
Abstract
The improvement of resistance to blast, a devastating fungal disease of rice, would support the sustainable production of one of the world's staple foods, yet the identification of genes for durable resistance in rice is a challenge owing to their complicated genetic control. Here we show that map-based cloning of Pi35 identifies multiple functional polymorphisms that allow effective control of the disease and that Pi35 is allelic to Pish, which mediates race-specific resistance to blast and encodes a protein containing a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs). Analysis using Pish–Pi35 chimeric genes demonstrated that multiple functional polymorphisms cumulatively enhance resistance and that an amino acid residue in a LRR of Pi35 is strongly associated with the gene's mediation of quantitative but consistent resistance to pathogen isolates in Japan, in contrast to Pish, which mediates resistance to only a single isolate. Our results reinforce the substantial importance of mining allelic variation for crop breeding.
Similar content being viewed by others


Introduction
Rice blast caused by the fungal pathogen Magnaporthe oryzae is a destructive disease of rice (Oryza sativa L.)1. The use of resistance (R) genes is an effective way to control it. Extensive studies have identified more than 90 R genes for resistance to M. oryzae from diverse genetic resources2. Most of them are race specific and all of those genes for race-specific resistance that have been cloned so far encode proteins containing a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs), constituting a major class of R genes in plants3,4; an exception is Pid2, which encodes a receptor-like kinase5. Race-specific resistance induces strong defense responses that are characterized by a hypersensitivity reaction (HR)6 and are explained by the gene-for-gene theory7, yet this resistance is rapidly overcome by the pathogen8,9. Plant breeders are thus obliged to find further R genes and introduce them to cultivars over and over, resulting in an evolutionary “arms race” between the crop and the pathogen10.
In contrast, resistance conferred by quantitative trait loci (QTLs) is characterized by a susceptible infection type, usually without race specificity or gene-for-gene interaction11. In general, cultivars carrying resistance QTLs maintain their resistance for a long time. Therefore, QTL-mediated resistance is considered to be more durable than race-specific resistance, possibly because of decreased selection pressure against the pathogen. Intensive genetic mapping has highlighted several chromosomal regions harboring beneficial QTLs12,13. From these QTLs, Pi21 is the first gene for resistance to M. oryzae cloned in rice14. However, on account of their small phenotypic effects, identification of the genes underlying QTLs is still a challenge. For example, qBR4-2, a gene complex comprising 3 loci that cumulatively enhance resistance to M. oryzae, presents difficulties in cloning of individual resistance alleles15. This complexity underlies the complicated genetic control of QTL-mediated resistance to M. oryzae16,17. In addition, the low resolution of genetic maps makes it difficult for plant breeders to introduce resistance QTL alleles from a donor without penalty, on account of linkage drag: the co-introduction of undesirable agricultural traits with the resistance14.
Pi35 is a QTL found in the Japanese breeding line Hokkai 188. As this line has maintained consistent resistance in natural field conditions since 196118, it has been used as a source of QTL-mediated resistance in the Japanese rice breeding program. Pi35 has been roughly mapped to a region on chromosome 1, where race-specific resistance genes Pi37 and Pish are located19,20. These 2 genes are typical R genes that encode NBS-LRR proteins. Their tandem arrangement implies a duplication event in this region, as in other resistance gene complexes21,22,23 and in the Pi9 and Pik gene complexes on chromosomes 6 and 11, respectively24,25,26,27,28. These examples indicate that NBS-LRR genes are an important component in the evolution of race-specificity in plant resistance. However, direct evidence that explains the linkage between NBS-LRRs and QTL-mediated blast resistance is limited.
We cloned Pi35 by a map-based cloning strategy to enhance our understanding of the molecular basis for QTL-mediated resistance and to clarify the relationship between the QTL and race-specific genes in the chromosomal region.
Results
Fine-scale genetic mapping of Pi35 locus
To delimit the Pi35 locus, we screened recombinants in a mapping population in which the locus segregated. In field tests, we evaluated 56 pairs of inbred lines from 3112 F5 plants, which include lines with recombination events within or around Pi35 and non-recombinant control lines. Data for the lines that delimit the position of the Pi35 locus are presented (Fig. 1a). The mean lesion area of the lines carrying the resistance allele was <12%, similar to that of the donor parent, Hokkai 188, whereas the mean lesion area of the lines carrying the susceptible allele was >60%, similar to that of the susceptible parent, Danghang-Shali. We concluded that Pi35 lies in the 59.2-kb region between markers P33688 and S010 (Fig. 1a).

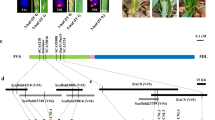
Genetic mapping and physical maps of Pi35.
(a) Graphical genotypes of recombinant inbred lines used to delimit the Pi35 locus. Red bars indicate chromosomes derived from the resistant cultivar Hokkai 188; blue, from the susceptible cultivar Danghang-Shali. Positions are based on the International Rice Genome Sequencing Project 1.0 pseudomolecules of the Nipponbare genome. Underlined markers are dominant; the marker in parentheses is not polymorphic and is used for confirming the presence of amplified product. The location of the Pi35 locus, indicated at the bottom, is based on phenotypic data obtained by field test, tabulated on the right. Data are means ± SD. Asterisks illustrate results of 2-tailed t-tests: ***P < 0.001; n.s., not significant, P > 0.05. (b) Sequence comparison of the Pi35 region between the susceptible cultivar Nipponbare and the resistant cultivar Hokkai 188. Positions of gene loci described in RAP-DB are indicated by boxes: white boxes, non-coding regions; green boxes, coding regions. The names of the genes that encode proteins containing a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs) are in bold. Blue vertical bars indicate DNA sequence variations between Hokkai 188 (resistant) and Nipponbare (susceptible); red triangles indicate those that cause amino acid changes. The similarity of the Danghang-Shali genome to Nipponbare is determined from the presence of PCR products at 18 loci obtained by using 14 primer pairs listed in Supplementary Table S1 and the results are indicated at the bottom; circles, the presence of PCR products; crosses, the absence of PCR products.
Candidate gene for Pi35
In the new Os-Nipponbare-Reference-IRGSP-1.0 genome assembly29, we delimited the Pi35 locus to a region where the Rice Annotation Project30 identified 8 gene loci (Fig. 1b). Four of these loci (Os01g0781100, Os01g0781200, Os01g0781700 and Os01g0782100) show similarity to previously reported disease resistance proteins containing NBS-LRRs. Comparison of the candidate region between Hokkai 188 with Nipponbare revealed overall similarity, but a 20.1-kb deletion in Hokkai 188 resulted in the loss of 2 loci, including Os01g0781700, which corresponds to Pi3719 (Fig. 1b). We found 17 sequence variations, of which only 6 in Os01g0782100, which corresponds to Pish20, showed variation in the deduced amino acid sequence; this left Os01g0782100 as the candidate for Pi35 (Fig. 1b). PCR analysis identified that the candidate region of the susceptible cultivar, Danghang-Shali, had no similarity with those of cultivars, Hokkai 188 or Nipponbare except around the P33688 locus and downstream of the S009 locus (Fig. 1b).
Characterization of Pi35 allele
Leaves of KS-Pi35, a near-isogenic line carrying Pi35 in the genetic background of a susceptible cultivar (KS) (Fig. 2a), had a smaller lesion area than KS in a greenhouse inoculation test (Fig. 2b). Pi35 had a consistent effect against all 6 isolates, which represent widely distributed races of M. oryzae and against isolate Kyu77-07A avirulent to KS carrying Pish31, although the Pi35-triggered resistance was incomplete compared with that triggered by Pish (Fig. 2b, c). The results confirm empirical observations: plants with Pish show HR-inducing resistance to Kyu77-07A, but are highly susceptible in field tests and in inoculation tests to other Japanese isolates. In contrast, plants with Pi35 do not show HR-inducing resistance against any of the isolates tested, but lesions against virulent races were smaller than in plants carrying Pish.

Characterization of the resistant Pi35 allele.
(a) Chromosome map of near-isogenic line KS-Pi35. Gray boxes, chromosomes from the susceptible cultivar Koshihikari (KS); red box, from the resistant donor cultivar Hokkai 188 (H188). Positions are based on the International Rice Genome Sequencing Project 1.0 pseudomolecules of the Nipponbare genome. “Del.” represents a 20.1-kb deletion compared with Nipponbare. (b) Lesion area of 30-day–old plants inoculated with each of 7 M. oryzae isolates. Pi35 allele is from H188. The Pish allele from KS shows resistant infection type accompanying hypersensitive reaction only against isolate Kyu77-07A. (c) Representative diseased leaves of KS-Pi35 and KS that were challenged by either Ina86-137 or Kyu77-07A. (d) Expression profiles of PR2 (AK070677) and Os01g0782100 after inoculation of isolates Ina86-137 (solid) and Kyu77-07A (dashed) into 30-day–old KS (blue) and KS-Pi35 (red) plants. Leaves were sampled at 0, 3, 6, 12, 24, or 48 h after inoculation (hpi). Expression of PR2 and Pi35 was standardized to that of mock inoculation. Data are means ± SEM (n = 3). Total RNA was extracted from leaves and reverse-transcribed and qPCR was performed. Primer pairs are listed in Supplementary Table S2.
The expression of pathogenesis-related gene 2 (PR2, AK070677) was increased in plants carrying Pi35 at 48 h after inoculation with the avirulent isolate Kyu77-07A or the virulent isolate Ina86-137 and in plants carrying Pish challenged by Kyu77-07A but not by Ina86-137 (Fig. 2d). As the level of Pi35 expression was the same as that of Pish before and after inoculation (Fig. 2d), we speculate that genetic variation in the coding region of Os01g0782100 controls the differences in lesion area and in the level of PR2 expression.
Natural variations at Os01g0782100
Variations were found in the NBS-domain-containing region and in the LRR region between the Pish allele from Nipponbare and the Pi35 allele from Hokkai 188 (Fig. 3a). Sequence analysis of the region of Os01g0782100 downstream of the BamHI site identified 6 haplotypes on the basis of 4 amino acid variations in LRRs among Asian cultivated rice32 (Fig. 3b, Supplementary Fig. S1). In field tests, only the Hokkai 188 type had a significantly smaller average lesion area than the control among backcrossed lines carrying the respective Os01g0782100 haplotypes in the genetic background of a susceptible cultivar (Fig. 3b).

Natural variations at the Os01g0782100 locus.
(a) Differences in DNA sequences (and amino acid sequences in parentheses) between the Pish allele from Nipponbare (black) and the Pi35 allele from Hokkai 188 (red). Positions of respective variations from the start codon are indicated. The gene is divided into 2 regions by the BamHI site: the NBS region and the LRR region. The NBS region contains coiled-coil (CC), nucleotide-binding adaptor shared by APAF-1, R proteins and CED-4 (NB-ARC) and two LRRs; the LRR region contains 9 LRRs. (b) Sequence analysis of the LRR region of Os01g0782100 identifies 6 haplotypes on the basis of 4 amino acid variations (boxed letters) among rice accessions. The Hokkai 188 type is absent in a core collection of 69 cultivars that represent the genetic diversity of Asian cultivated rice. Bars indicate the average lesion areas of backcrossed lines (n = 3) carrying the respective Os01g0782100 haplotypes (from indicated donor cultivars) evaluated in a field test. Asterisks illustrate the results of two-tailed t-tests: ***P < 0.001; * P ≤ 0.05; n.s., not significant, P > 0.05. Error bars indicate SD.
Complementation testing and molecular characterization of Pi35
Transforming a 13.2-kb genomic fragment containing Os01g0782100 from Hokkai 188 (Fig. 1b) into a susceptible cultivar conferred resistance to multiple isolates in T0 plants (Fig. 4a) and in progeny tests (Fig. 4b, “HH-Pi35”). To clarify whether variation in the NBS region or in the LRR region alters resistance to M. oryzae, we produced chimeric gene constructs between Pish and Pi35, introduced them into the susceptible cultivar and inoculated plants with virulent Ina 86-137 or with Kyu77-07A, avirulent to plants carrying Pish. Replacement of the LRR region of Pish with that of Pi35 resulted in the loss of race-specific resistance to the avirulent isolate, but conferred QTL-mediated resistance to both avirulent and virulent isolates (Fig. 4b, “NH-Pi35”). Out of 4 amino acid residues in the LRR region of Pi35 that differ from those in Pish, an Asp residue at position 1054 from the start codon significantly reduced the lesion area against Ina 86-137, (Fig. 4b, “HN1054D-Pi35”). This Glu to Asp substitution (E1054D) was still not sufficient to confer the full effect of Pi35 against Ina 86-137; however, it had a deleterious effect with Kyu77-07A. The Asp residue was strongly associated with the level of resistance to M. oryzae in the test with introgression lines (Fig. 3b). Plants carrying the chimeric Pi35-NBS/Pish-LRR gene (HN-Pi35) showed a non-significantly smaller lesion area than those carrying Pish (NN-Pi35) (Fig. 4b).

Complementation testing and molecular characterization of Pi35.
(a) Lesions of T0 Aichiasahi plants carrying a Pi35 construct (HH-Pi35) or an empty vector, 7 d after inoculation with 4 M. oryzae isolates: Ina 86-137, Ai 79-142, P-2b, or Ai 74-134. Data are means ± SD (n = 5). The photos below the graph show the phenotypic differences between the two. (b) Lesions on T2 or T3 Aichiasahi plants carrying Pi35, Pish, or chimeric gene constructs, 7 d after inoculation with Ina 86-137 or Kyu77-07A. “E → D” indicates substitution of a single amino acid residue Glu with Asp at position 1054 in the 7th LRR. Five single-copy lines derived from independent T0 plants were used; inoculation tests were performed 3 times. Controls: Pi35, near-isogenic line for Pi35 (KS-Pi35); Pish, Koshihikari (KS); Vector, T3 Aichiasahi plants carrying empty vector. Data are means ± SEM. Green bars followed by the same letters are not significantly different according to Tukey's HSD test at the 5% level.
Discussion
We found that Pi35 is involved in QTL-mediated resistance and is a member of the NBS-LRR class, a major class of R genes3,4. A weak allele of an R gene is considered to be a component of plant defense10,12,33, yet direct evidence that links NBS-LRRs and QTL-mediated resistance is limited. On account of the small gene effects, isolating genes associated with QTL-mediated resistance is time-consuming and more difficult than isolating genes for race-specific resistance. We identified Pi35 at the Os01g0782100 locus by fine-scale genetic mapping, complementation testing and progeny testing and showed that Pi35 is allelic to Pish, which is responsible for race-specific resistance to M. oryzae20.
The important finding of our study is that a combination of multiple functional polymorphisms in the gene confers a practical level of resistance. Although the effects of individual polymorphisms other than E1054D appeared to be non-significant, this substitution confers only about half of the effect of the Pi35 allele, suggesting that the rest is due to a cumulative effect of other polymorphisms. Race-specific resistance is easily overcome by rapid evolution of the fungal pathogen8,9. Enhancing resistance by combining multiple variations in a gene might confer a more durable form of resistance, owing to decreased selection pressure against the pathogen than by a single functional polymorphism that confers race-specific resistance and might allow us to avoid an evolutionary arms-race between a crop and its pathogens10.
Molecular characterization of the Pi35 and Pish alleles allowed us to dissect sequence variations associated with QTL-mediated and race-specific resistance, respectively. The NBS domain is considered to be involved in signal transduction34,35, while the LRR region plays a critical role in the determination of race specificity36,37. Recent evidence shows that the NBS domain also determines race specificity19. Our data confirm that the LRR region is the primary determinant of Pish-mediated resistance. In addition, the LRR region of Pi35 significantly reduced the lesion area provoked by isolates both avirulent and virulent to cultivars with Pish. Among the 4 amino acid residues of Pi35 that differ from Pish in the LRR region, Asp at position 1054 significantly reduced the lesion area. The high susceptibility of an introgression line with the same amino acid residues in the LRR region as in Pi35 except for the Asp (Padi Kuning allele in Fig. 3b) supports the hypothesis that this residue is important to the defense response to widely distributed pathogen races in Japan. The low frequency of accessions with Asp at this position among Asian cultivated rice accessions implies that the mutation occurred recently. Our experimental data suggest that a single mutation event can offer the opportunity to generate a beneficial QTL allele.
Dynamic structural changes in genomic regions containing R genes are considered to be the basis for the evolution of disease resistance in plants in the context of the co-evolution of plants and their pathogens38,39,40,41. Previous studies identified an array of R genes and uncovered their evolutionary processes on the basis of sequence comparison among genotypes in several species. Pi35 is located in a cluster of 4 R gene loci, Os01g0781100, Os01g0781200, Os01g0781700 and Os01g0782100. It is proposed that Os01g0781100 was the ancestral form, Os01g0781200 was generated next and either Os01g0781700 or Os01g0782100 was generated last19,20. Since Hokkai 188 is likely to carry a genotype without the final gene duplication and lacks Os01g0781700, our results support the hypothesis that Os01g0781700 is the most recent locus20. The observations from the previous studies and here suggest that the 2 newer loci contribute to resistance to M. oryzae, but that the older 2 do not. Pi35, a rare allele at Os01g0782100, might be more adaptive in regions where rice is severely damaged by blast. Therefore, the R gene cluster on rice chromosome 1 strongly supports the idea that multiple duplication and substitution events generate new R genes38,39,40.
Our results show that an allele of a typical R gene can confer a more durable form of resistance to M. oryzae, reminding us of the importance of mining allelic variation for use in crop breeding42,43. The fact that even a single amino acid substitution in an R protein can alter disease resistance in a quantitative manner might explain the continuous phenotypic variations that researchers find in diverse genetic resources or mapping populations16,17. QTL-mediated resistance is often undetectable on account of the race-specific resistance that is induced by pathogen races that are avirulent to cultivars carrying some R genes. As well as differential fungal isolates44,45, advanced-backcrossed lines46,47 are useful for the identification and fine-scale mapping of beneficial QTLs from diverse resources, on account of their efficient incorporation into elite genetic backgrounds without linkage drag14. A near-isogenic line for Pi35 (KS-Pi35) is being evaluated in field trials to confirm the absence of undesirable agronomic traits; monitoring of this line will confirm or deny the durability of Pi35-mediated resistance. Such research projects allow us to develop more durable crops for sustainable food production by combining multiple QTLs into elite genetic backgrounds.
Methods
Fine-scale genetic mapping and development of near-isogenic line for Pi35
The resistance QTL Pi35 was located around the marker locus RM1216 on chromosome 1 by an analysis using F3 lines of the cross between the susceptible indica cultivar Danghang-Shali and the resistant japonica breeding line Hokkai 18818. A preliminary genetic mapping using progeny lines from a previous study and the addition of genetic markers allowed us to delimit the Pi35 locus to an 892-kb region between marker loci P31065 and RM1003 on the rice reference genome. We used F5 plants in which resistance was segregated at this marker interval for fine-scale genetic mapping of Pi35. From 3112 F5 plants, we selected 56 fertile, normally growing plants among 124 recombinants between marker loci P31035 and RM1003 and evaluated their progeny lines (F6) by phenotype to determine the genotype at the Pi35 locus. A near-isogenic line for Pi35 was developed by 4 rounds of marker-assisted backcrossing using the susceptible japonica cultivar Koshihikari as the recurrent parent and the selection of minimum introgression in the BC4F2 and BC4F3 generations from nearly 800 individuals. Supplementary Table S1 shows PCR primers.
Construction of bacterial artificial chromosome library, sequencing and gene prediction
Megabase-size DNA was prepared from young leaves of Hokkai 188 as described48. A bacterial artificial chromosome (BAC) library was constructed by ligation of the megabase DNA with the pIndigoBAC vector (Epicentre) and transformation of BACs into DH10B Escherichia coli cells (Invitrogen)49. The library consisted of 19082 clones with an average insert size of 115 kb. Clones containing the Pi35 locus were screened with DNA markers P33688 and RM11744 (Supplementary Table S1) and a positive clone (HO188-07L09) was shotgun sequenced50.
Comparison of genome structure
A sequence of the BAC clone HO188-07L09 containing a 183-kb insert including Pi35 was aligned with the reference genome of japonica cultivar Nipponbare (IRGSP)29 by using the SEQUENCHER software (Gene Codes Co.). The structure of the susceptible Danghang-Shali's genome was analyzed with a set of 14 PCR primers designed from the Nipponbare genome (Supplementary Table S1) to identify Danghang-Shali genomic regions with similarity to the Nipponbare genome on the basis of the presence or absence of amplicons.
Detection of natural variation in Pi35
DNAs from 69 cultivars that represent the genetic diversity of Asian cultivated rice_ENREF_3532 were amplified and sequenced with the primer pairs listed (Supplementary Table S1). Deduced amino acid sequences were aligned with CLUSTALW software51. To confirm the effect of each haplotype on resistance to M. oryzae, we used 6 backcrossed lines, each carrying the target chromosome from 1 of 6 donors: Hokkai 188 (BC4F4), Padi Kuning (BC2F2), LAC23 (BC4F4), IR64 (BC4F4), Bleiyo (BC4F3), or Nipponbare (BC4F4). The lines for LAC23 (SL2207), IR64 (SL2003) and Nipponbare (SL602) were used in previous studies52,53,54.
Complementation test
A 13.2-kb SpeI–XhoI fragment containing the resistant Pi35 allele from the Hokkai 188 BAC clone HO188-07L09 (HH-Pi35) was inserted into the multiple cloning site of the binary vector pPZP2H-lac55 to create pPZ-HH-Pi35. pPZ-HH-Pi35 was introduced into the susceptible cultivar Aichiasahi by Agrobacterium-mediated transformation56. The presence of the transgene was confirmed by the presence of BamHI fragments obtained from the resistant Pi35 allele but not from susceptible allele (primer pair 5′-AAGCGTAAAAGACTCTAGATTC-3′/5′-GACTCTAGATTCTCAAAATAGG-3′). We inoculated 131 T0 plants carrying HH-Pi35 and 60 T0 plants carrying the empty vector with each one of 4 M. oryzae isolates (Ina 86-137, Ai 79-142, P-2b, or Ai 74-134).
Chimeric gene analysis
The region between the start codon and a unique BamHI site (which we designate as NBS) encodes the NBS domain and 2 LRRs, whereas the region between the BamHI site and the stop codon (which we designate as LRR) encodes 9 LRRs. The NBS region contains 2 amino acid variations and the LRR region contains 4 variations between the Pish allele from the susceptible Nipponbare and the Pi35 allele from the resistant Hokkai 188. To test the effect of the respective regions on resistance to M. oryzae, we produced 4 additional types of constructs: NN-Pi35 (both NBS and LRR regions from Nipponbare), HN-Pi35 (NBS from Hokkai 188 + LRR from Nipponbare), NH1054D-Pi35 (a substitution of the 1054th amino acid residue Glu in HN-Pi35 with the Hokkai 188–specific Asp by PCR-based site-directed mutagenesis57) and NH-Pi35 (NBS from Nipponbare + LRR from Hokkai 188). The chimeric genes were obtained by digestion with BamHI followed by ligation. The same procedure as used for the complementation test was used to create transgenic plants. Five T2 or T3 lines that originated from independent T0 plants and carried a single-copy transgene were selected on the basis of the relative amounts of the hygromycin phosphotransferase (HPT, K01193) and Pi35 genes, normalized to rice ubiquitin 2 (RUBQ2, AF184280)58, by using TaqMan probe–based quantitative real-time PCR (qPCR) (Supplementary Table S2). qPCR was performed with qPCR MasterMix (Eurogentec) in a 7900HT Fast Real-time PCR System (Applied Biosystems) according to the manufacturers' instructions. Five T3 lines that carried a single copy of HH-Pi35 were used for reference. Three controls were used for lesion area measurement: Pi35, near-isogenic line for Pi35 (KS-Pi35); Pish, Koshihikari (KS); vector, T3 Aichiasahi plants carrying the empty vector.
Expression analysis
Total RNA was isolated from 30-day–old plants at 0 (just after inoculation), 3, 6, 12, 24 and 48 h after inoculation by using an RNeasy Plant Mini Kit (Qiagen) and total RNA was reverse-transcribed with Oligo-dT18 primer and a First-Strand cDNA Synthesis Kit (GE Healthcare) according to the manufacturer's instructions. qPCR was performed as above with the primers listed in Supplementary Table S2. The expression level of RUBQ2 was used to standardize the RNA sample for each analysis. Expression of PR2 and Os01g0782100 was standardized to that of mock inoculation. RT-PCR and qPCR were performed at least 3 times for 3 independent samples per line.
Assessment of resistance to M. oryzae
Danghang-Shali and Hokkai 188 show a susceptible infection type, lacking HR, in inoculation tests using pathogenic races of M. oryzae widely distributed in Japan59. In fine-scale genetic mapping, we evaluated the resistance to M. oryzae of homozygous progeny of the recombinant plants (F6 lines) in an experimental field at the National Institute of Agrobiological Sciences (Tsukuba, Ibaraki), where the predominant fungal race is 037.3 and gained susceptible lesions on our plants. Plants were grown from 50 seeds per line in each of 3 independent experiments. The lesion area of 60- to 70-day–old plants was scored against a published reference scale14. The susceptible Aichiasahi was grown on either side of each line. In complementation testing and chimeric gene analysis, 30-day–old plants grown in the greenhouse and inoculated as described previously14 were used to assess resistance. The percentage of lesion area per leaf at 7 d after inoculation was scored against the reference scale. In chimeric gene analysis, 24 plants per line were used and the experiments were performed 3 times.
References
Ou, S. H. Rice Diseases, 2nd edn. (Commonwealth Agricultural Bureaux, 1985).
Monaco, M. K. et al. Gramene 2013: comparative plant genomics resources. Nucleic Acids Res. 42, D1193–D1199 (2013).
Martin, G. B., Bogdanove, A. J. & Sessa, G. Understanding the functions of plant disease resistance proteins. Annu. Rev. Plant Biol. 54, 23–61 (2003).
McHale, L., Tan, X., Koehl, P. & Michelmore, R. W. Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212 (2006).
Chen, X. et al. A B-lectin receptor kinase gene conferring rice blast resistance. Plant J. 46, 794–804 (2006).
Greenberg, J. T. & Yao, N. The role and regulation of programmed cell death in plant-pathogen interactions. Cell. Microbiol. 6, 201–211 (2004).
Flor, H. H. Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296 (1971).
Bonman, J. M., Khush, G. S. & Nelson, R. J. Breeding rice for resistance to pests. Annu. Rev. Phytopathol. 30, 507–528 (1992).
Kiyosawa, S. Genetic and epidemiological modeling of breakdown of plant disease resistance. Annu. Rev. Phytopathol. 20, 93–117 (1982).
Jones, J. D. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Ezuka, A. Field resistance of rice varieties to rice blast disease. Rev. Plant Prot. Res. 5, 1–21 (1972).
Poland, J. A., Balint-Kurti, P. J., Wisser, R. J., Pratt, R. C. & Nelson, R. J. Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14, 21–29 (2009).
Kou, Y. & Wang, S. Broad-spectrum and durability: understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 13, 181–185 (2010).
Fukuoka, S. et al. Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325, 998–1001 (2009).
Fukuoka, S. et al. A multiple gene complex on rice chromosome 4 is involved in durable resistance to rice blast. Theor. Appl. Genet. 125, 551–519 (2012).
Rao, Z. M. et al. Genetic dissections of partial resistances to leaf and neck blast in rice (Oryza sativa L.). Yi Chuan Xue Bao 32, 555–565 (2005).
Wu, J. L. et al. Genetic control of rice blast resistance in the durably resistant cultivar Gumei 2 against multiple isolates. Theor. Appl. Genet. 111, 50–56 (2005).
Nguyen, T. T. et al. Pi35(t), a new gene conferring partial resistance to leaf blast in the rice cultivar Hokkai 188. Theor. Appl. Genet. 113, 697–704 (2006).
Lin, F. et al. The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics 177, 1871–1880 (2007).
Takahashi, A., Hayashi, N., Miyao, A. & Hirochika, H. Unique features of the rice blast resistance Pish locus revealed by large scale retrotransposon-tagging. BMC Plant Biol. 10, 175 (2010).
Wang, G. L. et al. Xa21D encodes a receptor-like molecule with a leucine-rich repeat domain that determines race-specific recognition and is subject to adaptive evolution. Plant Cell 10, 765–779 (1998).
Dixon, M. S. et al. The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84, 451–459 (1996).
Xiao, S. et al. Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science 291, 118–120 (2001).
Qu, S. et al. The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172, 1901–1914 (2006).
Zhou, B. et al. The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol. Plant Microbe Interact. 19, 1216–1228 (2006).
Zhai, C. et al. The isolation and characterization of Pik, a rice blast resistance gene which emerged after rice domestication. New Phytol. 189, 321–334 (2011).
Yuan, B. et al. The Pik-p resistance to Magnaporthe oryzae in rice is mediated by a pair of closely linked CC-NBS-LRR genes. Theor. Appl. Genet. 122, 1017–1028 (2011).
Hua, L. et al. The isolation of Pi1, an allele at the Pik locus which confers broad spectrum resistance to rice blast. Theor. Appl. Genet. 125, 1047–1055 (2012).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4 (2013).
Sakai, H. et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol. 54, e6 (2013).
Imbe, T. & Matsumoto, S. Inheritance of resistance of rice varieties to the blast fungus strains virulent to the variety “Reiho”. Japan. J. Breed. 35, 332–339 (1985).
Kojima, Y., Ebana, K., Fukuoka, S., Nagamine, T. & Kawase, M. Development of an RFLP-based rice diversity research set of germplasm. Breed. Sci. 55, 431–440 (2005).
Li, Z. K. et al. A “defeated” rice resistance gene acts as a QTL against a virulent strain of Xanthomonas oryzae pv. oryzae. Mol. Gen. Genet. 261, 58–63 (1999).
Traut, T. W. The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem. 222, 9–19 (1994).
Takken, F. L., Albrecht, M. & Tameling, W. I. Resistance proteins: molecular switches of plant defence. Curr. Opin. Plant Biol. 9, 383–390 (2006).
Michelmore, R. W. & Meyers, B. C. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 8, 1113–1130 (1998).
Parker, J. E. et al. The Arabidopsis downy mildew resistance gene RPP5 shares similarity to the toll and interleukin-1 receptors with N and L6. Plant Cell 9, 879–894 (1997).
Hulbert, S. H., Webb, C. A., Smith, S. M. & Sun, Q. Resistance gene complexes: evolution and utilization. Annu. Rev. Phytopathol. 39, 285–312 (2001).
Richly, E., Kurth, J. & Leister, D. Mode of amplification and reorganization of resistance genes during recent Arabidopsis thaliana evolution. Mol. Biol. Evol. 19, 76–84 (2002).
Meyers, B. C., Kozik, A., Griego, A., Kuang, H. & Michelmore, R. W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant cell 15, 809–834 (2003).
Hayashi, N. et al. Durable panicle blast-resistance gene Pb1 encodes an atypical CC-NBS-LRR protein and was generated by acquiring a promoter through local genome duplication. Plant J. 64, 498–510 (2010).
Bhullar, N. K., Street, K., Mackay, M., Yahiaoui, N. & Keller, B. Unlocking wheat genetic resources for the molecular identification of previously undescribed functional alleles at the Pm3 resistance locus. Proc. Natl. Acad. Sci. U. S. A. 106, 9519–9524 (2009).
Bhullar, N. K., Zhang, Z., Wicker, T. & Keller, B. Wheat gene bank accessions as a source of new alleles of the powdery mildew resistance gene Pm3: a large scale allele mining project. BMC Plant Biol. 10, 88 (2010).
Hayashi, N. Rice blast fungus. In MAFF Microorganism Genetic Resources Manual 18, 13–15 (2005).
Telebanco-yanoria, M. J. et al. A set of standard differential blast isolates (Magnaporthe grisea (Hebert) Barr.) from the Philippines for rice (Oryza sativa L.) resistance JARQ. 42, 23–34 (2008).
Ali, M. L., Sanchez, P. L., Yu, S.-B., Lorieux, M. & Eizenga, G. C. Chromosome Segment Substitution Lines: A Powerful Tool for the Introgression of Valuable Genes from Oryza Wild Species into Cultivated Rice (O. sativa). Rice 3, 218–234 (2010).
Fukuoka, S., Nonoue, Y. & Yano, M. Germplasm enhancement by developing advanced plant materials from diverse rice accessions. Breed. Sci 60, 509–517 (2010).
Zhang, H.-B., Zhao, X., Ding, X., Paterson, A. H. & Wing, R. A. Preparation of megabase-size DNA from plant nuclei. Plant J. 7, 175–184 (1995).
Osoegawa, K. et al. An improved approach for construction of bacterial artificial chromosome libraries. Genomics 52, 1–8 (1998).
Fleischmann, R. D. et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–512 (1995).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Abe, T. et al. Detection of QTLs to reduce cadmium content in rice grains using LAC23/Koshihikari chromosome segment substitution lines. Breed. Sci 63, 284–291 (2013).
Hori, K. et al. Detection of quantitative trait loci controlling pre-harvest sprouting resistance by using backcrossed populations of japonica rice cultivars. Theor. Appl. Genet. 120, 1547–1557 (2010).
Nonoue, Y. et al. Developement of reciprocal chromosome segment substitution lines between a japonica cultivar Koshihikari and an indica cultivar IR64 in rice. Breed. Res. 12 sup. 2–55 (2010).
Fuse, T., Sasaki, T. & Yano, M. Ti plasmid vectors useful for functional analysis of rice genes. Plant Biotech. 18, 219–222 (2001).
Toki, S. Rapid and efficient Agrobacterium mediated transformation in rice. Plant Mol. Biol. Rep. 16, 16–21 (1997).
Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 (1989).
Matsubara, K. et al. Ehd3, encoding a plant homeodomain finger-containing protein, is a critical promoter of rice flowering. Plant J. 66, 603–612 (2011).
Naito, H., Iwano, M., Fujita, Y. & Ashizawa, T. Distribution of pathogenic races of rice blast fungus in Japan in 1994. Misc. Pub. Natl. Agric. Res. Cent. 39, 1–92 (1999).
Acknowledgements
This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation QTL2002, Genomics-based Technology for Agricultural Improvement, LCT0013, Development of mitigation and adaptation techniques to global warming in the sectors of agriculture, forestry and fisheries, rice—2001). We thank Dr. H. Hayashi, National Institute of Agrobiological Sciences (Tsukuba, Japan), for kindly providing M. oryzae isolate Kyu77-07A. We thank editors from ELSS, Inc. (http://elss.co.jp/en/) for editing the manuscript.
Author information
Authors and Affiliations
Contributions
S.F. designed the experiments and wrote the manuscript. T.T.T.N. and S.K. developed the mapping population. S.-I.Y., K.O. and K.S. performed transformation experiments. S.-I.Y., M.R. and S.F. developed transformant lines. S.F., N.Y. and Y.F. performed phenotyping in glasshouse inoculation tests. M.R. and S.F. performed phenotyping in field tests. S.-I.Y. and S.F. performed genotyping for cloning Pi35. T.M. performed BAC clone analysis. S.-I.Y., K.N. and S.F. performed sequencing analysis. Y.U. performed expression analysis and copy number measurement in transformants. M.Y. provided advice on the experiments and the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary tables and figure
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Fukuoka, S., Yamamoto, SI., Mizobuchi, R. et al. Multiple functional polymorphisms in a single disease resistance gene in rice enhance durable resistance to blast. Sci Rep 4, 4550 (2014). https://doi.org/10.1038/srep04550
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04550
This article is cited by
-
Race specific and non-specific resistance to Magnaporthe oryzae and QTL mapping in wild introgression lines using the standard differential system
Tropical Plant Pathology (2024)
-
Bulk segregant analysis coupled with transcriptomics and metabolomics revealed key regulators of bacterial leaf blight resistance in rice
BMC Plant Biology (2023)
-
Resistance QTLs controlling leaf and neck blast disease identified in a doubled haploid rice population
Euphytica (2023)
-
Broadening the horizon of crop research: a decade of advancements in plant molecular genetics to divulge phenotype governing genes
Planta (2022)
-
RXam2, a NLR from cassava (Manihot esculenta) contributes partially to the quantitative resistance to Xanthomonas phaseoli pv. manihotis
Plant Molecular Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
