
Abstract
Biomolecular signaling is of utmost importance in governing many biological processes such as the patterning of the developing embryo where biomolecules regulate key cell-fate decisions. In vivo, these factors are presented in a spatiotemporally tightly controlled fashion. Although state-of-the-art microfluidic technologies allow precise biomolecule delivery in time and space, long-term (stem) cell culture at the micro-scale is often far from ideal due to medium evaporation, limited space for cell growth or shear stress. To overcome these challenges, we here introduce a concept based on hydrogel microfluidics for decoupling conventional, macro-scale cell culture from precise biomolecule delivery through a gel layer. We demonstrate the spatiotemporally controlled neuronal commitment of mouse embryonic stem cells via delivery of retinoic acid gradients. This technique should be useful for testing the effect of dose and timing of biomolecules, singly or in combination, on stem cell fate.
Similar content being viewed by others

Introduction
Spatiotemporally variable signaling cues control cellular and multicellular behavior in many crucial biological systems. A case in point is the concentration- and time-dependent display of biomolecules termed morphogens that locally control key cell-fate decisions such as lineage commitment during the patterning of the early embryo1. Insights gained from developmental biology studies have also spurred advances in differentiating pluripotent stem cell such as embryonic stem cells (ESC) into various specialized cell types2. However, typical ESC differentiation protocols, relying on the formation of cell aggregates termed embryoid bodies (EB), expose cells to bulk culture conditions that crudely recapitulate the intricate biomolecule display found during embryogenesis. Although this method allows at least to some extent the temporal recapitulation of embryonic gene expression patterns in vitro, developing EBs lack an embryo-like spatial organization3. The delivery of graded biomolecules by traditional methods such as micropipettes or the Boyden chamber is rather limited in mimicking natural biomolecule presentation.
The emergence of microfluidic technology has revolutionized the generation of in vitro model systems by affording very precise, picoliter-scale fluid handling, parallelization of experiments and minimization of reagent consumption4,5. Microfluidic chips also offer unprecedented means to systematically probe the role of microenvironmental signals on stem cell fate in high-throughput6 and with exquisite spatiotemporal resolution7,8. Application of such microsystems have for example allowed to polarize single EBs9 or direct neural progenitor differentiation by soluble gradients10.
However, state-of-the-art microfluidic culture systems are often not ideal for long-term (stem) cell culture11,12. Firstly, since cells are continuously exposed to fluid flow in most microsystems, the constant removal of autocrine signals and shear stress may be problematic13, even though more complex shear-free systems have been reported14,15,16,17,18,19. Secondly, microsystems are typically made of poly(dimethylsiloxane) (PDMS), a material that, despite its many advantages for microfabrication and its excellent gas permeability20, suffers from susceptibility to liquid evaporation, protein adsorption from the medium21, leaching of non-reacted compounds and hydrophobic recovery22,23. Thirdly, it is not trivial to functionalize PDMS surfaces with biomolecules in order, for example, to mimic some of the natural interactions of stem cells with their microenvironment that are often critical in maintaining stem cell function24. To overcome some of these issues, researchers have started to incorporate hydrogels into PDMS microfluidic chips20, for example to generate gradients within 3D scaffolds25 or shield cells from flow and shear stresses26,27. In some cases, PDMS has been entirely replaced by hydrogels to generate ‘biomicrofluidic’ networks3,20,28 or scaffolds for tissue engineering21,29,30. However, despite these advances, key challenges related to micro-scale cell culture remain: The limited space available for cell growth and tissue development or the difficulty to perform routine cell culture manipulation such as passaging cells or remove cells from chips for downstream analyses have not yet been overcome31.
We reasoned that a simple hybrid system combining traditional cell culture in multiwell plates with microfluidic biomolecule delivery might offer a powerful means to tackle these challenges. We consequently designed a microfluidic hydrogel chip comprising an engineered culture substrate that could be used for ‘macro-scale’ cell culture, as well as embedded microchannels that could be used for the precise spatiotemporal biomolecule delivery through the gel layer ( Fig. 1 ). Here we report the design and characterization of this open access hydrogel microfluidic system for both adherent or aggregate (i.e. spheroid)-based cell culture and provide proof-of-principle for its application for the spatiotemporally controlled manipulation of mouse ESCs fate via delivery of a morphogen gradient. Since the microchip can be fabricated from different hydrogel types whose bulk and surface properties can be engineered, our hydrogel chip technology should be amenable to probe and manipulate the fate of multiple cell types.

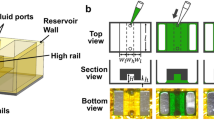
Hydrogel microfluidic concept.
(a), Schematic representation of hydrogel microfluidic inserts based on 24-well plate format. Hydrogel microfluidic inserts placed in individual wells can be used for cell culture and connected to a perfusion system for precise biomolecule delivery and cell-based assays. (b), Schematic representation of chip application for cell-based assays. Adherent culture on flat or spheroid-based (e.g. EB) culture on topographically structured gels is feasible. Precise delivery of biomolecule is achieved by molding microchannels at the bottom of the hydrogel chip for diffusion through the gel (box).
Results and discussion
Design and fabrication of the hydrogel microchip
To reduce the concept shown in Figure 1 to practice, we first created a PDMS mold by standard photo- and soft-lithography processes ( Fig. 2a and ESI Fig. S1a,b , scheme 1) allowing the fabrication of molds of various shapes, sizes and types of micro-scale features (e.g. microchannels or micropillars). In the simplest prototype, each chip comprises a cell culture surface (h = 5 mm, ø = 6 mm) and two embedded individually addressable microchannels (h = 400 μm, w = 200 μm). The hydrogel chip was formed by injecting a reactive gel precursor solution into the PDMS mold ( Fig. 2a step 1 and ESI Fig. S1a,b , scheme 2), resulting in a hydrogel chip upon crosslinking ( Fig. 2a and ESI Fig. S1a,b , scheme 3). Upon completion of cross-linking, the hydrogel chips were retrieved from the mold, placed at the bottom of a standard multi-well plate and immersed in water. Hydrogel chips were stored at 4°C for at least a day prior to use.

Fabrication of hydrogel chips and application to ESC culture.
(a), Fabrication of hydrogel microfluidic inserts. 1. Mold assembly and hydrogel precursor injection. 2. Gelation, mold disassembly and hydrogel microfluidic insert recovery. 3. Transfer into multiwell plate. A photograph of a chip is shown. 4. Immersion in culture media, cell seeding and connection to perfusion system for cell-based assays. Photograph of hydrogel chips with embedded microchannel visualized by food dye. (b), Schematic representation of enzymatically cross-linked gelatin hydrogel system. (c), Adherent mouse ESC culture on hydrogel chip surface. Colony morphology and expression of Oct4 (GFP) suggests maintenance of mouse ESC pluripotency. (d), Micrographs of topographically structured hydrogel chips for spheroid-based cultures. Scale bar, 400 μm (e), Micrographs of cell aggregates formed by seeding and centrifuging 100, 400, 1000 or 4000 cells per microwell and subsequent overnight culture. Scale bar, 100 μm (f), Graphical representation of size distribution of cell aggregate obtained by seeding 100, 400, 1000 and 4000 cells/microwell. Uniform aggregates are obtained with the AggreWellTM-based patterned gel surface.
ESC culture on the hydrogel chip surface
To target adherent cell culture applications, in particular those that include pluripotent stem cells, we reasoned that gelatin-based hydrogels might provide an adequate substrate. Most mammalian cells express integrins that bind gelatin domains (e.g. integrin αvβ3 binding RGD sites32) and pluripotent stem cells are routinely expanded on gelatin-coated substrates where they form adherent colonies.
We therefore molded a gelatin-based hydrogel chip by cross-linking a 10% (w/v) solution of porcine gelatin using bacterial transglutaminase (TG)28,33 ( Fig. 2b ). This enzymatic cross-linking scheme was chosen to stabilize the physically cross-linked and temperature-sensitive gelatin network, avoiding destruction of the relatively fragile features during demolding. Indeed, TG-cross-linked gelatin gels sustained mouse ESC adhesion and self-renewal under standard culture conditions, as demonstrated by efficient colony formation and maintenance of Oct4 expression ( Fig. 2c ).
To target in vitro culture applications that are based on cell aggregates such as EBs or neurospheres (i.e. free-floating colonies of neural stem/progenitor cells34), we sought to generate a surface topography that could be used to enhance, for example, homogeneity of EB differentiation or bias lineage commitment depending on EB size35. To this end, an AggreWellTM chip (STEMCELL Technologies) served as a template to fabricate the desired topography on a PDMS cylinder that could then be inserted into the mold ( ESI Fig. S1b ). A microwell array containing 100 microwells per chip was successfully molded into an agarose hydrogel surface ( Fig. 2d ). Highly uniformly sized ESC aggregates were obtained upon gentle centrifugation of cell suspensions of variable seeding density (ranging from 100 to 4000 cells) per aggregate loaded on the surface of the hydrogel chip ( Fig. 2e,f and ESI Fig. S2 ). These aggregates were cultured over several days on the hydrogel chip surface ( ESI Fig. S2 ) and can be easily removed for downstream assays such as differentiation ( ESI Fig. S2 ). Therefore, our hydrogel chip surface is well suited for ESC expansion cultures.
Hydrogel microfluidics: Characterization of biomolecule delivery through the hydrogel chip
We next characterized the delivery of biomolecules from microchannels embedded in the hydrogel slab ( Fig. 3 ). In a first approach, simple pipette tips inserted into the channel inlets and outlets were used as reservoirs to deliver biomolecules through the chip by hydrostatic pressure ( Fig. 3a ). Solutions of fluorescein isothiocyanate (FITC) and red-fluorescent bovine serum albumin (DsRED-BSA) as model biomolecules were loaded at 0.1 mgmL−1 into the reservoirs. Time-lapse fluorescence microscopy showed that the molecules initially filling the microchannels rapidly diffuse outwards until depletion. This resulted in the generation of anti-parallel overlapping gradients ( Fig. 3b,c ). Intensity profiles show transient gradients with decreasing intensity over time in good accordance with a finite element analysis ( ESI Fig. S3a ).

Characterization of spatiotemporally controlled biomolecule delivery.
(a), Schematic representation of hydrogel chip perfusion by pressure difference. (b), Characterization of transient FITC gradient generation by depletion. Fluorescent micrographs from a time series of a FITC (25 μL, 0.1 mg mL−1) gradient generated by depletion. Graphical representation of the transient gradient profiles obtained by depletion. (c), Characterization of transient DsRED-BSA (25 μL, 0.1 mg mL−1) gradient generation by depletion. (d), Schematic representation of hydrogel microfluidic inserts perfusion with a syringe pump. (e), Characterization of transient FITC (0.1 mg mL−1) gradient generated by continuous perfusion (0.01 mL min−1). Continuous perfusion yields gradients with increasing intensities over time. (f), Characterization of stable DsRED-BSA (0.1 mg mL−1) gradient generated by perfusion with a syringe pump at defined intervals (one minute at 0.01 mL min−1 every 30 minutes). The micrographs and respective intensity profiles demonstrate that stable gradient profiles can be generated by replenishing the biomolecule solution within the microchannel at given time intervals. Gradient profiles were measured across a single plane (right above the hydrogel surface), as depicted by the red dashed line in the schematic representation of the device. Scale bar, 200 μm.
In a second approach, to ensure continuous replenishment of factors, we used commercial syringe pumps to perfuse the hydrogel microchips with the model molecules at a flow rate of 0.01 μLmin−1 ( Fig. 3d ). These experiments showed rapid diffusion and the generation of gradients of increasing intensity over time ( Fig. 3e and ESI Fig. S4 ), again in accordance with predictions from simulations ( ESI Fig. S3b ). Importantly, by dispensing biomolecules in a pulsatile fashion, stable long-term gradients were also generated. Intensity profiles initially increase over a period of three hours before remaining stable over extended periods of time ( Fig. 3f ). Together, these data show that by fluid handling in microchannels embedded in the hydrogel chip, biomolecules can be delivered to build up transient or stable gradients.
Spatiotemporally controlled neural differentiation of ESCs via graded retinoic acid delivery
To validate this versatile cell culture platform, we tested the spatiotemporally controlled induction of neurogenic differentiation of ESC under serum-free culture conditions (N2B27 medium36) by delivery of a gradient of the morphogen retinoic acid (RA) ( ESI Fig. S5 ). To probe induction of neurogenic differentiation in situ, we used a Sox1-GFP reporter ESC line36. Uniform EBs composed of circa 400 ESCs were exposed to RA gradients for two, six, eight or twelve hours and kept in culture for an additional three days ( Fig. 4 ). Bright-field and fluorescent images were acquired in mosaïque mode at day four of differentiation. Image analysis was performed to quantify Sox1 expression in individual EBs on the entire microwell array. Stitched images show a striking position- and thus concentration-dependent effect of RA on EB size and Sox1-expression with EBs close to the RA source showing strongest neural induction ( Fig. 4a–d and ESI Fig. S6 ). EBs close to the RA source are significantly larger (216 ± 41 μm) than far away from the source (124 ± 42 μm) ( ESI Fig. S7 ). The difference in size is likely due to differential cell proliferation that precedes commitment. The GFP expression profiles along the RA gradient was found to transition from a relative flat line for short exposure times (two and six hours, Fig. 4e,f ), whereas the profile became increasingly steep for longer exposures (eight and twelve hours, Fig. 4g,h ). Statistical analysis revealed a significant dose dependency of GFP expression for RA gradients delivery for six hours or longer.

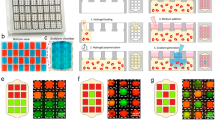
Analysis of induction of neuronal differentiation within individual cell aggregates by delivery of retinoic acid gradients.
(a), Micrographs of cell aggregates exposed to retinoic acid gradients within a single hydrogel microfluidic insert after four days of culture. (b), Mask generated from bright-field micrographs. (c), Overlay of the mask onto fluorescent micrographs of GFP expression within individual cell aggregates. d, Graphical representation of normalized GFP expression of individual cell aggregates (standard score). (e–h), Normalized GFP intensity along the RA gradient after two (e), six (f), eight (g) and twelve (h) hours of exposure. Graphs show box plot (max, quartile, median, min), comparison by t-test Bonferroni correction for multiple comparisons – * p < 0.05, ** p < 0.005, ***p < 0.001. Scale bar, 100 μm.
These data suggest that the temporal modulation of RA delivery influences neurogenic induction in our in vitro model. It is well known that morphogen signaling in vivo is highly complex, not only spatially but also temporally1. For example, recent in vivo visualization of RA in zebrafish embryos via genetically encoded probes for RA allowed for the first time to detect short lived (< six hours) RA gradients (ranging from two to six nM) during embryogenesis37. Although the RA gradient ranges (0.01 to 100 nM) and exposure times tested here are different, species differences could explain the requirement of higher RA concentration and longer exposure times for neurogenic commitment of mouse ESC.
Conclusions
We present a simple yet powerful concept to deliver biomolecules in a tightly controlled fashion to stem cells cultured in a conventional, macro-scale format. Our chip design allows the decoupling of cell culture from microfluidic manipulation, which we think solves a key problem related to microfluidic long-term cell culture. By selecting different hydrogel systems for the chip fabrication, we demonstrate that adherent and spheroid-type ESC culture is possible. For example, the introduction of a surface topography allows the formation of an array of uniformly sized EBs that can be cultured over up to at least a week, not different from conventional assays. We believe that the direct access to cells on the substrate is important because cells can be easily retrieved to perform downstream analyses such as flow cytometry or PCR.
The fluid processing within the embedded microchannels affords precise spatiotemporal delivery of biomolecules comparable to other microfluidic systems. The possibility to fabricate virtually any microchannel layout can be exploited to generate more complex dynamic microenvironments reminiscent of the morphogen display during embryogenesis. We believe that this approach will for example be useful for testing the effect of the dose and timing of morphogens, singly or in combination, on stem cell fate, for example to model in vitro more complex developmental processes. The simplicity of the platform and the interface with traditional multiwell plates will hopefully enable wide adoption in biology laboratories.
Methods
Microfabrication
Standard photo- and soft lithography processes were used to fabricate customized PDMS molds ( Fig. 2a , scheme 1). In short, the bottom piece of the PDMS molds were fabricated as follow: (i) a PDMS bearing the desired microchannel layout was fabricated by injection molding against a SU8 on silicon master (made in advance in a clean room, alternatively stacked layers of tape could be cut to create the desired microstructure). Note the custom-made PMMA mold we use to fabricate these chip comprise a cylinder (h = 5 mm, ø = 6 mm) to fabricate a macro-well on the opposite side of the microstructures. (ii) This PDMS chip was plasma treated for two minutes, immersed in Sigmacote® (Sigma) for two minutes and dried overnight. (iii) Uncured PDMS was then poured onto the Sigmacoted PDMS chip for PDMS/PDMS replica molding. (iv) After baking the PDMS chip was removed from the larger PDMS slab that will become the bottom part of our mold for hydrogel chip fabrication. For microwell array fabrication, PDMS pieces from an AggreWellTM (Stem Cell Technologies) were plasma-treated, immersed in Sigmacote® (Sigma) for two minutes, dried overnight and used for PDMS/PDMS molding. The resulting PDMS pieces were punched (ø = 6 mm) and inserted into the top part of the PDMS mold (the PDMS chip mentioned above).
Hydrogel microfluidic chip fabrication
A detailed fabrication procedure can be found in the electronic supplementary information (ESI), Figure S1. Briefly, for adherent cell culture, the PDMS mold was assembled ( Fig. 2a , Scheme 1) and a gelatin solution (10% w/v, porcine high strength, Fluka) supplemented with bacterial transglutaminase (1 UmL−1 TG, Zedira) was injected with a syringe. The mold was then placed in a humidified incubator at 37°C for five hours ( Fig. 2a , scheme 2), cooled to 4°C and the cross-linked chips carefully recovered from the mold and placed in a well plate ( Fig. 2a , scheme 3). The wells containing the chips were filled with PBS, placed at 65°C for 30 minutes to arrest the enzymatic reaction and then stored at 4°C before use in cell culture ( Fig. 2a , scheme 4–5). Chips for spheroid culture were fabricated similarly (details in ESI Fig. S1b ). The custom-made mold was assembled to bear the micropillar array and a solution of agarose (2% w/v) injected with a syringe. The mold was placed for ten minutes at 4°C before recovery from the PDMS mold. To ensure tight microchannels, a thin slab of agarose (2% w/v) was heat-bonded (3 sec at 71°C) at the bottom of the agarose chip. After cooling the construct to room temperature for 10 minutes, it was placed in a well plate, immersed in PBS and stored at 4°C before use in cell culture.
Characterization of biomolecule diffusion
Fluorescent time-lapse microscopy (Zeiss Axio Observer, Metamorph) was used to characterize gradient generation and evolution over time. Depletion gradients of model biomolecules (FITC and DsRED-BSA, 0.1 mg mL−1) were obtained by filling (10 μL) custom-made reservoirs (pipette tips inserted in inlets and outlets) with gel-loading tips. In the case of connection to a syringe pump, 1 mL syringes (Omnifix) were used with tygon tubings (ø = 1.35 mm). The tubings were directly inserted into the inlets of the hydrogel microfluidic chips. Another open tube was inserted at each outlet to remove excess liquid waste.
ESC culture
Mouse ESCs were maintained in standard feeder-free culture condition (DMEM, Gibco, sodium pyruvate, non-essential amino acids, beta mercaptoethanol and supplemented with 15% fetal bovine serum, Hyclone and Leukemia inhibitory factor).
Formation of cell aggregates
Centrifugation of cells in microwell array was used to generate uniformly sized cell aggregates. ESC were passaged and resuspended in N2B27 medium36. Cell suspension containing 2 × 106, 8 × 106, 2 × 107 and 8 × 107 cells per mL were gently dispensed (50 μL) onto the central cell culture area of the chips to yield 100, 400, 1000 or 4000 cells per well, respectively. The plates were centrifuged at 1000 rpm for three minutes and placed overnight in a humidified incubator at 37°C. Microscopy (Zeiss Axio Observer) and image analysis (Metamorph) was used to quantify the resulting cell aggregates.
Induction of EB differentiation by RA gradients
Mouse ESC aggregates were formed as mentioned above to yield ca. 400 cells per aggregate. The plate was incubated overnight. Reservoirs (pipette tips) were inserted into the inlets and outlets of the chips. A solution of retinoic acid (25 μL, 10−6 M) was injected into the reservoirs with a gel-loading tip. This solution was exchanged every two hours over a period of 2, 6, 8 or 12.
References
Gurdon, J. B. & Bourillot, P. Y. Morphogen gradient interpretation. Nature 413, 797–803 (2001).
Murry, C. E. & Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 132, 661–680 (2008).
Nishikawa, S. I., Jakt, L. M. & Era, T. Embryonic stem-cell culture as a tool for developmental cell biology. Nat Rev Mol Cell Biol. 8, 502–507 (2007).
El-Ali, J., Sorger, P. K. & Jensen, K. F. Cells on chips. Nature 442, 403–411 (2006).
Ashton, R. S., Keung, A. J., Peltier, J. & Schaffer, D. V. Progress and Prospects for Stem Cell Engineering. Annu Rev Chem Biomol Eng., Vol 2 2, 479–502 (2011).
Kobel, S. & Lutolf, M. P. Biomaterials meet microfluidics: building the next generation of artificial niches. Curr Opin Biotechnol. 22, 690–697 (2011).
Kim, S., Kim, H. J. & Jeon, N. L. Biological applications of microfluidic gradient devices. Integr Biol (Camb). 2, 584–603 (2010).
Park, J. Y., Takayama, S. & Lee, S. H. Regulating microenvironmental stimuli for stem cells and cancer cells using microsystems. Integr Biol (Camb). 2, 229–240 (2010).
Fung, W. T., Beyzavi, A., Abgrall, P., Nguyen, N. T. & Li, H. Y. Microfluidic platform for controlling the differentiation of embryoid bodies. Lab Chip 9, 2591–2595, 10.1039/B903753e (2009).
Park, J. Y. et al. Differentiation of Neural Progenitor Cells in a Microfluidic Chip-Generated Cytokine Gradient. Stem Cells 27, 2646–2654 (2009).
Paguirigan, A. L. & Beebe, D. J. Microfluidics meet cell biology: bridging the gap by validation and application of microscale techniques for cell biological assays. Bioessays 30, 811–821 (2008).
Kshitiz, Kim, D. H., Beebe, D. J. & Levchenko, A. Micro- and nanoengineering for stem cell biology: the promise with a caution. Trends in Biotechnol. 29, 399–408 (2011).
Toh, Y. C. & Voldman, J. Fluid shear stress primes mouse embryonic stem cells for differentiation in a self-renewing environment via heparan sulfate proteoglycans transduction. FASEB J 25, 1208–1217 (2011).
Mosadegh, B. et al. Uniform cell seeding and generation of overlapping gradient profiles in a multiplexed microchamber device with normally-closed valves. Lab Chip 10, 2959–2964 (2010).
Park, E. S., Brown, A. C., DiFeo, M. A., Barker, T. H. & Lu, H. Continuously perfused, non-cross-contaminating microfluidic chamber array for studying cellular responses to orthogonal combinations of matrix and soluble signals. Lab Chip 10, 571–580 (2010).
Sip, C. G., Bhattacharjee, N. & Folch, A. A modular cell culture device for generating arrays of gradients using stacked microfluidic flows. Biomicrofluidics 5 (2011).
Cate, D. M., Sip, C. G. & Folch, A. A microfluidic platform for generation of sharp gradients in open-access culture. Biomicrofluidics 4, 44105 (2010).
Kawada, J., Kimura, H., Akutsu, H., Sakai, Y. & Fujii, T. Spatiotemporally controlled delivery of soluble factors for stem cell differentiation. Lab Chip 12, 4508–4515 (2012).
Torisawa, Y. S. et al. Microfluidic platform for chemotaxis in gradients formed by CXCL12 source-sink cells. Integr Biol (Camb) 2, 680–686 (2010).
Domachuk, P., Tsioris, K., Omenetto, F. G. & Kaplan, D. L. Bio-microfluidics: Biomaterials and Biomimetic Designs. Adv Mat 22, 249–260 (2010).
Ling, Y. et al. A cell-laden microfluidic hydrogel. Lab Chip 7, 756–762 (2007).
Regehr, K. J. et al. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 9, 2132–2139 (2009).
Berthier, E., Young, E. W. K. & Beebe, D. Engineers are from PDMS-land, Biologists are from Polystyrenia. Lab Chip 12, 1224–1237 (2012).
Morrison, S. J. & Spradling, A. C. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132, 598–611 (2008).
Saadi, W. et al. Generation of stable concentration gradients in 2D and 3D environments using a microfluidic ladder chamber. Biomed Microdevices 9, 627–635 (2007).
Cheng, S. Y. et al. A hydrogel-based microfluidic device for the studies of directed cell migration. Lab Chip 7, 763–769 (2007).
Tan, D. C. W., Yung, L. Y. L. & Roy, P. Controlled microscale diffusion gradients in quiescent extracellular fluid. Biomed Microdevices 12, 523–532 (2010).
Paguirigan, A. & Beebe, D. J. Gelatin based microfluidic devices for cell culture. Lab Chip 6, 407–413 (2006).
Cabodi, M. et al. A microfluidic biomaterial. JACS 127, 13788–13789 (2005).
Choi, N. W. et al. Microfluidic scaffolds for tissue engineering. Nat Mater 6, 908–915 (2007).
Toh, Y. C., Blagovic, K. & Voldman, J. Advancing stem cell research with microtechnologies: opportunities and challenges. Integr Biol (Camb). 2, 305–325 (2010).
Davis, G. E. Affinity of Integrins for Damaged Extracellular-Matrix - Alpha-V-Beta-3 Binds to Denatured Collagen Type-I through Rgd Sites. Biochem Bioph Res Co 182, 1025–1031 (1992).
Paguirigan, A. L. & Beebe, D. J. Protocol for the fabrication of enzymatically crosslinked gelatin microchannels for microfluidic cell culture. Nat Protoc 2, 1782–1788 (2007).
Reynolds, B. A. & Weiss, S. Generation of Neurons and Astrocytes from Isolated Cells of the Adult Mammalian Central-Nervous-System. Science 255, 1707–1710 (1992).
Ungrin, M. D., Joshi, C., Nica, A., Bauwens, C. & Zandstra, P. W. Reproducible, Ultra High-Throughput Formation of Multicellular Organization from Single Cell Suspension-Derived Human Embryonic Stem Cell Aggregates. Plos One 3, (2008).
Ying, Q. L., Stavridis, M., Griffiths, D., Li, M. & Smith, A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotech 21, 183–186 (2003).
Shimozono, S., Iimura, T., Kitaguchi, T., Higashijima, S. & Miyawaki, A. Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 496, 363 (2013).
Acknowledgements
We thank Austin Smith for providing the Sox1-GFP knock-in (46C) reporter mouse ESC line. We thank Yannick Devaud for testing hydrogels for microfluidic fabrication and Sonia Hallen for help with optimizing ESC aggregate culture and differentiation. This work was funded by the EU FP7 Large-scale integrating project ‘PluriMes’ (http://www.plurimes.eu/) as well as a EURYI award to M.P.L.
Author information
Authors and Affiliations
Contributions
S.C. and M.P.L. conceived the study, interpreted results and wrote the manuscript. S.C. performed all experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported license. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cosson, S., Lutolf, M. Hydrogel microfluidics for the patterning of pluripotent stem cells. Sci Rep 4, 4462 (2014). https://doi.org/10.1038/srep04462
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04462
This article is cited by
-
A guide to the organ-on-a-chip
Nature Reviews Methods Primers (2022)
-
Recent Advancements in Engineering Strategies for Manipulating Neural Stem Cell Behavior
Current Tissue Microenvironment Reports (2020)
-
High-throughput cell migration assay under combinatorial chemical environments by a novel 24-well-plate based device
Biomedical Microdevices (2020)
-
User-defined morphogen patterning for directing human cell fate stratification
Scientific Reports (2019)
-
Low Cost and Lithography-free Stamp fabrication for Microcontact Printing
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
