
Abstract
The Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) has been reported to have both tumor-promoting and tumor-suppressing roles in tumorigenesis. However, the role of SHP-2 in tumor immunity remains unclear. Here we observed progressively lower levels of phosphorylated SHP-2 in tumor-associated CD4+ T cells during melanoma development in a murine model. Similarly, the levels of phosphorylated SHP-2 in the CD4+ T cells of human melanoma specimens revealed a decrease paralleling cancer development. The CD4+ T cell-specific deletion of SHP-2 promoted melanoma metastasis in mice. Furthermore, SHP-2 deficiency in CD4+ T cells resulted in the increased release of inflammatory cytokines, especially IL-6 and the enhanced accumulation of tumor-promoting myeloid-derived suppressor cells (MDSCs) in tumor-bearing mice. An IL-6-neutralizing antibody reduced MDSC accumulation and inhibited tumor growth in CD4+ T-cell-specific SHP-2-knockout mice. Our results suggest that SHP-2 in CD4+ T cells plays an important role in preventing melanoma progression and metastasis.
Similar content being viewed by others


Introduction
SHP-2 is a ubiquitously expressed cytoplasmic protein tyrosine phosphatase that contains two Src-homology 2 domains and functions as a signaling regulator1. The tyrosine phosphorylation of SHP-2 is crucial for its function. SHP-2 exerts both positive and negative regulatory activities on cytokine receptor signal transduction and it also acts as an important mediator of inhibitory receptor signaling. The dysregulation of SHP-2 function or expression has been implicated in the pathogenesis of human diseases, including cancer, but its involvement in cancer progression and metastasis is controversial2. Because activating mutations of the SHP-2-coding gene PTPN11 are associated with leukemogenesis, PTPN11/shp-2 was previously identified as a proto-oncogene3. In contrast, more recent studies have demonstrated that the loss of SHP-2 promotes hepatocellular carcinoma and that decreased SHP-2 expression was detected in a subfraction of human hepatocellular carcinoma patient samples, suggesting that SHP-2 also acts as a tumor suppressor4,5. The opposing functions of SHP-2 may depend on cellular context6. This possibility drives us to seek further insight into the pro- or antitumor roles of SHP-2 in immune cells.
CD4+ T cells contribute to tumor regression by orchestrating immune responses that target the tumor7. However, CD4+ T cells are also associated with tumor-promoting immune responses when they are present in a tumor microenvironment that favors polarized protumorigenic inflammatory states, indicating that CD4+ T cells have a dual role in tumor development8. A number of studies have demonstrated the involvement of SHP-2 in the negative regulation of immune responses through its effects on JAK-STAT activation and inhibitory receptor signaling pathways9,10. SHP-2 might also be a critical regulator of T cell development and function10,11. T cells from transgenic mice harboring a dominant negative SHP-2 mutant (SHP-2 C/S) in a T cell lineage-specific manner express higher levels of activation markers such as CD69 and CD44 and SHP-2 C/S CD4+ T cells secrete more IL-4 and IL-10 than control cells following antigenic stimulation12. Cytotoxic T-lymphocyte antigen-4 (CTLA-4) or programmed death-1 (PD-1) is known to recruit and activate SHP-2, which contributes to the negative regulation of T cell activation13,14. SHP-2 can also downregulate T cell adhesion because the overexpression of SHP-2 C/S enhances the adhesion of Jurkat T cells to fibronectin in response to TCR-mediated stimulation15. Despite these key regulatory functions of SHP-2 in T cells, the link between SHP-2 in tumor-associated CD4+ T cells and tumor progression is not fully understood.
In this study, we evaluated the role of SHP-2 expressed by tumor-associated CD4+ T cells in tumor progression and metastasis using murine melanoma metastasis models. We found that the SHP-2 in CD4+ T cells exerted tumor-suppressing effects on melanoma. The selective deletion of SHP-2 in CD4+ T cells potentiated melanoma progression and promoted metastasis in mice by increasing the amount of circulating IL-6. This increase enhanced the accumulation and function of tumor-promoting MDSCs. Importantly, this link between SHP-2 inactivation and tumor progression was confirmed in human melanoma specimens and was found to depend on the degree of malignancy. This study provides the first evidence for a role of SHP-2 in tumor immunity.
Results
The inactivation of SHP-2 in CD4+ T cells accompanies the development of melanoma in a malignancy-dependent fashion
Tumor-associated T cells usually exhibit an exhausted phenotype and impaired effector function during melanoma progression16,17. When C57BL/6 mice were injected with B16BL6 cells into the right hind footpads, we found that PD-1 and CTLA-4 expression were significantly and temporally upregulated in CD4+-gated tumor-draining lymph node (TDLN) cells and tumor-infiltrating leukocytes (TILs) compared with normal CD4+ lymph node cells (day 0) (Fig. 1a). In contrast, the production of IFN-γ in the CD4+-gated population of TDLN cells showed a time-dependent decrease after a transient increase on day 5 (supplementary Fig. S1).

Phosphorylated SHP-2 was progressively downregulated in CD4+ T cells during melanoma development in mice.
B16BL6 cells (1 × 106) were injected subcutaneously into the right hind footpads of C57BL/6 mice. Draining popliteal lymph nodes and tumors were resected on days 0, 5, 14 and 21 after tumor cell inoculation (n = 8–12 mice per group). (a) The expression of PD-1 and CTLA-4 on CD4+ tumor draining lymph node (TDLN) cells or tumor-infiltrating leukocytes (TIL). All the experiments were done three times. Data shown are mean ± s.e.m. (b, c) The levels of phosphorylated SHP-2 (pSHP-2) in CD45.2+ CD4+ TDLN cells or TILs. Data on scatter plots represent experimental replicates for pooled TDLN cells or TILs. (d) pSHP-2 expression in purified CD4+ TDLN cells was detected by western blotting. *P < 0.05, **P < 0.01.
To assess the status of SHP-2 activation in these tumor-associated CD4+ T cells, we monitored their levels of phosphorylated SHP-2 (pSHP-2) during melanoma progression. Surprisingly, the level of pSHP-2 was greatly decreased in a time-dependent manner in CD45.2+ CD4+ T cells from both the TDLN and TIL cell populations (Fig. 1b, c). Similar results were obtained in purified CD4+ TDLN cells by western blotting (Fig. 1d). The levels of pSHP-2 in these cells decreased progressively, even though the total SHP-2 expression level did not change significantly. These results implicate a link between the inactivation of SHP-2 and tumor progression.
We further used an in vitro co-culture system to evaluate the effects of tumor cells on SHP-2 activation in CD4+ T cells. After the co-culture of murine tumor cells with lymph node cells, both melanoma B16BL6 cells and Lewis lung carcinoma LLC cells downregulated the expression of pSHP-2 in CD4+ T cells in a time-dependent manner (Fig. 2a). Decreased pSHP-2 expression was also found in human CD4+ T cells after co-cultured with human melanoma A375 or A875 cells (Fig. 2b). These results were identical to the phenomenon observed in vivo. More importantly, the levels of pSHP-2 in human melanoma specimens were confirmed to be markedly decreased in the CD4+ T cells paralleling cancer development, shown as decreased percentage of pSHP-2 high (++) expression level samples which is 75% in normal tissues or benign tumors, 52.1% in primary malignant tumors and 40% in metastatic tumors (Fig. 2c). These results indicate that decreases in pSHP-2 depend on the degree of malignancy.

Phosphorylated SHP-2 was downregulated in CD4+ T cells after co-culture of tumor cells or in human specimens of patients with malignant melanoma.
(a) B16BL6 or LLC cells were co-cultured at a ratio of 1:10 with LN cells from C57BL/6 mice for the indicated time. The levels of pSHP-2 in CD45.2+ CD4+ LN cells were detected. (b) A375 or A875 cells were co-cultured at a ratio of 1:10 with human PBMC from healthy donors. The levels of pSHP-2 in human CD4+ T cells were detected. All the experiments were done three times. Data shown are mean ± s.e.m. (c) pSHP-2 expression patterns in CD4+ cells were analyzed in human melanoma specimens using tissue microarrays. Representative immunofluorescent staining of human melanoma specimens with anti-pSHP-2 and CD4 specific antibodies is shown. Bar, 100 μM. *P < 0.05, **P < 0.01.
SHP-2 deficiency in CD4+ T cells leads to a transient delay followed by an eventual escalation of melanoma growth
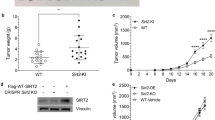
To address the possible link between SHP-2 expression in tumor-associated CD4+ T cells and tumor progression, we investigated the growth and metastasis of melanoma using CD4+ T cell-specific SHP-2 knockout (cSHP-2 KO) mice. The cSHP-2 KO mice were born at the expected frequency and were morphologically indistinguishable from their control littermates. B16BL6 cells were inoculated into the right flanks of WT or cSHP-2 KO mice to assess tumor growth and spontaneous metastasis over a relatively long period. Tumor growth was modestly but significantly inhibited in cSHP-2 KO mice during only the first two weeks after B16BL6 cell inoculation (Fig. 3a). To confirm the delayed tumor growth at the early stage, the tumors were removed from the cSHP-2 KO mice on day 14. The average weight of tumors from KO mice was significantly less than that from WT mice (Fig. 3b). However, tumor growth was not significantly inhibited after day 14. At the late stage of tumor progression, the tumors from cSHP-2 KO mice were comparable in size to those from WT mice. Additionally, the survival rate of KO and WT tumor-bearing mice did not differ (Fig. 3c).

SHP-2 deficiency in CD4+ T cells first delayed but then potentiated melanoma growth in mice.
B16BL6 cells (2 × 105) were injected subcutaneously into the right flanks of WT and cSHP-2 KO mice. (a) Tumor volumes were monitored and recorded (n = 8–10 mice per group). (b) Tumors excised on day 14 were weighed (n = 6 mice per group). All the experiments were done three times. Data shown are mean ± s.e.m. (c) The survival rates of each group were recorded and shown as Kaplan-Meier curves (n = 8–10 mice per group). (d) Tumor tissues excised on day 14 were stained with H&E, an antibody specific for PCNA and TUNEL reagents. The data shown are representative of three experiments. Bar, 200 μM (HE); 100 μM (PCNA, left); 20 μM (PCNA, right); 200 μM (TUNEL). *P < 0.05.
We examined the histopathologic differences between the two groups in the tumor tissues excised on day 14. Hematoxylin and eosin (H&E) staining showed obvious cell hyperplasia in tumor tissues from cSHP-2 KO mice (Fig. 3d). Positive immunostaining for proliferating cell nuclear antigen (PCNA) confirmed the increase in cell proliferation. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays revealed lower levels of apoptosis in cSHP-2 KO tumors than in WT tumors. These results indicate that the SHP-2 deficiency in CD4+ T cells eventually potentiated melanoma progression despite the delay in melanoma growth during the early stage.
SHP-2 deficiency in CD4+ T cells promotes melanoma metastasis
We also investigated the effects of SHP-2 deficiency in CD4+ T cells on melanoma metastasis, which primarily occurs during the late stage of tumor progression. The tumor burden in the lungs was examined 32 days after B16BL6 cell inoculation. H&E staining showed extensive pulmonary metastasis in the cSHP-2 KO mice (Fig. 4a). Over 70% of the cSHP-2 KO mice developed lung metastases. This incidence of metastasis was approximately 3-fold greater than the incidence observed in the WT control group (Fig. 4b). Specifically, the CD4+ T cell-specific deletion of SHP-2 dramatically increased the number of metastatic foci in the lungs. Next, we examined the expression of prometastatic and angiogenic genes in tumor tissues excised on day 14. Immunostaining revealed that CD31+ blood vessels in cSHP-2 KO mice were modestly increased compared with the WT controls (Fig. 4c). The expression levels of the CCL2, MMP9 and VEGF genes were also significantly upregulated in cSHP-2 KO tumors (Fig. 4d).

SHP-2 deficiency in CD4+ T cells led to increased angiogenesis and metastasis in mice.
B16BL6 cells (2 × 105) were injected subcutaneously into the right flanks of WT and cSHP-2 KO mice. (a, b)The lungs were isolated on day 32. The total number of metastatic lesions was counted (n = 8–10 mice per group). (a) Representative H&E-stained lung sections. Arrows indicate metastatic foci in the lungs. Bar, 200 μM. (b) The proportion of mice with lung metastases in each group (left) and the numbers of metastatic foci in the lungs of each mouse (right). (c, d) Tumor tissues were excised on day 14 (n = 6 mice per group). (c) Representative tumor tissue sections stained with an antibody specific for CD31 (left) and quantification of CD31 expression levels (right). Bar, 50 μM. Data shown are the density mean within that tumor area ± s.e.m. (d) Gene expression in tumor tissues was quantified by real-time PCR. GAPDH was used as an invariant control. (e, f) B16F10 cells (2 × 105) were injected into the tail veins of WT and cSHP-2 KO mice. On day 20, the lungs and livers were isolated, sectioned and stained with H&E (n = 8–10 mice per group. (e) Representative lung, liver and H&E-stained lung sections. Bar, 200 μM. (f) The number of macroscopically visible metastases on the lung and liver surfaces was quantified. (g) Firefly luciferase-expressing B16F10 cells (2 × 105) were injected into the tail veins of WT and cSHP-2 KO mice (n = 4–5 mice per group). On day 20, the mice were analyzed by live bioluminescence imaging. All the experiments were done three times. Data shown are mean ± s.e.m. *P < 0.05.
To confirm the effects of SHP-2 deficiency in CD4+ T cells on melanoma metastasis, we used a passive metastasis model in which mice are inoculated with B16F10 cells via injection into the tail vein. The lungs and livers were isolated 20 days after injection. Consistent with the previous results, the cSHP-2 KO mice developed extensive lung and liver metastases (Fig. 4e, f). Live bioluminescence imaging revealed that tumors generated in cSHP-2 KO mice by the implantation of firefly luciferase-expressing B16F10 cells metastasized to the lungs and liver at an increased rate (Fig. 4g).
SHP-2 deficiency in CD4+ T cells causes an enhanced IL-6 signaling in tumor-bearing mice
Because SHP-2 plays a negative role in JAK-STAT signaling, which affects a number of cytokines, we investigated whether CD4+ T cell-specific SHP-2 deletion results in increased cytokine levels in tumor-bearing mice. B16BL6 cells were inoculated into the right flanks of WT and cSHP-2 KO mice. The serum levels of Th1/Th2/Th17 cytokines were measured on day 14. We detected dramatically and significantly increased IL-6 levels in cSHP-2 KO mice, as well as upward trends in the levels of IL-10, TNF-α, IL-17, IFN-γ and IL-4 (Fig. 5a). We also noted that serum IL-6 levels were higher on day 32 in the cSHP-2 KO mice compared with the WT controls, indicating that SHP-2 ablation led to sustained IL-6 release (Fig. 5b). Further analysis of the inflammatory gene expression in tumor tissues excised on day 14 revealed that the expression levels of IL-6, IL-17, TNF-α and IFN-γ were elevated in cSHP-2 KO mice compared with the WT controls (Fig. 5c). Consistent with these results, immunostaining revealed that phosphorylated STAT3 levels were enhanced in tumors from cSHP-2 KO mice compared with tumors from WT controls (Fig. 5d). Similar results were obtained for tumor tissues by western blotting (Fig. 5e).

SHP-2 deficiency in CD4+ T cells increased the levels of inflammatory cytokines in tumor-bearing mice.
B16BL6 cells (2 × 105) were injected subcutaneously into the right flanks of WT and cSHP-2 KO mice. (a) Serum cytokine levels were measured using a CBA assay on day 14 (n = 6 mice per group). (b) The IL-6 serum level was measured by ELISA on day 32 (n = 6 mice per group). (c–f) Tumor tissues were excised on day 14 (n = 6 mice per group). (c) Cytokine gene expression in tumor tissues was quantified by real-time PCR on day 14. GAPDH was used as an invariant control.(d) Representative tumor tissue sections stained with an antibody specific for pTyr705 STAT3 (left) and quantification of pTyr705 STAT3 expression levels (right). Bar, 50 μM. Data shown are the density mean ± s.e.m. (e) The protein levels of pTyr705 STAT3 in tumor tissues were detected by western blotting. The relative band densities were calculated using actin as an invariant control. (f) Single-cell suspensions of CD45.2− cells and CD45.2+ cells separated from tumor tissues excised on day 14 were cultured for 48 h. The IL-6 levels in supernatants were measured using a CBA assay. All the experiments were done three times. Data shown are mean ± s.e.m. *P < 0.05, **P < 0.01.
To determine the source of IL-6 in tumor-bearing cSHP-2 KO mice, we separated CD45.2+ cells (leukocytes) and CD45.2− cells (tumor cells) from tumor tissues that were excised on day 14 and incubated the separated cells for 48 h. Conditional SHP-2 depletion caused greater IL-6 secretion by CD45.2+ leukocytes, whereas a very low and comparable amount of IL-6 was produced by CD45.2− tumor cells from WT and cSHP-2 KO mice, suggesting that tumor-associated leukocytes were the primary source of IL-6 (Fig. 5f).
SHP-2 deficiency in CD4+ T cells induces functional MDSC accumulation in tumor-bearing mice
To understand how SHP-2 deficiency in CD4+ T cells promotes melanoma progression, we investigated the profile of leukocytes in tumor-bearing mice. B16BL6 cells were inoculated into the right flanks of WT and cSHP-2 KO mice. When examining the leukocytes (CD45.2+) present in the tumor tissues on day 14, we found that the percentage of CD11b+ Gr-1+ myeloid-derived suppressor cells (MDSCs) in cSHP-2 KO mice was markedly increased compared with that of the WT controls (Fig. 6a, b). An increase in the MDSC population was also observed in the spleens of cSHP-2 KO mice on day 14 (Fig. 6c). In contrast, there was no significant difference between the two groups in the proportions of other cell populations (CD4+ T cells, CD8+ T cells, Tregs and macrophages) present in the tumors, lymph nodes and spleens (Fig. 6a and supplementary Fig. S2).

SHP-2 deficiency in CD4+ T cellsincreased functional MDSC accumulation in tumor-bearing mice.
(a–d) B16BL6 cells (2 × 105) were injected subcutaneously into the right flanks of WT and cSHP-2 KO mice (n = 6 mice per group). (a) The profile of CD45.2-gated leukocytes in tumor tissues on day 14 was analyzed by flow cytometry. The data are presented as the percentage of CD11b+ Gr-1+ MDSCs among CD45.2-gated leukocytes in (b) tumor tissues and (c) the spleen on day 14. (d) Gene expression was quantified by real-time PCR in MDSCs that had been purified from spleens on day 14 and stimulated with LPS for 12 h. GAPDH was used as an invariant control. (e–g) MDSCs were purified from the spleens of tumor-bearing mice and then co-injected with B16BL6 tumor cells into naive C57BL/6 mice (n = 6 mice per group). (e) The tumor volumes were monitored and recorded. *P < 0.05 vs. controls co-injected with MDSCs from WT mice. (f) Tumors excised on day 14 were weighed. (g) Gene expression was quantified by real-time PCR in tumor tissues on day 14. GAPDH was used as an invariant control. All the experiments were done three times. Data shown are mean ± s.e.m. *P < 0.05, **P < 0.01.
Next, we examined the function of MDSCs that had been purified from the spleen on day 14. Quantitative analyses showed that the gene expression levels of arginase-1 (Arg-1) and MMP9, which are recognized as mediators of MDSC-mediated immunosuppression, were significantly upregulated in LPS-stimulated MDSCs from cSHP-2 KO mice compared with LPS-stimulated MDSCs from WT controls (Fig. 6d). The co-injection of B16BL6 cells with MDSCs isolated from tumor-bearing cSHP-2 KO mice significantly promoted tumor growth and the expression of angiogenic genes in tumors compared with the co-injection of B16BL6 cells with MDSCs from WT tumor-bearing mice (Fig. 6e–g). Together, these exvivo and in vivo results indicated that conditional SHP-2 deletion enhanced MDSC function in tumor-bearing mice in addition to promoting MDSC accumulation.
IL-6 blockade reduces MDSC accumulation and inhibits tumor growth in T cell-specific SHP-2-deficient mice
An increased number of MDSCs in the spleen, blood and tumors of cancer patients and tumor-bearing animals is a hallmark of tumor-promoting immune responses18,19. Furthermore, IL-6-STAT3 signaling plays a pivotal role in the induction of MDSCs20. To assess the role of IL-6 in the promotion of tumor growth in cSHP-2 KO mice, the mice were inoculated with B16BL6 cells and treated intraperitoneally with a neutralizing anti-IL-6 mAb or a control rat IgG. The IL-6 neutralizing antibody significantly inhibited tumor growth in cSHP-2 KO mice (Fig. 7a, b). Further analysis of the tumor tissues showed that apoptosis was increased, while PCNA and CD31 expression was decreased, in cSHP-2 KO mice treated with the anti-IL-6 neutralizing mAb compared with those treated with the control rat IgG (Fig. 7c). As expected, the IL-6 neutralizing mAb significantly reduced MDSC accumulation in the spleen and tumor tissues (Fig. 7d, e).

An IL-6 neutralizing antibody inhibited tumor growth and angiogenesis and reduced MDSC accumulation in cSHP-2KO mice.
cSHP-2 KO mice were inoculated with B16BL6 tumor cells and injected intraperitoneally with a neutralizing anti-IL-6 mAb or a control rat IgG (25 μg/mouse) on days 0, 4, 8 and 12 (n = 4 per group). (a) The tumor volumes were monitored and recorded. *P < 0.05 vs. controls treated with rat IgG. (b) Tumors excised on day 14 were weighed. (c) Tumor tissue sections were stained with TUNEL and antibodies specific for PCNA and CD31. Bar, 200 μM (TUNEL); 25 μM (PCNA); 50 μM (CD31). The data shown are representative of three experiments. (d–e) The profile of CD45.2-gated leukocytes in tumor tissues on day 14 was analyzed by flow cytometry. The data are presented as the percentage of CD11b+ Gr-1+ MDSCs among CD45.2-gated leukocytes in (d) the spleen and (e) tumor tissues on day 14. All the experiments were done three times. Data shown are mean ± s.e.m. *P < 0.05.
Discussion
In the present study, we found that decreased expression of phosphorylated SHP-2 in the tumor-associated CD4+ T cells paralleled melanoma progression. These cells exhibited an exhausted phenotype and an impaired effector function. This impairment was characterized by concomitant increases in the levels of PD-1 and CTLA-4 as well as a decrease in the production of IFN-γ. This finding implicates the link between the inactivation of SHP-2 in CD4+ T cells and the failure of protective antitumor immune responses which leads to tumor progression. Nevertheless, SHP-2 is known to play dual roles in tumorigenesis and the opposing functions may be dependent on cellular context4,6,21. Numerous studies demonstrate that the differentiation state, phenotype and effector functions of CD4+ T cells can be regulated by the tumor microenvironment, which in part determines whether a pro- or antitumor immune program is favored8,22. Thus, it is inferred that the SHP-2 in CD4+ T cells could be inactivated by the tumor microenvironment to favor tumor progression. Accordingly, both murine and human tumor lines caused the downregulation of pSHP-2 expression in CD4+ T cells after co-culture. More importantly, such a decrease was confirmed in human melanoma tissues and found to parallel with the degree of malignancy. Furthermore, the selective deletion of SHP-2 in CD4+ T cells greatly potentiated melanoma progression and promoted metastasis in mice, demonstrating that the SHP-2 in CD4+ T cells plays a key role in antitumor immune function.
In an attempt to identify the mechanism by which the loss of SHP-2 in CD4+ T cells potentiates melanoma progression, we detected the circulating inflammatory cytokines in tumor-bearing cSHP-2 KO mice. Numerous studies have demonstrated an interplay between cancer and inflammation23,24. Specifically, tumor growth and metastasis are promoted by inflammatory signals from the surrounding environment. Although the loss of SHP-2 in CD4+ T cells mildly hindered early melanoma development, the delay in tumor growth was transient and any tumor suppression was subsequently overwhelmed by the ultimate progression of melanoma. This transient delay may be attributed to the tumor-suppressing effects of certain inflammatory cytokines that are usually inhibited by the anti-inflammatory action of SHP-2. Ultimately, however, the tumor can make more efficient use of the protumor inflammatory environment to usurp antitumor immune mechanisms. DeNardo and colleagues demonstrated that when CD4+ T cells were present in a Th2-type tumor microenvironment, they promoted the pulmonary metastasis of mammary adenocarcinomas8. In our study, the tumors excised at the onset of accelerated melanoma growth (day 14) were found to be resistant to apoptosis and have increased expression of PCNA and CD31, as well as high expression of the genes CCL2, MMP9 and VEGF. These findings are indicative of a proangiogenic and prometastatic microenvironment. On day 14, dramatically and significantly increased IL-6 levels were detected in the sera and tumor tissues of cSHP-2 KO mice, while tumor free KO mice have comparable serum IL-6 levels to control littermates (Data not shown). The high serum IL-6 levels were sustained up to day 32. These results suggest that IL-6 signaling may be sustained throughout the progression of the tumor. Accordingly, the levels of pSTAT3 were higher in tumors from cSHP-2 KO mice compared with those from WT mice. Therefore, inflammation in the tumor microenvironment may subvert an adaptive immune response in cSHP-2 KO mice.
The negative regulatory role of SHP-2 in JAK-STAT signaling can be responsible for enhanced inflammatory signals. Consistent with our results, the hepatocyte-specific deletion of SHP-2 resulted in a marked increase in the strength of the inflammatory IL-6/STAT3 signal, which facilitated the development of hepatocellular carcinoma4. STAT3 was also hyperactivated in the colonic epithelium from mice with an intestinal epithelial cell-SHP-2 deletion25. Nguyen and colleagues investigated the impact of SHP-2 in T-cell biology by generating shp-2flox/flox: Lck-Cre/+ mice and found that conditional deletion of SHP-2 in thymocytes reduced expansion of CD4+ T cells and suppressed TCR signals26. In this study, however, normal numbers and distributions of T-cell subset were present in peripheral lymphoid tissues from the cSHP-2 KO (shp-2flox/flox: CD4-Cre/+) mice (supplementary Fig. S3a). And enhanced secretion of some inflammatory cytokines, such as IL-17, was produced by primary CD4+ T cells following TCR stimulation (supplementary Fig. S3b). Similar results were obtained in T–cell specific SHP-2 C/S transgenic mice in which SHP2(C/S) was cloned into the hCD2 minigene cassette12. Indeed, SHP-2 deficiency in CD4+ T cells was also found to aggravate murine experimental colitis in cSHP-2 KO mice (our unpublished data).
The tumor inflammatory microenvironment leads to the recruitment of inflammatory cells, such as MDSCs, TAM and regulatory T cells27,28,29. In this study, a significant increase in the percentage of CD11b+ Gr-1+ MDSCs was observed in the tumor tissues from cSHP-2 KO mice, while the percentages of other populations (CD4+ T cells, CD8+ T cells, Tregs and macrophages) present in the lymph nodes, spleens and tumors were not significantly changed. MDSC accumulation was found not only in the spontaneous melanoma metastasis model but also in the passive metastasis model, in which tumor cells are injected into the tail vein to mimic the metastatic events that occur after tumor cells enter the circulatory system (supplementary Fig. S4). In addition, exvitro and in vivo assays indicated that CD4+ T cell-specific SHP-2 deficiency enhanced the tumor-promoting function of MDSCs in tumor-bearing mice. In contrast, no difference was detected in the function of TAM between the cSHP-2 KO and WT mice (supplementary Fig. S5). It has been well documented that various cytokines, chemokines and growth factors including IL-6, IL-10, TGF-β are needed to drive MDSC migration into tumor lesions and to keep their suppressive phenotype in tumor-bearing host30,31,32. An accumulation of functionally active MDSC in melanoma lesions and lymphoid organs was reported to be strongly associated with T-cell anergy33. Therefore, increased levels of inflammatory cytokines, especially IL-6, could induce the accumulation of MDSCs in the tumor and spleen. An IL-6 neutralizing antibody reduced MDSC accumulation and inhibited tumor growth in cSHP-2 KO mice, suggesting that MDSCs, IL-6 and tumor progression are linked in the context of CD4+ T cell-specific SHP-2 deletion.
SHP-2 has previously been considered as a potential drug target for cancer therapy34,35. Recent studies indicate that the inhibition of SHP-2 can enhance the anti-melanoma activity of IFNα-2b in human melanoma cells36. Our study, however, provides the first evidence to suggest that the loss of SHP-2 activity in CD4+ T cells promotes melanoma development and metastasis. This finding highlights the tumor-suppressing role of SHP-2 in melanoma and may have clinical implications for therapies against melanoma.
Methods
Mice
C57BL/6 mice (6 to 8 weeks old) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences. CD4+ T cell-specific SHP-2 knockout (cSHP-2 KO) mice (C57BL/6) were generated by crossing shp-2flox/flox mice with CD4-Cre transgenic mice37,38. Shp-2flox/flox littermates were used as control mice (WT). Genotyping and sequence confirmation were performed by PCR analyses of tail genomic DNA as previously described39. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Nanjing University. This study was carried out in accordance with the recommendations in the guidelines of the Ministry of Science and Technology of China (2006) and the related ethical regulations of Nanjing University. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Human blood samples
Human peripheral blood mononuclear cells (PBMC) were isolated from healthy donors using lymphocyte-monocyte separation medium, washed three times in PBS and suspended in complete RPMI 1640 medium. Informed consents were obtained from the donors involved. The study was approved by the Ethic Committee of Nanjing University.
Cell culture
B16BL6 and B16F10 mouse metastatic melanoma cells, LLC mouse Lewis lung carcinoma cells and A375 and A875 human melanoma cells were purchased from the American Type Culture Collection and firefly luciferase-expressing B16F10 melanoma cells were purchased from Cold Spring Biotech Corp. All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Life Technologies), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Melanoma metastasis models
For the spontaneous metastasis model, B16BL6 cells (1 × 106 cells in 20 μL PBS) were injected into the right hind footpads of C57BL/6 mice as described previously40. Draining popliteal lymph nodes and tumors were resected on days 0 (the day of tumor cell inoculation), 5, 14 and 21. In another protocol, B16BL6 cells (2 × 105 cells in 100 μL of PBS) were injected into the right flank of WT or cSHP-2 KO mice. The tumor volumes were measured every 2 days and calculated using the following formula:0.5236 × L1 × (L2)2, where L1 and L2 are the long and short axes of the tumor, respectively. For the passive metastasis model, B16F10 cells (2 × 105 cells in 100 μL of PBS) were injected via the tail vein into WT mice or cSHP-2 KO mice as described previously41. At the end of the experiments, the lung and liver tissues were examined to identify metastases. Tissues with metastases were either photographed for gross morphology or further analyzed by histology and immunostaining. For IL-6 neutralization experiments in vivo, mice were treated with 25 μg of an anti-IL-6 antibody (eBioscience), which was injected intraperitoneally every 4 days during the course of the experiment, starting on the day of tumor inoculation.
Isolation of tumor-infiltrating lymphocytes
Melanoma tissues were minced and digested with 0.5 mg/mL collagenase IV (Sigma-Aldrich) and 0.01 mg/mL DNaseI (Roche) in RPMI 1640/5% FCS for 1 h at 37°C. The cell suspension was then filtered through a 70-μm nylon mesh, layered on a Percoll gradient (30%–70%) and centrifuged at 800 × g for 20 min. The separated tumor-infiltrating lymphocyte (TIL) fraction was washed twice before use. CD45.2+ cells represent the total leukocyte population.
Flow cytometry
Fluorescently labeled antibodies were purchased from eBioscience (CD4, CD8, CD3, CD45.2, CD11b, Gr-1 and IFN-γ) or BD Biosciences (PD-1, CTLA-4 and SHP-2 (pY542)). Splenocytes, lymph node cells and TILs were incubated with specific surface-binding antibodies for 15–20 min at room temperature. For anti-CTLA-4 or SHP-2 (pY542) mAb staining, cells were pretreated with Cytofix/Cytoperm Kit reagents (BD Biosciences). For intracellular IFN-γ staining, cells were stimulated with 100 ng/ml PMA (Sigma-Aldrich) and 500 ng/ml ionomycin (Sigma-Aldrich) in the presence of monensin (BD Pharmingen) for 4 h. The washed cells were then fixed and permeabilized using a Cytofix/Cytoperm Kit after labeling with surface marker antibodies, followed by anti-IFN-γ mAb staining. Regulatory T cells were detected using a Mouse Regulatory T cell Flow CytometryStaining Kit (eBioscience). Samples were analyzed by flow cytometry on a FACScan flow cytometer (Becton Dickinson).
Cytokine analysis by ELISA and CBA assay
Sera obtained from tumor-bearing mice were stored at −80°C until use. The IL-6 level was measured using a specific ELISA kit from eBioscience according to the manufacturer's instructions. The levels of IL-2, IL-17, TNF-α, IFN-γ, IL-6 and IL-4 were determined using a cytometric bead array (CBA) cytokine assay kit as recommended by the manufacturer (BD Pharmingen).
Histology and immunostaining
Human malignant melanoma tissue microarray (#ME1004a and ME2082b) were purchased from US Biomax, Inc, containing primary malignant melanoma, metastatic malignant melanoma, nevus tissue, adjacent normal skin tissues and normal skin tissues. Mouse melanoma, liver and lung tissues were embedded in paraffin or snap-frozen in Tissue-Tek OCT medium (Sakura Finetek). Paraffin-embedded sections were stained with hematoxylin and eosin (H&E) using standard protocols. A TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche). For pulmonary nodule enumeration, the number of metastatic foci in 6 H&E-stained lung sections was counted in a blinded fashion. For the immunohistochemical analyses, paraffin-embedded or frozen sections were stained with antibodies against PCNA (Santa Cruz Biotechnology), pTyr705 STAT3 (Cell Signaling Technology) or CD31 (BD Pharmingen), as previously described39. Protein expression was quantified by measuring the total area of the tumor sections using Image Pro Plus 6 (Media Cybernetics) and then counting the density mean within that tumor area. For the immunofluorescence analyses, positive expression was based upon the score (0–300) that was a product of intensity of stain. Low expression were characterized as −/+. High expression (++) had scores between 150–300.
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Invitrogen Life Technologies). Quantitative PCR was performed with the ABI Prism 7000 Sequence Detection System (Applied Biosystems) using SYBR Green I dye (Biotium, Inc.) and threshold cycle numbers were obtained using ABI Prism 7000 SDS software version 1.0. The amplification conditions were 1 cycle of 94°C for 5 min followed by 35 cycles of 94°C for 30 s, 59°C for 35 s and 72°C for 45 s. The primer sequences used in this study are listed in Supplemental Table 1.
Western blotting
Western blotting was carried out as described previously40.
Adoptive transfer experiments
B16BL6 cells (2 × 105 cells in 100 μL PBS) were injected into the right flanks of WT or cSHP-2 KO mice. After 14 days, CD45.2+CD11b+Gr-1+ MDSCs were isolated from the spleens of tumor-bearing mice by FACS (Becton Dickinson). The isolated MDSCs (2 × 105/mouse) were co-injected with B16BL6 cells (1 × 105/mouse) into C57BL/6 mice and tumor growth was monitored.
Live bioluminescence imaging
Firefly luciferase-expressing B16F10 cells (2 × 105 cells in 100 μL of PBS) were injected via the tail vein into WT mice or cSHP-2 KO mice. Total tumor cell loading was assessed by bioluminescence imaging using an IVIS imaging system (Xenogen). Tumor-bearing mice were anesthetized and D-luciferin (15 mg/kg) was injected into the peritoneal cavity. Several images were taken over a 10–20 min period and analyzed using Living Image Software (Xenogen).
Statistical analysis
Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by the Student's two-tailed t-test. The Kaplan-Meier method was used to evaluate the survival results. P < 0.05 was considered significant.
References
Neel, B. G., Gu, H. & Pao, L. The Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28, 284–293 (2003).
Chong, Z. Z. & Maiese, K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation and injury. Histol Histopathol 22, 1251–1267 (2007).
Mohi, M. G. & Neel, B. G. The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev 17, 23–30 (2007).
Bard-Chapeau, E. A. et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19, 629–639 (2011).
Jiang, C. et al. The tumor suppressor role of Src homology phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J Cancer Res Clin Oncol 138, 637–646 (2012).
Li, S., Hsu, D. D., Wang, H. & Feng, G. S. Dual faces of SH2-containing protein-tyrosine phosphatase Shp2/PTPN11 in tumorigenesis. Front Med 6, 275–279 (2012).
Perales, M. A. & Wolchok, J. D. CD4 help and tumor immunity: beyond the activation of cytotoxic T lymphocytes. Ann Surg Oncol 11, 881–882 (2004).
DeNardo, D. G. et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009).
Purdy, A. K. & Campbell, K. S. SHP-2 expression negatively regulates NK cell function. J Immunol 183, 7234–7243 (2009).
Salmond, R. J. & Alexander, D. R. SHP2 forecast for the immune system: fog gradually clearing. Trends Immunol 27, 154–160 (2006).
Lorenz, U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228, 342–359 (2009).
Salmond, R. J., Huyer, G., Kotsoni, A., Clements, L. & Alexander, D. R. The src homology 2 domain-containing tyrosine phosphatase 2 regulates primary T-dependent immune responses and Th cell differentiation. J Immunol 175, 6498–6508 (2005).
Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H. & Riley, J. L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 173, 945–954 (2004).
Parry, R. V. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25, 9543–9553 (2005).
Kwon, J. et al. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. EMBO J 24, 2331–2341 (2005).
Ahmadzadeh, M. et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 (2009).
Wang, S. F. et al. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PLoS One 6, e17621 (2011).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9, 162–174 (2009).
Marigo, I., Dolcetti, L., Serafini, P., Zanovello, P. & Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 222, 162–179 (2008).
Ostrand-Rosenberg, S. & Sinha, P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182, 4499–4506 (2009).
Aceto, N. et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med 18, 529–537 (2012).
de Visser, K. E., Eichten, A. & Coussens, L. M. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6, 24–37 (2006).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Yu, H., Kortylewski, M. & Pardoll, D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7, 41–51 (2007).
Coulombe, G. et al. Epithelial tyrosine phosphatase SHP-2 protects against intestinal inflammation in mice. Mol Cell Biol 33, 2275–2284 (2013).
Nguyen, T. V., Ke, Y., Zhang, E. E. & Feng, G. S. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol 177, 5990–5996 (2006).
Koebel, C. M. et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29, 235–271 (2011).
Bunt, S. K. et al. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67, 10019–10026 (2007).
Tu, S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
Zhao, F. et al. Activation of p38 mitogen-activated protein kinase drives dendritic cells to become tolerogenic in ret transgenic mice spontaneously developing melanoma. Clin Cancer Res 15, 4382–4390 (2009).
Meyer, C. et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A 108, 17111–17116 (2011).
Dawson, M. I. et al. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J Med Chem 51, 5650–5662 (2008).
Irandoust, M., van den Berg, T. K., Kaspers, G. J. & Cloos, J. Role of tyrosine phosphatase inhibitors in cancer treatment with emphasis on SH2 domain-containing tyrosine phosphatases (SHPs). Anticancer Agents Med Chem 9, 212–220 (2009).
Win-Piazza, H. et al. Enhanced anti-melanoma efficacy of interferon alfa-2b via inhibition of Shp2. Cancer Lett 320, 81–85 (2012).
Zhang, E. E., Chapeau, E., Hagihara, K. & Feng, G. S. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A 101, 16064–16069 (2004).
Dong, X., Li, J., Li, S., Zhang, J. & Hua, Z. C. A novel genotyping strategy based on allele-specific inverse PCR for rapid and reliable identification of conditional FADD knockout mice. Mol Biotechnol 38, 129–135 (2008).
Wu, X. et al. Selective Sequestration of STAT1 in the Cytoplasm via Phosphorylated SHP-2 Ameliorates Murine Experimental Colitis. J Immunol 189, 3497–3507 (2012).
Wang, L. et al. An anticancer effect of curcumin mediated by down-regulating phosphatase of regenerating liver-3 expression on highly metastatic melanoma cells. Mol Pharmacol 76, 1238–1245 (2009).
Wu, X. et al. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am J Pathol 164, 2039–2054 (2004).
Acknowledgements
This work was supported by the Science Fund for Creative Research Groups (No. 81121062), the National Natural Science Foundation of China (Nos. 90913023, 31370900, 81173070, 91129728), National Science & Technology Major Project (No. 2012ZX09304-001) and Jiangsu Province Clinical Science and Technology Project (Clinical Research Center, BL2012008).
Author information
Authors and Affiliations
Contributions
T.Z., W.G., Y.Y., W.L., L.G. and Y.Shen. performed the experiments, T.Z., W.G. and Y.Shen. analysed the data and prepared the figures. Y.G., Y. S., L.W., X.W., Z.H. and Y.K. provided expertise and the mouse lines. Q.X., Y.Shen. and Y.Sun. conceived, directed the study and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental materials
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhang, T., Guo, W., Yang, Y. et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Sci Rep 3, 2845 (2013). https://doi.org/10.1038/srep02845
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02845
This article is cited by
-
SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses
Nature Immunology (2023)
-
Targeting protein phosphatases in cancer immunotherapy and autoimmune disorders
Nature Reviews Drug Discovery (2023)
-
Endothelial deletion of SHP2 suppresses tumor angiogenesis and promotes vascular normalization
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
