
Abstract
1,2,4-butanetriol (BT) is an important bulk chemical mainly used for producing the superior energetic plasticizer (1,2,4-butanetriol trinitrate) in propellant and explosive formulations. BT is commercially produced by chemical synthesis from petroleum-based feedstocks; until recently a costly biosynthetic route from xylose or arabinose was reported. Here we designed a novel biosynthetic pathway for BT from malate, for the purpose of using glucose as an alternative and cheaper substrate in future. This biosynthetic pathway was achieved through six sequential enzymatic reactions. Following tests of several combinations of enzymes for the pathway, five enzymes including malate thiokinase, succinate-semialdehyde dehydrogenase, 4-hydroxybutyrate dehydrogenase, 4-hydroxybutyrate CoA-transferase and bifunctional aldehyde/alcohol dehydrogenase were finally chosen. All enzyme genes were expressed on two compatible plasmids in E. coli and their functions verified separately. Following assembly of two functional modules, BT was detected in the fermentation broth upon addition of malate, proving BT can be biosynthesized from malate. Furthermore, BT was detected in the fermentation using glucose as the sole carbon source, suggesting that such novel BT biosynthetic pathway has created the possibility for the production of BT from the cheaper substrate glucose.
Similar content being viewed by others

Introduction
1,2,4-butanetriol (BT) is an important chemical mainly used to produce 1,2,4-butanetriol trinitrate (BTTN), an energetic plasticizer in propellant and explosive formulations. BTTN is generally considered as a safe substitute for the traditional nitroglycerin because of its advantages in shock sensitivity, thermostability and volatility1. Chemical synthesis of BT is not cost-competitive because of the harsh reaction conditions (high temperature and high pressure) and low yield2. Although there is no natural biosynthetic pathway available for BT, the assembly of four enzymatic reactions into two microorganisms enabled conversion of pentose (xylose or arabinose) to BT1. The yields of BT in these bioconversion processes reached 0.175 mol/mol xylose and 0.189 mol/mol arabinose, respectively. A subsequent international patent claimed that BT production from xylose could be achieved in a single strain, resulting in a BT titer of 18 g/L and a yield of 0.55 mol/mol xylose3.
Because the substrate cost is typically the main determining factor in the commercial viability of a bioconversion process, we are seeking to develop an alternative route for BT biosynthesis, with the expectation of replacing the expensive substrate (xylose/arabinose) with a much cheaper one. Glucose has been widely used as a cheap feedstock for producing bulk chemicals4,5,6,7,8,9,10 and can be efficiently metabolized through the tricarboxylic acid (TCA) cycle. Malate, one of the TCA intermediates, shares a similar structure with BT except for the two terminal groups (two carboxyl groups in malate, versus two hydroxyl groups in BT). This inspired us to construct a non-natural biosynthetic pathway for production of BT from malate. To test the feasibility of this new idea, we screened potential enzyme candidates capable of reducing a carboxyl group to a hydroxyl group. Those that showed potential catalytic activity towards the desired reactions were chosen. The genes encoding the selected enzyme candidates were then assembled into two modules and heterologously expressed in E. coli. Production of BT from malate was achieved by verifying the function of individual modules followed by a further verification upon assembly of the two functional modules together.
Methods
Strains and plasmids
E. coli DH5α and E. coli GM2163 strains were used for plasmid construction, while E. coli BL21(DE3) strain was used for expression of genes. Luria-Bertani (LB) media were used for routine cultures. 50 μg/mL of kanamycin (Kan), 5 μg/mL of tetracycline (Tetra), and/or 25 μg/mL of chloramphenicol (Cm) were added for maintenance of the appropriate plasmid. All genes encoding the selected enzyme candidates were synthesized by Genewiz company (Beijing, China). The codon preference was optimized using an online tool for E. coli expression system (http://www.jcat.de/). The NCBI accession number, reported catalytic reaction and insertion sites of the synthesized genes are listed in Table 1. It is noteworthy that the BclI site is sensitive to methylation, therefore the corresponding plasmid must be prepared in E. coli GM2163, which is deficient in both DNA adenine methyltransferase (dam) and DNA cytosine methyltransferase (dcm). Two modules were constructed by using two different but compatible plasmids as backbones to accommodate the respective genes. The first module was based on plasmid pET-30a where enzyme-encoding genes for the first three steps of the biosynthetic pathway (Fig. 1) were cloned, each with a separate T7 promoter placed upstream of the gene. The second module was based on plasmid pACYC184 where enzyme-encoding genes for the last three steps of the 6-step pathway were cloned, each with a separate T7 promoter upstream and a shared T7 terminator downstream.

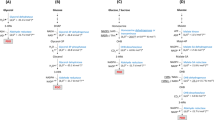
Novel designed pathway (left) and equations of every reaction (right).
The designed 1,2,4-butanetriol (BT) biosynthetic pathway from malate were divided into two modules at the junction of 2,4-dihydroxybutyrate (DHB). In module 1, one of the terminal carboxyl groups of malate was firstly activated with CoA and then reduced to an aldehyde group and then further reduced to a hydroxyl group, to yield DHB. Similarly, the other terminal carboxyl group of DHB was reduced to a hydroxyl group to yield the final product BT in module 2. The standard Gibbs free energy change of every reaction (reaction delta G) was shown in the left designed pathway, which was the difference of standard Gibbs free energy of formation (formation delta G) between products and substrates in the right every equation. This artificial six-step pathway was thermodynamically feasible as the sum of reaction delta G from six reactions was negative (−15 kcal/mol).
Fermentation
Small-scale fermentations were conducted in anaerobic bottles containing 20 ml modified M9 minimal salts medium (6.78 g/L Na2HPO4, 3.0 g/L KH2PO4, 0.5 g/L NaCl, 1.0 g/L NH4Cl, 1 mM MgSO4, 0.1 mM CaCl2, 10 mM NaHCO3, 20 g/L D-glucose, 100 mM MOPS, 10 μg/ml thiamine and the appropriate antibiotics). Recombinant E. coli BL21(DE3) strains containing the plasmids of module 1 and/or module 2, were grown under microaerobic conditions. Such conditions were achieved by piercing the septum with a small needle after flushing capped anaerobic bottles with nitrogen for 5 min6. When the culture had reached its mid-exponential growth phase (OD600 = 0.4–0.6), coinciding with the addition of an appropriate concentration of IPTG to induce protein expression, the substrate (malate or DHB) was also added. All cultures were grown at 37°C and repeated in triplicate.
GC-MS analysis
Gas chromatography-mass spectrometry (GC-MS) was conducted using a method modified slightly from the literature6 and is described as follows. 20 mL of internal standard (10 mM cyclohexanol solution diluted in dimethylformamide) was added to 100 μL filtered and dried fermentation liquid sample, followed by addition of 100 μL derivatization reagent (N,O-bis(trimethylsilyl) triflouro-acetimide with 1% trimethylchlorosilane) and incubated at 70°C for 30 min. Subsequently, the mixture was centrifuged (15,000 × g) for 5 min. The supernatant was immediately tested by GC-MS. 2 mL was injected without using a split ratio on an Agilent HP-5 MS capillary column (30 m length) using helium as the carrier gas. The inlet temperature and flow rate were set at 280°C and 1.0 mL/min, respectively. The oven temperature gradient program was set as follows: initially held at 80°C for 1.5 min, raised by 3°C/min to 140°C (held for 0 min) and finally fast ramped to 280°C at 50°C/min (held for 10 min). The total run time was about 44 min. For better separation of products, the GC-MS condition was modified as follows: initially held at 80°C for 1.5 min, raised by 2°C/min to 140°C (held for 0 min) and finally fast ramped to 280°C at 50°C/min (held for 10 min). The MS conditions for identification of BT and other products were as follows: for full-scan mode, 30–500 m/z mass-range; for selected ion monitoring (SIM) of BT, the selected ions were set as 103, 129, 219 and 232 m/z; for SIM of 2,4-dihydroxybutyrate (DHB), the selected ions were set as 103, 129, 189 and 219 m/z; for SIM of cyclohexanol, the selected ions were set as 75, 129, 157 and 172 m/z. Quantification was based on the peak area ratios of the different compounds to standard chemicals, using cyclohexanol as an internal standard.
Results
De novo design of a novel BT biosynthetic pathway from malate
The novel BT biosynthetic pathway acts as a metabolic shunt from E. coli central metabolism. Following scrutiny of all the central metabolites of E. coli, malate in the TCA cycle was selected based on its structural similarity to BT and its efficient production from glucose.
Malate differs from BT by only two terminal carboxyl groups, hence conversion of malate to BT requires reduction of only these two terminal carboxyl groups to hydroxyl groups. One carboxyl group can be reduced to a hydroxyl group through the addition of coenzyme A (CoA) followed by formation of an aldehyde group and then of the hydroxyl group, which are sequentially catalyzed by CoA synthetase, aldehyde dehydrogenase and alcohol dehydrogenase. When one considers the substrate specificity of enzymatic reactions, the two terminal carboxyl groups of malate should be converted separately by different enzymes sets. For this reason, the biosynthetic pathway of BT from malate was designed to compose of six enzymatic reactions, as shown in Fig. 1. The standard Gibbs free energy of formation of every non-natural product was estimated by the group contribution method11. The calculated total standard Gibbs free energy change of six reactions for biosynthesis of BT from malate was −15 kcal/mol, suggesting that the designed six-step pathway was thermodynamically feasible (Fig. 1).
For the newly designed six-step non-natural BT biosynthetic pathway, candidate enzymes (listed in Table 1) were selected. The selection was based on the following criteria (in order of importance): (1) similarity of the reactions catalyzed by the candidate enzymes to the desired reactions, as shown in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database or in published literature; (2) ability to be actively expressed in E. coli; and (3) homology of species with E. coli. Enzymes from gram-negative strains were preferred to those from gram-positive ones for example.
Construction of the novel BT biosynthetic pathway from malate in E. coli
To facilitate the construction and functional validation of the six-step BT biosynthetic pathway, the entire pathway were divided into two modules. Each of the modules contained three reactions and was joined through the intermediate of the pathway, 2,4-dihydroxybutyrate (DHB). As shown in Fig. 1, module 1 will convert malate to DHB via the reduction of one terminal carboxyl group to a hydroxyl group and module 2 will convert DHB to BT via another similar reduction reaction of the other terminal carboxyl group to a hydroxyl group.
Two compatible plasmids, pET-30a and pACYC184, were used to construct the two modules following codon-optimization of each enzyme gene. As illustrated in Table 1, a total of thirteen homologous and heterologous enzymes were selected for the six steps. For module 1, five malate thiokinase genes (mtkABs) for the first step, one succinate-semialdehyde dehydrogenase gene (sucD) for the second step and one 4-hydroxybutyrate dehydrogenase gene (4hbD) for the third step were assembled into pET-30a. The five recombinant plasmids subsequently generated were 30a-mtkABM. loti-sucD-4hbD, 30a-mtkABM. petroleiphilum-sucD-4hbD, 30a-mtkABM. extorquens-sucD-4hbD, 30a-mtkABN. europaea-sucD-4hbD and 30a-mtkABM. radiotolerans-sucD-4hbD. For module 2, one 4-hydroxybutyrate CoA-transferase gene (abfT-2) and one cinnamoyl-CoA:phenyllactate CoA-transferase gene (fldA) for the fourth step, one coenzyme A acylating aldehyde dehydrogenase gene (ald) and a methylmalonate-semialdehyde dehydrogenase gene (msdh) for the fifth step, a alcohol dehydrogenase gene (eutG) for the sixth step and a bifunctional aldehyde/alcohol dehydrogenase gene (adhE2) for both the fifth and sixth steps were assembled into pACYC184. The resultant six recombinant plasmids were 184-abfT-2-ald-eutG, 184-abfT-2-msdh-eutG, 184-fldA-ald-eutG, 184-fldA-msdh-eutG, 184-abfT-2-adhE2 and 184-fldA-adhE2.
The function of each module was tested by separately adding their respective substrates (malate and DHB) and detecting their products (DHB and BT) respectively. To test the function of module 1, 0.2 mM IPTG and 100 mM malate were added to the cultures of the six recombinant E. coli BL21(DE3) strains and incubated for 24 h. Five of the six cultures contained a pET-30a recombinant plasmid as mentioned above, respectively. The sixth was a control containing an empty pET-30a plasmid. Analysis by GC-MS showed that all five recombinant E. coli BL21(DE3) strains had the ability to produce DHB from malate with differing capabilities, but was not observed from the control strain E. coli BL21(DE3)/pET-30a (Fig. 2A). The highest production of DHB (6.4 mg/L) was detected in the culture of the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD (Fig. 2B–2D). It not only confirmed module 1 was functional, but also suggested that the malate thiokinase from Methylibium petroleiphilum PM1 was much more efficient than those from the other four microorganisms.

GC-MS analysis for functional validation of module 1 by converting malate to 2,4-dihydroxybutyrate (DHB).
(A), The relative titer of DHB produced from malate by different recombinant E. coli. I, II, III, IV and V represented the recombinant E. coli BL21(DE3)/30a-mtkABM. loti-sucD-4hbD, E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD, E. coli BL21(DE3)/30a-mtkABM. extorquens-sucD-4hbD, E. coli BL21(DE3)/30a-mtkABN. europaea-sucD-4hbD and E. coli BL21(DE3)/30a-mtkABM. radiotolerans-sucD-4hbD, respectively. CK was the control strain recombinant E. coli BL21(DE3)/pET-30a. (B) and (C), identification of DHB converted from malate by E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD. The retention time (RT) of DHB was 10.78 min (scan mode). (D), the RT of chemically synthesized DHB standard (WuXi AppTec, China) was 10.78 min.
Similarly, for testing of module 2, 0.2 mM IPTG and 100 mM DHB were added. GC-MS results of 24 h post-induction samples showed that only the recombinant E. coli BL21(DE3)/184-abfT-2-adhE2 and E. coli BL21(DE3)/184-fldA-adhE2 were able to produce BT from DHB (Fig. 3A) and the highest titer (55 mg/L) of BT was detected in the culture of the former recombinant strain (Fig. 3B–3D). Similarly to module 1, BT was not detected in the control strain E. coli BL21(DE3)/pACYC184, confirming that module 2 was functional. It also suggested that the AbfT-2 was more proficient than FldA, AdhE2 performed better than the simple combination of an aldehyde dehydrogenase and an alcohol dehydrogenase and that the native aldehyde dehydrogenases and alcohol dehydrogenases in E. coli did not perform well in this designed pathway.

GC-MS analysis for functional validation of module 2 by converting 2,4-dihydroxybutyrate (DHB) to 1,2,4-butanetriol (BT).
(A), The relative titer of BT produced from DHB by different recombinant E. coli. I, II, III, IV, V and VI, represented the recombinant E. coli BL21(DE3)/184-abfT-2-ald-eutG, E. coli BL21(DE3)/184-abfT-2-msdh-eutG, E. coli BL21(DE3)/184-fldA-ald-eutG, E. coli BL21(DE3)/184-fldA-msdh-eutG, E. coli BL21(DE3)/184-abfT-2-adhE2 and E. coli BL21(DE3)/184-fldA-adhE2, respectively. CK was the control strain recombinant E. coli BL21(DE3)/pACYC184. (B) and (C), identification of BT converted from DHB via E. coli BL21(DE3)/184-abfT-2-adhE2. The retention time (RT) of BT was 16.88 min (scan mode). (D), the RT of commercial BT standard (Sigma) was 16.88 min.
Because the two modules were demonstrated to be functional, we assembled them by co-transforming E. coli BL21(DE3) with the recombinant plasmids 30a-mtkABM. petroleiphilum-sucD-4hbD and 184-abfT-2-adhE2. The control strain was co-transformed with empty plasmids pET-30a and pACYC184. The cultures of these recombinant strains were provided with 0.25 mM IPTG and 100 mM malate and induced for 24 h. GC-MS analysis showed that 180 ng/L BT was produced in the culture of the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD/184-abfT-2-adhE2 (Fig. 4A and 4B), whereas it was undetectable for the control strain E. coli BL21(DE3)/pET-30a/pACYC184. This verified that BT can be produced from malate through the designed non-natural six-step BT biosynthetic pathway.

GC-MS analysis for proof of concept that 1,2,4-butanetriol (BT) can be biosynthesized from malate.
(A), detection of BT converted from malate by the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD/184-abfT-2-adhE2. (B), result of the commercial BT standard (Sigma). The retention time of BT was 17.52 min (selected ion monitoring (SIM) mode).
Additionally, the reduction of the two terminal carboxyl groups of malate by the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD/184-abfT-2-adhE2 was indeed in the sequence shown in Fig. 1. BT was unable to be detected in the cultures of the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD when added with IPTG and DHB (data not shown). Also, 3,4-dihydroxybutyrate was not detected in the cultures of the recombinant E. coli BL21(DE3)/184-abfT-2-adhE2 when added with IPTG and malate (data not shown). It suggested the reduction process should be highly dependent on the chemical structures and functional groups of the compounds and the specificities of enzymes.
However, the addition of malate to the cultures of the recombinant E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD/184-abfT-2-adhE2 led to the significant increase of succinate (Fig. 5A) and this strain also produced large amounts of non-natural 4-hydroxybutyrate (4-HB) and 1,4-butanediol (BDO), a level much higher than that of DHB and BT, respectively (Fig. 5B–5D). These suggested that a part of the malate was likely converted to succinate, thus did not enter the BT biosynthetic pathway and that the enzymes selected for BT biosynthesis from malate probably had greater activities and/or specificities towards intermediates for BDO biosynthesis, namely succinate, succinyl CoA, succinyl semialdehyde, 4-hydroxybutyrate, 4-hydroxybutyryl CoA and 4-hydroxybutyraldehyde.

The relative titer of succinates produced in different fermentation broth and GC-MS analysis for main byproducts produced from malate by the recombinant E. coli BL21(DE3)/30a-mtkABM.petroleiphilum-sucD-4hbD/184-abfT-2-adhE2.
(A), the bar labeled as succinate containing an asterisk represented the relative titer of succinate from fermentation broth without the addition of malate and the other bar represented that with addition of malate. (B), a large quantities of 4-hydroxybutyrate (4-HB) and succinate observed in the fermentation with addition of malate (Scan mode). (C) and (D), 1.5 mg/L BDO was detected in sample compared to the 1 mg/L commercial BDO standard (Sigma) (scan mode). The retention time (RT) of BDO was 10.98 min.
The novel pathway creates possibility of BT production from glucose
120 ng/L BT was found to be produced by E. coli BL21(DE3)/30a-mtkABM. petroleiphilum-sucD-4hbD/184-abfT-2-adhE2 using glucose as the sole carbon source without the need to supplement malate or 2,4-dihydroxybutyrate (Fig. 6A and 6B). This suggested the novel BT biosynthetic pathway constructed here has successfully laid the groundwork for future studies on the production of BT from the significantly cheaper substrate glucose.

GC-MS analysis for production of 1,2,4-butanetriol (BT) from glucose as a sole carbon source by the recombinant E. coli BL21(DE3)/30a-mtkABM.petroleiphilum-sucD-4hbD/184-abfT-2-adhE2.
(A), 120 ng/L BT was detected in sample (selected ion monitoring (SIM) mode). (B), detection of 100 ng/L commercial BT standard (Sigma) for accurate identification (SIM mode). It is noteworthy that this time the retention time (RT) of BT was 21.15 min that was greatly different from that in Fig. 4, because the heating program of GC-MS was modified for better separation of products (see Materials and Methods).
Discussion
The work outlined here is compelling evidence that an energetic material precursor 1,2,4-butanetriol could be produced from malate through the successful design and construction of a novel non-natural biosynthetic pathway, which has created the possibility of BT production from the much cheaper substrate glucose; representing an alternative biosynthesis for this non-naturally occurring product.
To produce BT from glucose, the major challenge of constructing and expressing a non-natural heterologous BT biosynthetic pathway from central metabolic intermediates should be overcome. Challenges that are met here are likely to be characteristic of the biological production of any non-natural chemical, as these compounds are not synthesized biologically in nature. Malate was chosen because in addition to its structural similarity to BT (a difference of only two terminal groups), it can be efficiently produced from glucose. For construction of the pathway, native E. coli enzymes, heterologous enzymes working on their native substrates and heterologous enzymes made to act on non-natural substrates, were selected after searching the literature of candidate enzymes, primarily based on the similarity of the reactions catalyzed by them to the desired reactions. Here the selected enzymes of module 1 can only reduce the 4-carboxyl group of malate, but are unable to carry out the reduction of the 1-carboxyl group of malate. For this reason, other enzymes were necessary to complete the entire structural conversion of malate. Finally, the selected enzymes of module 2 reduced the 1-carboxyl group of malate only after the reduction of the 4-carboxyl group of malate was performed by the enzymes of module 1, but was unable to produce 3,4-dihydroxybutyrate from malate. Therefore, the entire six-step pathway was designed and constructed to be highly dependent on the specificities of these selected enzymes.
This study gave clarification on the principles of BT production from malate, however, the titer of BT produced from malate was very low. The enzymes and cofactors were considered as two important limiting factors. The large amount of the non-natural but structurally similar 4-HB and BDO produced as byproducts, suggested the enzymes selected for BT biosynthesis in this study performed much more efficiently towards the intermediates in the similar reduction reaction for BDO from succinate and succinyl CoA. Therefore, the biosynthetic reactions of the competitive substrates succinate and succinyl CoA in E. coli would be blocked. Additionally, the bioconversion efficiency of module 1 was significantly lower than that of module 2 by about one order of magnitude (6.4 mg/L DHB produced in the module 1 versus 55 mg/L BT produced in the module 2, under the same conditions), suggesting the rate-limiting step probably existed in the module 1. Furthermore, it has been reported malate thiokinase can catalyze non-native substrate succinate with similar efficiency to its native substrate malate12, suggesting the first step was likely to be the bottleneck of the entire pathway. Therefore, more efficient enzymes, especially for the first step, should be screened. For the purpose of reducing the number of enzymes in the pathway and enhancing the efficiency of the bioconversions, we would next also like to screen a number of more specialized enzymes that can act on the two terminal carboxyl groups simultaneously. In terms of the availability of malate, not all malate enters the BT biosynthetic pathway. It has been reported malate can not only be converted to fumarate by native fumarate hydratase, which can further be reduced to succinate by native fumarate reductase, but also can be converted to pyruvate by native malate dehydrogenase under oxygen-limited cultivation conditions in E. coli5,13. Therefore, these competitive reactions of malate substrate would be blocked.
For metabolic balance of the six-step BT pathway, the combined use of multi-T7 promoters in this study for expression of each enzyme-coding gene, not only resulted in a substantial metabolic burden on E. coli, but also would probably create imbalance in the fluxes of each enzymatic reaction of the pathway, leading to the accumulation of intermediates, which might be toxic to E. coli. Therefore, the expression levels of individual enzymes would be modulated and the activities of rate-limiting enzymes would be improved by directed evolution. The biosynthesis of BT entails the reduction of a highly oxidized substrate malate into a more reduced target molecule. For the production of one BT molecule, the pathway will consume six expensive cofactors (four NADH molecules and two ATP molecules). The E. coli in this study that were not been metabolically engineered could not support the efficient running of the BT pathway. Construction of a novel strain is necessary, one in which the production of BT is the only way to balance redox and enable its oxygen-limited growth. For the better availability of cofactors, some NADH-consuming reactions of common fermentation side products should be blocked and some enzyme-coding genes should be mutated for better supply of NADH and ATP. Additionally, the microaerobic condition needs to be optimized so that sufficient NADH is available to drive the BT pathway and excess ATP generated beyond the capacity of the pathway is also available for cell growth, maintenance and product transport.
Furthermore, the production of BT from glucose as a sole carbon source was achieved in this study and the titer level of the produced BT was the same to that by addition of malate. It suggested the carbon flux from malate to BT was the major obstacle for the production of BT from glucose, which could be overcome by the optimization methods discussed above. It is also necessary to improve the malate production from glucose for the better performance of the entire glucose to BT pathway and this work has partially been done by other scholars in E. coli. To date, its highest titer and yield reported is 34 g/L and 1.42 mol/mol glucose, respectively5. Potential still exists therefore for improving the production of BT from malate or glucose.
References
Niu, W., Molefe, M. N. & Frost, J. W. Microbial synthesis of the energetic material precursor 1,2,4-butanetriol. J. Am. Chem. Soc. U S A. 125, 12998–12999 (2003).
Yamada-Onodera, K. et al. Production of optically active 1,2,4-butanetriol from corresponding racemate by microbial stereoinversion. J. Biosci. Bioeng. 103, 494–496 (2007).
Frost, J. W. & Niu, W. Microbial synthesis of D-1,2,4-butanetriol. International patent WO 2008/091288 A2 (2008).
Song, C. W., Kim, D. I., Choi, S., Jang, J. W. & Lee, S. Y. Metabolic engineering of Escherichia coli for the production of fumaric acid. Biotechnol. Bioeng. 10.1002/bit.24868 (2013).
Zhang, X., Wang, X., Shanmugam, K. T. & Ingram, L. O. L-malate production by metabolically engineered Escherichia coli. Appl. Environ. Microbiol. 77, 427–434 (2011).
Yim, H. et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat. Chem. Biol. 7, 445–452 (2011).
Shen, C. R. et al. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl. Environ. Microbiol. 77, 2905–2915 (2011).
Zhang, X. et al. Metabolic evolution of energy-conserving pathways for succinate production in Escherichia coli. Proc. Natl. Acad. Sci. U S A. 106, 20180–20185 (2009).
Atsumi, S. et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 10, 305–311 (2008).
Zhang, X., Jantama, K., Moore, J. C., Shanmugam, K. T. & Ingram, L. O. Production of L-alanine by metabolically engineered Escherichia coli. Appl. Microbiol. Biotechnol. 77, 355–366 (2007).
Jankowski, M. D., Henry, C. S., Broadbelt, L. J. & Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 95, 1487–1499 (2008).
Hersh, L. B. Malate adenosine triphosphate lyase. Separation of the reaction into a malate thiokinase and malyl coenzyme A lyase. J. Biol. Chem. 248, 7295–7303 (1973).
Jantama, K. et al. Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol. Bioeng. 101, 881–893 (2008).
Acknowledgements
This work was financially supported by the Innovative Program of the Chinese Academy of Sciences (Grant No. KSCX2-EW-G-5), Beijing Natural Science Foundation (Grant No. 5122025) and the China Postdoctoral Science Foundation (Grant No. 2012M510584).
Author information
Authors and Affiliations
Contributions
X.L. performed all experiments, wrote the main manuscript text and prepared all table and figures. Z.C., Y.L. and Y.Z. contributed in the design of experiments and research supervision. All authors contributed to the drafting of this manuscript and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Li, X., Cai, Z., Li, Y. et al. Design and Construction of a Non-Natural Malate to 1,2,4-Butanetriol Pathway Creates Possibility to Produce 1,2,4-Butanetriol from Glucose. Sci Rep 4, 5541 (2014). https://doi.org/10.1038/srep05541
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05541
This article is cited by
-
Efficient production of 1,2,4-butanetriol from corn cob hydrolysate by metabolically engineered Escherichia coli
Microbial Cell Factories (2024)
-
Progress in research on the biosynthesis of 1,2,4-butanetriol by engineered microbes
World Journal of Microbiology and Biotechnology (2024)
-
Rational protein engineering of a ketoacids decarboxylase for efficient production of 1,2,4-butanetriol from arabinose
Biotechnology for Biofuels and Bioproducts (2023)
-
Rebooting life: engineering non-natural nucleic acids, proteins and metabolites in microorganisms
Microbial Cell Factories (2022)
-
Engineering microbial pathways for production of bio-based chemicals from lignocellulosic sugars: current status and perspectives
Biotechnology for Biofuels (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
