viral replication and other internal promoters other than those in IGR. Also, our approach can be coupled with other new developments, such as expressing peptide aptamers that bind to geminiviral Rep protein to generate resistance to diverse geminiviruses51 and PTGS targeting the multifunctional AL2 (TrAP) gene that plays roles as a transcriptional activator, gene silencing suppressor and suppressor of basal defense5. Similarly, the exonic MYSV NP sequence of the MY-int-hpIGR-NP transgene that triggers PTGS and confers effective resistance against the tospovirus MYSV can be replaced by appropriate sequences for effective control of other tospoviruses and any other RNA viruses. Using MY-int-hpIGR-NP transgene, we have transformed also the real crop tomato and obtained several tomato lines concurrently resistant to the tospovirus MYSV and the begomovirus AYVV. These transgenic tomato lines are being analyzed under greenhouse conditions.
The concurrent double-resistance to whitefly-borne geminvirus and thrips-borne tospovirus, mediated by TGS at DNA and PTGS RNA level, respectively, does not involve protein production because of the appropriate engineering of the transgene. Moreover, the selection marker gene nptII can be successfully removed after selfing of the selected transgenic lines. Our approach also eases the biosafety concerns of toxicity and allergencity of transgenic proteins and avoids the potential biosafety and environmental risks from the selection marker. Thus, we have opened a new gate way for concurrent control of the globally devastating whitefly-borne DNA viruses and thrips-borne RNA viruses for many economically important crops.
Methods
Construction of two T-DNAs binary vector
We attempted to develop a two-T-DNA binary vector, in which the selection marker nptII gene and the target gene contain their own T-DNA border sequences. In this way, the selection marker and the target gene can be inserted at different loci of host genome and then segregated by selfing. All primers used in this study are listed in Supplementary Table 1. The selection marker nptII gene, nopaline synthase gene promoter (nos-P) and the right border (RB) sequence of T-DNA from pBI121vector (Clontech, Palo Alto, CA) were PCR amplified with the primer pair P-nptII-PstI-HindIII/M-RB-EcoRV and introduced into EcoRV/Pst I site of pBluescritpt II SK(-) (Agilent Technologies, California, USA) to generate p II SK-RB-nptII (Supplementary Fig. 1). Two intron sequences from A. thaliana, intron 1088 of gene At1G44960 (accession number NC_003070.9; base pairs 16999880–17000968) and intron H of gene AT3G18780 (accession number NC_003074.8; base pairs 6475076-6475527), were engineered as a spacer to separate the two T-DNAs. Intron1088 was PCR-amplified with the primer pair P-intron 1088- EcoRV/M-intron 1088 and intron H with LB fragment with the primer pair P-intron H/M-LB-intron H- Hind III-Xba I. The amplified intron 1088 and intron H- LB fragments were linked together by overlaping PCR with the primer pair P-intron 1088- EcoRV/M-LB-intron H-Hind III - Xba I. This amplified fragment was digested with EcoRV/Xba I and inserted into p II SK-RB-nptII vector to generate p II SK-LB-intron H-intron 1088-RB-nptII, which were then digested with Hid III and introduced into pBCo-DC-YFP binary vector52 to generate pk2T binary vector (see Supplementary Fig. 1).
Construction of target genes in two T-DNAs binary vector
In this study, we attempted to generate a construct to confer transgenic resistance to AYVV by the mechanism of TGS and to MYSV by PTGS. An intron fragment of gene At3947160 of A. thaliana (accession number AL133292.3, bps 79239–78574) was amplified by PCR with the primer pair P-AT int-NdeI/M-AT int-NheI. The amplified fragment was digested with NdeI and NheI and inserted into the vector pENTR™/D-TOPO (Invitrogen, Carlsbbad, CA, USA) to generate pEN-intron (Fig. 1a). The pEN-intron was mutated with the primer pair P-inBsSp/M-inBsSp to create BspEI and SpeI sites at 130–141 bp of the intron, in which the fragment of the antisense intergenic region (IGR) of Ageratum yellow vein virus (AYVV) amplified by the primer pair P-IGR-Xba I/M-IGR-Xma I by PCR was introduced by XmaI/Xba I digestion to generate pEN-int-antiIGR (Fig. 1a). The construct pEN-int-antiIGR was mutated with the primer pair P-inAvBs/M-inAvBs to generate AvrII and BspEI sites at bps 622-633 of pEN-int-antiIGR, in which the fragment of the sense IGR of AYVV PCR-amplified with the primer pair P-IGR-Xba/M-IGR-Xma I was introduced by XbaI/XmaI digestion to generate pEN-int-hpIGR, with a spacer of 96 bp from the middle part of the At3947160 intron (Fig. 1a).
An untranslatable nucleocapsid protein (NP) fragment of Melon yellow sport virus (MYSV) was amplified by RT-PCR from the total RNA extracted from a N. benthamiana plant infected with MYSV (accession number FJ386391.1) with the primer pair P-MYSV-Xba I/M-MYSV-Xma I and introduced into XbaI/Xma I sites of the pCR 2.1-TOPO vector (Invitrogen, Carlsbbad, CA, USA) to generate pTOPO- MYSV (Fig. 1a). The int-hpIGR fragment released from NdeI and NheI sites from pEN-int-hpIGR, was subsequently inserted to the pTOPO-MYSV to generate pTOPO-MY-int-hpIGR-NP, from which the MY-int-hpIGR-NP fragment was released by Xba I/XmaI digestion and introduced into the pk2T binary vector to generate pk2T-MY-int-hpIGR-NP (Fig. 1a).
In addition to the above construct that was attempted to induce both RdDM and PTGS for generating transgenic resistance to AYVV and MYSV, respectively, a construct intended to induce PTGS for generating transgenic resistance to AYVV was also constructed as a control. For this purpose, the IGR fragment of AYVV was amplified with the primer pair P-IGR-Xba I/M-IGR-Xma I by PCR and introduced into pK2T binary vector at Xba I/Xma I sites to generate pK2T-IGR (Fig. 1a).
A hairpin RNA construct was also used as a control for generating transgenic resistance to AYVV by the PTGS approach. The hairpin fragment of AYVV IGR in pEN-int-hpIGR was amplified by PCR with the primer pair P-IGR-Xba I/M-spacer-Xba I and introduced into PK2T-IGR via XbaI/Xba I sites to generate the pK2T-hpIGR (Fig. 1a).
Furthermore, an untranslatable construct was used as a control for generating transgenic resistance to MYSV by the PTGS approach. The untranslatable MYSV-NP coding sequence, with two stop codons at the 5′ end, was amplified by PCR with the primer pair P-MYSV-Xba I/M-MYSV-Xma I from pTOPO- MYSV and introduced into pK2T binary vector via Xba I/Xma I sites to generate pK2T-MYSV-NP (Fig. 1a).
A positive control was constructed for confirmation of the splicing of the At3947160 intron. The intron fragment released from NdeI and NheI sites from pEN-intron, was subsequently inserted into the pK2T-MYSV-NP vector to generate pK2T-MY -intron -NP (Fig. 1a).
Agroinfiltration and transformation of N. benthamiana plants
All the pK2T constructs were separately introduced into A. tumefaciens strain ABI. Individual colonies of the bacteria carrying each construct were cultured in the LB medium at 28°C for 16 hr and then subcultured in LB media with 10 mM 2-(N-morpholino) ethanesulfonic acid (MES) and 40 μM acetosyringone up to an OD600 of 0.5. The bacteria were then spun down and pellets were resuspended in 10 mM MgCl2 and 150 μM acetosyringone and kept at room temperature for 3 hr. With a 2 ml syringe without needle, the suspension was infiltrated into the intercellular space of leaves of N. benthamiana plants. Regeneration of transgenic plants from the infiltrated leaf tissues was conducted as previously described21.
Confirmation of splicing of MY-int-hpIGR-NP by reverse transcription polymerase chain reaction (RT-PCR)
Total RNAs were extracted using TRIzol® reagent (Invitrogen, Carlsbbad, CA, USA) from tobacco leaf tissues agroinfiltrated with individual constructs of pK2T-MYSV-NP, pK2T-MY-intron -NP and pK2T-MY-int-hpIGR-NP at 3 days after infiltration. The first-strand cDNA were synthesized with M-MLV reverse transcriptase using M-MYSV-Xma I primer (Epicentre, Madison, Wisconsin, USA) at 42°C for one hour. The PCR amplification was performed with Taq DNA polymerase (MDBio Inc., Taipei, Taiwan) using the primers P-MYSV-Xba I and M-MYSV-Xma I to amplify the corresponding MYSV-NP fragment. For confirming the fidelity of the spliced transcript, the RT-PCR amplified fragment from the tissues agroinfiltrated with pK2T-MY-int-hpIGR-NP was sequenced.
Evaluation of resistance to AYVV
The plantlets micropropagated from an individual shoot regenerated after transformation with each construct were considered an individual transgenic line. Rooted shoots of transgenic plantlets of each transgenic line were transplanted onto Florobella (Klasmann-Deilmann, Geeste, Germany) potting compost-sand mix (3:1) in a growth chamber for 4–5 days for hardening and then they were grown in temperature-controlled conditions (23–28°C) in a greenhouse for 2 weeks (5–6 leaf stage). The infectious clones used the Rolling Circle Amplification (RCA) products of genome of AYVV was digested and ligated into the PstI/BamHI site of the binary vector pCAMBIA0380 to generate infectious construct pAYVV53. The pAYVV harboring bacteria grown in LB medium containing 50 mg l−1 kanamycin and 50 mg l−1 streptomycin at 28°C for 16 hr was used at an OD600 of 0.1. The transgenic plants were injured at the junction of stem and petiole at three places with a needle (23G, 0.63*25 mm) of a syringe with bacterial suspension. Non-transgenic N. benthamiana plants were used as controls. All inoculated plants were kept in a temperature-controlled (23–28°C) greenhouse and symptom development was monitored daily up to 8 weeks. AYVV infection on all transgenic plants with symptoms was confirmed by PCR assay with P-AYVV-C4/M-AYVV-C4 primers specific to C4 gene.
Evaluation of resistance to MYSV
The transgenic tobacco plants (5–6 leaf stage) were also mechanically inoculated with MYSV and non-transgenic tobacco plants obtained by tissue culture were used as controls. The MYSV inoculum was prepared from virus-infected plants of N. benthamiana 8 days after mechanical inoculation, by grinding 1 g leaf tissue with 50 ml 0.01 M potassium phosphate buffer, pH 7.0. The test plants were inoculated by rubbing the carborundum (600 mesh)-dusted first and second fully expanded leaves. Inoculated plants were kept in a temperature-controlled greenhouse (23–28°C) and symptom development was monitored up to 5 weeks after inoculation. The accumulation of MYSV was assessed by indirect ELISA using the antiserum to the MYSV N protein37
Northern blotting analysis for siRNA detection
For small interfering (si) RNA detection, isolation and separation of total RNAs were performed as described previously54. The [α-32P]ATP-labeled probe was prepared from the IGR fragment using Primer-It II random primer labeling kit (Stratagne, LaJolla, CA), following manufacturer's instructions. After post-hybridization washing of the filter, autoradiography was performed by mounting an X-ray film (Hyperfilm Mp, Amershan Phamacia Biotech, UK) on the membrane at room temperature.
Southern blotting analysis
Fifteen μg of total DNA, extracted as described above, from non-transgenic plants or T0 plant of MY-int-hpIGR-NP transgenic lines, was digested with AseI that digests the construct once before the transgene sequences. The digested products were separated by electrophoresis on a 0.8% agarose gel and then transferred to nylon membrane by a POSIBLOTTM pressure blotter (Stratagene, CA, USA). The DNA bound to the nylon membrane was hybridized with the probe generated from the NP sequence of MY-int-hpIGR-NP construct by PCR amplification using specific primer pair (MYSV-Xba I /MYSV-N-350) and labeled with [α-32P] ATP by the Pimer-It II random primer labeling kit (Stratagene, CA, USA), following manufacturer's instructions. After post-hybridization washing of the filter, autoradiography was performed by mounting an X-ray film (Hyperfilm Mp, Amershan Phamacia Biotech, UK) on the membrane at room temperature.
Bisulfite sequencing analysis
To confirm the methylation status of the transgene, bisulfite sequencing was performed as previously described30, with required modifications. The genomic DNA of transgenic tobacco plant was extracted using the Plant Genomic DNA Purification Kit (Genemark, Taichung, Taiwan) and 500 ng of AseI-digested DNA was bisulfite-treated with the EZ DNA MethylationTM kit (Zymo Research, Irvine, CA, U.S.A.). The bisulfite-treated DNA was amplified by PCR with Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, U.S.A.). The 247-bp (IGR-247) and the 350-bp (IGR-350) fragments were amplified with the BisP1/BisM1 and BisP2/BisM2 primers from MY-int-hpIGR-NP and hpIGR constructs, respectively. As a positive control for full cytosine conversion, 500 ng of genomic DNA of non-transgenic tobacco was mixed with approximately 40 pg of AseI-cleaved plasmid DNA. For each sample, the resulted PCR products were directly sequenced three times and the sequences were analyzed using the software Vector NTI Advance 10.3 to assess the frequency of methylation.
Generation an AYVV auto-replicating shuttle vector with GFP marker
In order to assay whether siRNA induced RdDM targeting the IGR of AYVV to suppress the promoter activity, an auto-replicating shuttle vector with GFP driven by IGR promoter was constructed. The binary vector pBCO-DC-HA52 was amplified with primers P-BCO and M-BCO with KOD plus polymerase (TOYOBO, Osaka, Japan). The amplified fragment was self-ligated to generate the pBCO vector to remove the Cauliflower mosaic virus 35S promoter and HA sequence. The AYVV IGR was amplified from pAYVV with primers PAYIGR-E and MAYIGR-NX and the amplified fragment was digested with EcoRV and XmaI and then introduced into the EcoRV/Xma I -digested pBCO vector to generate pAYIGR. A GFP ORF was amplified from pEGFP1 (Clontech, Basingstoke, UK) with primers P-GFP and M-GFP and the amplified fragment was digested with NcoI/XmaI and inserted into the NcoI/XmaI-digested pAYIGR to get pAYIGR-GFP (a vector control in which only one IGR present and not auto-replicating). The C1, C2, C3 and IGR regions of AYVV was amplified from pAYVV with primers P-AYVV1016 and M-AYVV133 and the PCR product was digested with SnaBI/XmaI and inserted into the SnaBI/XmaI-digested pAYIGR-GFP to generate pAY-ST which contains IGR rightward promoter, GFP, C1, C2, C3 and C4 coding regions (see Supplementary Fig. 3). The C1 and IGR regions of AYVV was amplified from pAYVV with primers P-AYVV1465 and M-AYVV133 and the PCR product was digested with SnaBI/XmaI and inserted into the SnaBI/XmaI-digested pAYIGR-GFP to generate pAY-ST-d possessing IGR rightward promoter, GFP, C1 and C4 coding regions, but lacking C2 and C3 regions (see Supplementary Fig. 3). The pAY-ST and pAY-ST-d were separately transferred into A. tumefaciens ABI strain and tested for GFP expression in leaf tissues of N. benthamiana plants by agroinfiltration. Total DNA were extracted from tobacco leaves at 3 days after agroinfiltration with individual constructs of pAY-ST, pAY-ST-d and pAYIGR-GFP. The circularized unit-length replication of the constructs was analyzed by PCR from genomic DNA with primers P-GFP-625 and M-GFP-82 targeting at the 3′ and 5′ portions of the GFP-ORF, respectively, in opposite orientations (see Supplementary Fig. 3).
GFP fluorescence assay for IGR Promoter activity
Plants of transgenic lines MY-int-hpIGR-NP-6, 12 and hpIGR-3 resistant to AYVV, transgenic lines MY-int-hpIGR-NP-2 and hpIGR-19 susceptible to AYVV and non-transgenic control were inoculated with an auto-replicating AYVV-derived construct pAY-ST in which a green fluorescent protein (GFP) is driven by the IGR rightward promoter, by agroinfiltration at an OD600 of 0.01 into leaves of N. benthamiana plants. At 1.5 or 2.5 days after agroinfiltration, the intensity of GFP fluorescence was recorded from the infiltrated tissues by a Molecular Imaging System under Kodak 4000MM image station (Eastman Kodak Co., Rochester, NY) and quantified using a KODAK 1D Image Analysis Software (Eastman Kodak Co). The ANOVA analysis was conducted by measuring four independent repeats for each sample.
References
Vanderschuren, H., Stupak, M., Futterer, J., Gruissem, W. & Zhang, P. Engineering resistance to geminiviruses - review and perspectives. Plant Biotechnol J 5, 207–220 (2007).
Prins, M. & Goldbach, R. The emerging problem of tospovirus infection and nonconventional methods of control. Trends Microbiol 6, 31–35 (1998).
Pappu, H. R., Jones, R. A. & Jain, R. K. Global status of tospovirus epidemics in diverse cropping systems: successes achieved and challenges ahead. Virus Res 141, 219–236 (2009).
Hanley-Bowdoin, L., Bejarano, E. R., Robertson, D. & Mansoor, S. Geminiviruses: masters at redirecting and reprogramming plant processes. Nat Rev Microbiol 11, 777–788 (2013).
Raja, P., Wolf, J. N. & Bisaro, D. M. RNA silencing directed against geminiviruses: post-transcriptional and epigenetic components. Biochim Biophys Acta 1799, 337–351 (2010).
Mansoor, S. et al. Cotton leaf curl disease is associated with multiple monopartite begomoviruses supported by single DNA beta. Arch Virol 148, 1969–1986 (2003).
de Haan, P., Kormelink, R., de Oliveira Resende, R., van Poelwijk, F., Peters, D. & Goldbach, R. Tomato spotted wilt virus L RNA encodes a putative RNA polymerase. J Gen Virol 72, 2207–2216 (1991).
Kormelink, R., Storms, M., Van Lent, J., Peters, D. & Goldbach, R. Expression and subcellular location of the NSM protein of tomato spotted wilt virus (TSWV), a putative viral movement protein. Virology 200, 56–65 (1994).
Kikkert, M., Verschoor, A., Kormelink, R., Rottier, P. & Goldbach, R. Tomato spotted wilt virus glycoproteins exhibit trafficking and localization signals that are functional in mammalian cells. J Virol 75, 1004–1012 (2001).
Kormelink, R., Kitajima, E. W., De Haan, P., Zuidema, D., Peters, D. & Goldbach, R. The nonstructural protein (NSs) encoded by the ambisense S RNA segment of tomato spotted wilt virus is associated with fibrous structures in infected plant cells. Virology 181, 459–468 (1991).
Takeda, A. et al. Identification of a novel RNA silencing suppressor, NSs protein of Tomato spotted wilt virus. FEBS Lett 532, 75–79 (2002).
Matzke, M., Kanno, T., Daxinger, L., Huettel, B. & Matzke, A. J. RNA-mediated chromatin-based silencing in plants. Curr Opin Cell Biol 21, 367–376 (2009).
Tang, G. siRNA and miRNA: an insight into RISCs. Trends Biochem Sci 30, 106–114 (2005).
Ruiz-Ferrer, V. & Voinnet, O. Roles of plant small RNAs in biotic stress responses. Annu Rev Plant Biol 60, 485–510 (2009).
Duan, C. G., Wang, C. H. & Guo, H. S. Application of RNA silencing to plant disease resistance. Silence 3, 5 (2012).
de Haan, P. et al. Characterization of RNA-mediated resistance to tomato spotted wilt virus in transgenic tobacco plants. Biotechnology (N Y) 10, 1133–1137 (1992).
Jan, F. J., Fagoaga, C., Pang, S. Z. & Gonsalves, D. A single chimeric transgene derived from two distinct viruses confers multi-virus resistance in transgenic plants through homology-dependent gene silencing. J Gen Virol 81, 2103–2109 (2000).
Catoni, M., Lucioli, A., Doblas-Ibanez, P., Accotto, G. P. & Vaira, A. M. From immunity to susceptibility: virus resistance induced in tomato by a silenced transgene is lost as TGS overcomes PTGS. Plant J 75, 941–953 (2013).
Hassani-Mehraban, A., Brenkman, A. B., van den Broek, N. J., Goldbach, R. & Kormelink, R. RNAi-mediated transgenic Tospovirus resistance broken by intraspecies silencing suppressor protein complementation. Mol Plant Microbe Interact 22, 1250–1257 (2009).
Prins, M., Kikkert, M., Ismayadi, C., de Graauw, W., de Haan, P. & Goldbach, R. Characterization of RNA-mediated resistance to tomato spotted wilt virus in transgenic tobacco plants expressing NS(M) gene sequences. Plant Mol Biol 33, 235–243 (1997).
Kung, Y. J. et al. Multiple artificial microRNAs targeting conserved motifs of the replicase gene confer robust transgenic resistance to negative-sense single-stranded RNA plant virus. Mol Plant Pathol 13, 303–317 (2012).
Chellappan, P., Vanitharani, R., Pita, J. & Fauquet, C. M. Short interfering RNA accumulation correlates with host recovery in DNA virus-infected hosts and gene silencing targets specific viral sequences. J Virol 78, 7465–7477 (2004).
Hou, Y. M., Sanders, R., Ursin, V. M. & Gilbertson, R. L. Transgenic plants expressing geminivirus movement proteins: abnormal phenotypes and delayed infection by Tomato mottle virus in transgenic tomatoes expressing the Bean dwarf mosaic virus BV1 or BC1 proteins. Mol Plant Microbe Interact 13, 297–308 (2000).
Sinisterra, X. H., Polston, J. E., Abouzid, A. M. & Hiebert, E. Tobacco plants transformed with a modified coat protein of tomato mottle begomovirus show resistance to virus infection. Phytopathology 89, 701–706 (1999).
Zhang, P., Vanderschuren, H., Futterer, J. & Gruissem, W. Resistance to cassava mosaic disease in transgenic cassava expressing antisense RNAs targeting virus replication genes. Plant Biotechnol J 3, 385–397 (2005).
Yang, Y., Sherwood, T. A., Patte, C. P., Hiebert, E. & Polston, J. E. Use of Tomato yellow leaf curl virus (TYLCV) Rep Gene Sequences to Engineer TYLCV Resistance in Tomato. Phytopathology 94, 490–496 (2004).
Mette, M. F., Aufsatz, W., van der Winden, J., Matzke, M. A. & Matzke, A. J. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19, 5194–5201 (2000).
Zilberman, D., Cao, X. & Jacobsen, S. E. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299, 716–719 (2003).
Wierzbicki, A. T., Ream, T. S., Haag, J. R. & Pikaard, C. S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet 41, 630–634 (2009).
Dalakouras, A., Moser, M., Zwiebel, M., Krczal, G., Hell, R. & Wassenegger, M. A hairpin RNA construct residing in an intron efficiently triggered RNA-directed DNA methylation in tobacco. Plant J 60, 840–851 (2009).
Dale, P. J., Clarke, B. & Fontes, E. M. Potential for the environmental impact of transgenic crops. Nat Biotechnol 20, 567–574 (2002).
Tuteja, N., Verma, S., Sahoo, R. K., Raveendar, S. & Reddy, I. N. Recent advances in development of marker-free transgenic plants: regulation and biosafety concern. J Biosci 37, 167–197 (2012).
Komari, T., Hiei, Y., Saito, Y., Murai, N. & Kumashiro, T. Vectors carrying two separate T-DNAs for co-transformation of higher plants mediated by Agrobacterium tumefaciens and segregation of transformants free from selection markers. Plant J 10, 165–174 (1996).
Wong, S. M., Swanson, M. M. & Harrison, B. D. A geminivirus causing vein yellowing of Ageratum conyzoides in Singapore. Plant Pathol 42, 137–139 (1993).
Tan, P. H., Wong, S. M., Wu, M., Bedford, I. D., Saunders, K. & Stanley, J. Genome organization of ageratum yellow vein virus, a monopartite whitefly-transmitted geminivirus isolated from a common weed. J Gen Virol 76, 2915–2922 (1995).
Yeh, S. D., Lin, Y. C., Cheng, Y. H., Jih, C. L., Chen, M. J. & Chen, C. C. Identification of tomato spotted wilt-like virus on watermelon in Taiwan. Plant Disease 76, 835–840 (1992).
Chen, T. C. et al. Serological relationship between Melon yellow spot virus and Watermelon silver mottle virus and differential detection of the two viruses in cucurbits. Arch Virol 155, 1085–1095 (2010).
Lin, C. Y., Ku, H. M., Chiang, Y. H., Ho, H. Y., Yu, T. A. & Jan, F. J. Development of transgenic watermelon resistant to Cucumber mosaic virus and Watermelon mosaic virus by using a single chimeric transgene construct. Transgenic Res 21, 983–993 (2012).
Turina, M., Tavella, L. & Ciuffo, M. Tospoviruses in the Mediterranean area. Adv Virus Res 84, 403–437 (2012).
Qian, L., Vu, M. N., Carter, M. & Wilkinson, M. F. A spliced intron accumulates as a lariat in the nucleus of T cells. Nucleic Acids Res 20, 5345–5350 (1992).
Wierzbicki, A. T., Haag, J. R. & Pikaard, C. S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135, 635–648 (2008).
Daxinger, L. et al. A stepwise pathway for biogenesis of 24-nt secondary siRNAs and spreading of DNA methylation. EMBO J 28, 48–57 (2009).
Cao, X. et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol 13, 2212–2217 (2003).
Aufsatz, W., Mette, M. F., van der Winden, J., Matzke, A. J. & Matzke, M. RNA-directed DNA methylation in Arabidopsis. Proc Natl Acad Sci U S A 99, 16499–16506 (2002).
Pelissier, T., Thalmeir, S., Kempe, D., Sanger, H. L. & Wassenegger, M. Heavy de novo methylation at symmetrical and non-symmetrical sites is a hallmark of RNA-directed DNA methylation. Nucleic Acids Res 27, 1625–1634 (1999).
Dalakouras, A., Tzanopoulou, M., Tsagris, M., Wassenegger, M. & Kalantidis, K. Hairpin transcription does not necessarily lead to efficient triggering of the RNAi pathway. Transgenic Res 20, 293–304 (2011).
Poornima Priyadarshini, C. G., Ambika, M. V., Tippeswamy, R. & Savithri, H. S. Functional characterization of coat protein and V2 involved in cell to cell movement of Cotton leaf curl Kokhran virus-Dabawali. PLoS One 6, e26929 (2011).
Regnard, G. L., Halley-Stott, R. P., Tanzer, F. L., Hitzeroth, I. I. & Rybicki, E. P. High level protein expression in plants through the use of a novel autonomously replicating geminivirus shuttle vector. Plant Biotechnol J 8, 38–46 (2010).
Jones, L., Ratcliff, F. & Baulcombe, D. C. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr Biol 11, 747–757 (2001).
Zrachya, A. et al. Production of siRNA targeted against TYLCV coat protein transcripts leads to silencing of its expression and resistance to the virus. Transgenic Res 16, 385–398 (2007).
Reyes, M. I., Nash, T. E., Dallas, M. M., Ascencio-Ibanez, J. T. & Hanley-Bowdoin, L. Peptide aptamers that bind to geminivirus replication proteins confer a resistance phenotype to tomato yellow leaf curl virus and tomato mottle virus infection in tomato. J Virol 87, 9691–9706 (2013).
Wu, H. W., Lin, S. S., Chen, K. C., Yeh, S. D. & Chua, N. H. Discriminating mutations of HC-Pro of zucchini yellow mosaic virus with differential effects on small RNA pathways involved in viral pathogenicity and symptom development. Mol Plant Microbe Interact 23, 17–28 (2010).
Wu, C. Y., Yang, S. H., Lai, Y. C., Lin, N. S., Hsu, Y. H. & Hu, C. C. Unit-length, single-stranded circular DNAs of both polarity of begomoviruses are generated in Escherichia coli harboring phage M13-cloned begomovirus genome with single copy of replication origin. Virus Res 125, 14–28 (2007).
Niu, Q. W. et al. Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat Biotechnol 24, 1420-1428 (2006).
Acknowledgements
This study was supported by the Ministry of Education, Taiwan, R.O.C. under the ATU plan; and NCHU-UCD Plant and Food Biotechnology programs from National Science Council under the programs NSC101-2911-I-005-301 and NSC102-2911-I-005-301.
Author information
Authors and Affiliations
Contributions
C.F.Y., K.C.C. and S.D.Y. designed the experiment. C.F.Y., K.C.C., Y.L.H. and W.C.C. performed the experiment. Y.H.C. contributed experiment materials. C.F.Y., J.A.J.R. and S.D.Y. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary file
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Yang, CF., Chen, KC., Cheng, YH. et al. Generation of Marker-free Transgenic Plants Concurrently Resistant to a DNA Geminivirus and a RNA Tospovirus. Sci Rep 4, 5717 (2014). https://doi.org/10.1038/srep05717
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05717
This article is cited by
-
CRISPR/Cas9 for plant genome editing: accomplishments, problems and prospects
Plant Cell Reports (2016)
-
Conferring resistance to geminiviruses with the CRISPR–Cas prokaryotic immune system
Nature Plants (2015)
-
Untranslatable tospoviral NSs fragment coupled with L conserved region enhances transgenic resistance against the homologous virus and a serologically unrelated tospovirus
Transgenic Research (2015)
-
Immunity to tomato yellow leaf curl virus in transgenic tomato is associated with accumulation of transgene small RNA
Archives of Virology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
