
Abstract
Mechanical forces are critical to modulate cell spreading, contractility, gene expression and even stem cell differentiation. Yet, existing tools that can apply controllable subcellular forces to a large number of single cells simultaneously are still limited. Here we report a novel ultrasound tweezing cytometry utilizing ultrasound pulses to actuate functionalized lipid microbubbles covalently attached to single live cells to exert mechanical forces in the pN - nN range. Ultrasonic excitation of microbubbles could elicit a rapid and sustained reactive intracellular cytoskeleton contractile force increase in different adherent mechanosensitive cells. Further, ultrasound-mediated intracellular cytoskeleton contractility enhancement was dose-dependent and required an intact actin cytoskeleton as well as RhoA/ROCK signaling. Our results demonstrated the great potential of ultrasound tweezing cytometry technique using functionalized microbubbles as an actuatable, biocompatible and multifunctional agent for biomechanical stimulations of cells.
Similar content being viewed by others
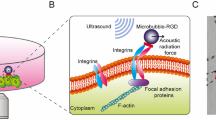
Introduction
Mechano-sensitivity or -responsiveness to extracellular biomechanical signals is a fundamental characteristic that controls the function of many types of mammalian cells. However, the molecular mechanism of such mechanotransduction processes is still elusive1,2. Although shear stresses and stretch forces applied to adherent mammalian cells can induce cellular responses such as reorganization of actin cytoskeleton and changes of intracellular contractile force, it is difficult to identify cell membrane receptors responsible for force transmission and converting external mechanical signals into intracellular biochemical events at a subcellular resolution3,4. Optical5 and magnetic tweezers6,7 have been commonly employed to apply local subcellular forces using functionalized microbeads attached to cell membrane via ligand-receptor binding. Optical tweezer can apply forces typically in the piconewton (pN) range, which is suitable for manipulation of single molecules but not large enough to induce cellular functional responses8,9. Further, optical tweezer can only apply stimuli to one single cell at a time and thus is prohibitive for large-scale, high-throughput cellular functional assays involving many single cells simultaneously. Magnetic tweezer can apply local subcellular pulling force as well as twisting stress in the range of pN to nanonewton (nN) by actuating magnetic beads functionalized with specific membrane receptor ligands10. Magnetic tweezer has been successfully applied to mammalian cells to regulate gene expression and even stem cell differentiation11,12.
Given the importance of mechanical forces to modulate mechanoresponsive behaviors of cells, as well as the need of new cellular bioengineering tools for high-throughput multiparametric screening and translational applications, it is highly desirable to develop new tools with expanded capabilities that can apply controllable mechanical forces with a subcellular precision on a large number of live single cells simultaneously.
Here we report a novel acoustic tweezing cytometry technique that utilizes ultrasound excitation of membrane-bound gaseous microbubbles to generate controllable subcellular mechanical stimulations to live single cells. Microbubbles are highly responsive to ultrasound excitation owing to a large difference in acoustic impedance between gas inside bubble and surrounding liquid media. Oscillatory positive and negative pressure of an ultrasound field readily induces microbubble expansion and contraction (stable cavitation, resulting in fluid microstreaming), and/or violent collapse (inertial cavitation, which can generate high-speed fluid micro-jet that can penetrate cell membrane) if the pressure amplitude is high enough13,14,15. In addition to rapid volumetric expansion/contraction and collapse of microbubbles, an ultrasound field can also generate a directional force on the bubble16,17,18, recognized as the acoustic radiation force, which can compress the bubble against cell membrane to exert a mechanical force on the cell. The acoustic radiation force resulted from ultrasound stimulated microbubbles can lead to rupture of cell membrane18.
Stabilized microbubbles encapsulated by lipids or a thin protein layer have been recently developed successfully as contrast agents for ultrasound imaging in clinical diagnostic radiological applications19,20. Recently, lipid-stabilized microbubbles coated with streptavidin have been developed to enable functionalization of bubbles with specific ligands on the bubble shell encapsulating the gas core. Functionalized bubbles allow selective or targeted attachments of bubbles to mammalian cells via specific ligand-receptor binding21,22,23, enabling in vivo ultrasound molecular imaging by recognizing molecular markers associated with specific diseases including inflammation and angiogenesis24,25,26,27. In addition, ultrasound excitation of functionalized lipid microbubbles have been exploited for delivering cell-membrane impermeable DNAs, drugs and other therapeutic compounds from extracellular space into cells28,29 by transiently disrupting cell membrane via stable30,31 or inertial cavitation (sonoporation)32. Ultrasound-induced microbubble activities can generate localized yet significant mechanical impact on cells30,32,33.
In this study, we developed a novel ultrasound tweezing cytometry utilizing ultrasound-excitable microbubbles targeted to cell membrane to apply spatiotemporally controlled subcellular mechanical forces to live single cells. By applying ultrasound pulses with appropriate amplitude and duration, functionalized lipid microbubbles attached to individual cells were actuated to exert subcellular mechanical forces in the pN - nN range to live single cells. Specifically, using NIH/3T3 fibroblasts and human mesenchymal stem cells (hMSCs) as mechanosensitive cell models, we showed that ultrasonic excitation of lipid microbubbles attached on cell membrane through integrin-mediated adhesions could elicit a rapid and sustained reactive intracellular cytoskeleton contractile force increase. Such ultrasound-mediated intracellular cytoskeleton contractility enhancement was dose-dependent and required an intact actin cytoskeleton structure as well as RhoA/ROCK signaling for intracellular transmission and conversion of mechanical signals. Our results demonstrated for the first time the potential of ultrasound tweezing cytometry technique using targeted microbubbles as an actuatable, biocompatible and multifunctional agent for biomechanical stimulation of mechano-sensitive and -responsive cells.
Results
Ultrasound tweezing cytometry to control subcellular contractile response
To study ultrasound-mediated changes in intracellular cytoskeleton contractility, we utilized an elastomeric micropost array we have recently developed as subcellular live-cell force sensors to quantify intracellular contractile forces34. The micropost array consists of an array of uniformly spaced, vertical, elastomeric posts fabricated using photolithography and replica molding with silicone elastomer polydimethylsiloxane (PDMS) ( Fig. 1A ). After adhesive proteins are coated using microcontact printing on the post tips, cells are able to adhere, spread out and exert contractile forces that deflect underlying posts. Each post, therefore, functions as a cantilever, capable of measuring local cellular traction force exerted at the post tip. For tip deflections δ that are small compared to the post height L, the PDMS post can be modeled as a linearly elastic beam subjected to pure bending. Thus, intracellular contractile force F applied at the tip of the PDMS post can be calculated as F = 3πEd4δ/(64L3), where E is the elastic modulus of PDMS and d is the post diameter. In this study, NIH/3T3 fibroblasts and hMSCs were first seeded as single cells on fibronectin-coated PDMS microposts. Individual lipid microbubbles with a mean diameter of 2.3 μm and functionalized with Arg-Gly-Asp (RGD) peptides using avidin-biotin conjugation chemistry were then attached to the cells through specific binding to integrins on cell membrane ( Supplementary Fig. S1 ).

Modulation of subcellular cytoskeleton contractility by ultrasound (US) excitation of microbubbles attached to cell membrane ("acoustic tweezing cytometry").
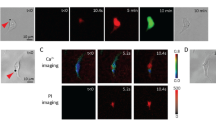
(A) A schematic showing US excitation of microbubbles (MB) attached via RGD-integrin binding to the membrane of a cell seeded on the PDMS micropost array. (B&C) Single NIH/3T3 fibroblasts with one MB (arrow head) attached to cell membrane and subjected to 10 sec (B) or 20 sec (C) US stimulations. Subcellular contractile force distributions (top panels) and pseudocolor ratio images of intracellular free calcium concentration ([Ca2+]) (bottom panels) before (t < 0 min) and after US application were shown. Scale bars, 10 μm. (D) Temporal evolution of total cytoskeleton contractile force (normalized to contractility before US excitation). (E) Fluorescence ratio representing [Ca2+] as a function of time. Data in D and E are shown as mean ± s.e.m. n = 31 for 10 sec US exposure and n = 10 for 20 sec US stimulation. Control groups without MBs attached and treated with (n = 8) or without (n = 10) US were included for comparison. (F) No intracellular propidium iodide (PI) was detected in NIH/3T3 fibroblasts with MBs attached after US stimulation. Calcein AM essay showed the same cell could convert calcein AM into green fluorescent calcein and confine it within cell cytoplasm. US parameters used in Fig. 1: US frequency of 1.25 MHz, acoustic pressure of 0.05 MPa, PRF of 1 Hz and duty cycle of 5%.
We first applied low amplitude ultrasound pulses with a peak negative pressure of 0.05 MPa, center frequency of 1.25 MHz, pulse repetition frequency (PRF) of 1 Hz and duty cycle of 5% (i.e., pulse duration of each individual pulse was 50 ms) to stimulate individual NIH/3T3 fibroblasts with one microbubble attached to cell membrane for a total period of 10 sec. A gradual and sustained increase of intracellular cytoskeleton contractile force in NIH/3T3 fibroblasts was observed immediately after ultrasound application ( Fig. 1B and Supplementary Fig. S2A ). Total cytoskeleton contractility of NIH/3T3 fibroblasts increased up to 148.5% ± 3.6% (mean ± standard error of mean) (n = 31; Supplementary Fig. S2A ) 30 min after ultrasound stimulation compared to pre-ultrasound contractile levels. Cytoskeleton contractile response to ultrasound excited microbubbles appeared to be dose-dependent ( Fig. 1C&D ). For ultrasound stimulation for 20 sec with other ultrasound parameters remained unchanged, the total cytoskeleton contractility of NIH/3T3 fibroblasts at 30 min after ultrasound exposure increased to 163.7% ± 5.0% (mean ± standard error of mean) of pre-ultrasound contractile levels (n = 10; Fig. 1C&D and Supplementary Fig. S2B ), significantly higher than the contractile force increase observed for 10 sec ultrasound exposure (p = 0.02 from a student's t-test). NIH/3T3 fibroblasts examined in control groups without microbubbles attached or without ultrasound treatments exhibited no observable change in cytoskeleton contractile force ( Fig. 1D and Supplementary Fig. S2C& D ).
Ratiometric calcium imaging using calcium indicator Fura-2-acetoxymethyl ester (Fura-2AM) showed that mechanical stimulations of cells with ultrasound conditions used in Fig. 1 caused no change in intracellular calcium ion concentrations ([Ca2+]i), indicating an absence of calcium signaling initiation ( Fig. 1B, C&E ). Further, no intracellular entry of cell-membrane impermeant propidium iodide (PI) was detected, indicating that ultrasound stimulation in Fig. 1 did not cause cell membrane disruption ( Fig. 1F ). Calcein AM-based cell viability assay performed 30 min after ultrasound stimulations showed that NIH/3T3 fibroblasts were able to convert non-fluorescent calcein AM into green fluorescent calcein and confine it inside cell cytoplasm, indicating cell viability ( Fig. 1F ).
To demonstrate that cytoskeleton contractile response to ultrasound tweezing was not cell-type specific, we applied the same ultrasound stimulation used in Fig. 1 for 10 sec to single live hMSCs. Total cytoskeleton contractility of hMSCs increased up to 148.2% ± 9.8% (mean ± standard error of mean) (n = 8) 30 min after ultrasound exposure compared to pre-ultrasound contractile levels ( Supplementary Fig. S3 ), an enhancement comparable in magnitude to that observed for NIH/3T3 fibroblasts.
Taken together, our results in Fig. 1 and Supplementary Fig. S3 demonstrated a sustained cytoskeleton contractility increase in both NIH/3T3 fibroblasts and hMSCs resulted directly from ultrasonic excitation of cell membrane bound lipid microbubbles without membrane disruption or calcium signaling initiation. Our results were consistent with previous studies using optical and magnetic tweezers, where mechanical stimulations of microbeads functionalized with RGD peptides attached on cell membranes lead to cytoskeleton contractility increase at adhesion contacts between microbeads and cell membrane12.
Ultrasound excitation of microbubbles with membrane disruption
To investigate how cytoskeleton contractile force would respond to ultrasound excitation of microbubbles with membrane disruption, we applied ultrasound pulses with a greater acoustic pressure (0.13 MPa) and shorter pulse duration (8 μs) to excite microbubbles attached to cell membrane. (These parameters have been shown previously to be capable of generating sonoporation of cells with localized membrane disruption28.) Under such ultrasound stimulations, cytoskeleton contractile force of NIH/3T3 fibroblasts immediately decreased before slowly recovering to the contractile level prior to ultrasound treatments (top panel in Fig. 2A&B ). Transient calcium influx was simultaneously observed in ultrasound-stimulated cells, as indicated by a rapid increase of intracellular Fura-2 fluorescence intensity after ultrasound stimulations (bottom panel in Fig. 2A&C ). For NIH/3T3 fibroblasts in a control group without microbubbles attached, cytoskeleton contractile force and [Ca2+]i remained unchanged under the same ultrasound perturbation ( Fig. 2B&C ). Further, PI uptake into cells at subcellular locations where lipid microbubbles were attached was detected after ultrasound application ( Fig. 2D ), suggesting transient cell membrane disruption by ultrasonic excitation of lipid bubbles. Calcein AM-based cell viability assay performed 30 min after ultrasound application confirmed cell viability ( Fig. 2D ).

Transient disruption of cell membrane by acoustic tweezing cytometry induced transient calcium influx and rapid cytoskeleton contractile force decrease.
(A) Brightfield image (left) showing a single NIH/3T3 fibroblast attached with one MB (arrow head) before US stimulation (t < 0 min). Subcellular cytoskeleton contractile force distributions (top panel) and pseudocolor ratio images of [Ca2+] (bottom panel) before (t < 0 min) and after US exposure were included. Scale bar, 10 μm. (B) Temporal evolution of total cytoskeleton contractile force (normalized to contractility before US excitation). (C) Fluorescence ratio representing [Ca2+] as a function of time for NIH/3T3 fibroblasts exposed to US excitation. Data in B & C are shown as mean ± s.e.m. n = 10 for cells attached with one MB and n = 10 for cells without MBs attached. (D) Excitation of microbubble resulted in transient pore formation on cell membrane, as indicated by emergence of PI fluorescence at the MB attachment location (arrow head). Calcein viability assay performed 30 min after US excitation showed that the cell was able to convert non-fluorescent calcein AM into green fluorescent calcein and confined it within cell cytoplasm. US parameters used in Fig. 2: acoustic pressure of 0.13 MPa and duration of 8 μs.
Subcellular distribution of cytoskeleton contractile force response
We next examined the effect of the attachment location of microbubble on subcellular distribution of reactive cytoskeleton contractile force increase. We divided NIH/3T3 fibroblasts examined in Fig. 1 into two groups based on the location of microbubble on cell membrane. In the first group (n = 15), each cell had a microbubble attached to the cell periphery region (i.e. outer 1/3 region from cell centroid) (see Methods), while in the other group (n = 16), each cell had a bubble attached to the cell central area. Contractile force maps obtained by summation of the contractile force change for each cell with the bubble location aligned ( Supplementary Fig. S4 ) showed that when the microbubble was attached to cell periphery, cytoskeleton contractile force enhancement was concentrated in the local subcellular area surrounding the bubble ( Supplementary Fig. S4A ). In contrast, when the bubble was located near cell centroid, contractile force increase was distributed evenly across entire cell area ( Supplementary Fig. S4B ). It should be noted that the total contractile force increase for the two cell groups was of a similar magnitude ( Supplementary Fig. S4C ). The dependence of subcellular distribution of reactive cytoskeleton contractile force increase on bubble attachment location was likely due to subcellular non-uniform distribution of actin cortex, known to concentrate on cell periphery to transmit and distribute reactive intracellular contractile force.
Actin, myosin II and RhoA/ROCK signaling required for cytoskeleton contractile response to ultrasound tweezing
The actin cytoskeleton consists of multiple molecular structures and components, with actin microfilaments and myosin II molecules as primary determinants for intracellular force transmission properties. RhoA/ROCK signaling is also critically involved in mechanotransduction and regulation of intracellular cytoskeleton contractility2,35,36. To investigate functional roles of actin polymerization, myosin II activity and RhoA/ROCK signaling in regulating ultrasound-mediated contractile force enhancement, we treated NIH/3T3 fibroblasts with small molecule inhibitors to target specifically actin polymerization (cytochalastin D, an inhibitor of actin polymerization), myosin II activity (blebbistatin, a myosin II ATPase inhibitor) and RhoA/ROCK signaling (Y27632, a ROCK inhibitor) ( Supplementary Fig. S5 ). NIH/3T3 fibroblasts attached with microbubbles were first treated with individual drugs for 30 min before stimulated by ultrasound. Cytoskeleton contractile forces of drug-treated NIH/3T3 fibroblasts decreased rapidly within the first 10 min after the onset of drug treatments before reaching an equilibrium contractile state at the end of the 30-min period (data not shown). More specifically, after 30-min treatments with cytochalastin D, blebbistatin and Y27632, cytoskeleton contractile forces of live single NIH/3T3 cells decreased by 70.1% ± 4.5% (n = 5), 71.2% ± 9.3% (n = 5) and 60.0% ± 4.5% (n = 4), respectively. Interestingly, drug-treated NIH/3T3 fibroblasts did not show any contractile force response after the same ultrasound stimulations used in Fig. 1 (acoustic pressure 0.05 MPa, center frequency 1.25 MHz, PRF 1 Hz, duty cycle 5% and total stimulation 10 sec), supporting that cytoskeleton integrity, myosin II activity and RhoA/ROCK signaling were all required for ultrasound induced cellular biomechanical responses.
Shear stress and microbubble displacement induced by ultrasound tweezing
To investigate the biomechanical mechanism(s) underlying ultrasonic bubble excitation-mediated cytoskeleton contractile force enhancement, we employed high-speed imaging to examine in situ bubble activities driven by ultrasound exposures. We observed that microbubbles exhibited translational movements or displacements relative to their original location on cell membrane when stimulated using ultrasound. As shown in Fig. 3A and Supplementary Video 1 , using the same ultrasound condition as in Fig. 1 (acoustic pressure 0.05 MPa, center frequency 1.25 MHz, PRF 1 Hz and duty cycle 5%), a microbubble initially bound to cell membrane was pushed away from and then pulled back to its original position repeatedly during ultrasound stimulation and such periodic bubble displacement corresponded temporarily with pulsed ultrasound exposures. During ultrasound “pulse-on” periods, the microbubble was pushed away in the ultrasound propagation direction by the primary acoustic radiation force acting on the bubble ( Fig. 3A&B ). Assuming a plane ultrasound field, the maximum acoustic radiation force on a bubble with a radius of 2.3 μm was estimated to be 17.0 nN (see Methods), comparable in magnitude to mechanical forces generated by magnetic tweezers10. During ultrasound “pulse-off” periods, the microbubble was pulled back partially or fully sometimes to its original position, indicating a biomechanical recovery process dictated by elastic properties of the bubble-focal adhesion-cytoskeleton mechanical linkage after release of external acoustic force.

In situ microbubble activities induced by acoustic tweezing cytometry.
(A) Time-lapse brightfield images showing one MB (arrow head) attached to a single NIH/3T3 fibroblast during 10 sec pulsed US stimulation. US was applied at t = 0 min, with a peak negative pressure amplitude of 0.05 MPa, PRF of 1 Hz and duty cycle of 5%. Scale bars, 12 μm. (B) Translational displacement of MB during the 10 sec US stimulation. (C) Temporal evolution of MB radius during ultrasound application. (D) Positive correlation between the maximum relative cytoskeleton contractile force change 30 min after US treatments and the accumulated MB translational displacement (n = 13 and R2 = 0.72).
Besides bubble displacement, the rapid volumetric expansion and contraction of microbubbles driven by ultrasound excitation can generate fluid flow (microstreaming) and thus shear stress on cells37,38,39. Shear stress exerted on cells were estimated from the peak fluid flow velocity near the bubble measured using the Micron-resolution Particle Image Velocimetry (micro-PIV) (see Methods; the peak velocity was 20.15 ± 3.92 mm/s (n = 28) corresponding to a maximum shear stress exerted on cell membrane of 7.05 ± 1.41 Pa). Note that our calculation was based on the peak value of a pulsatile shear stress field surrounding a microbubble during the 10-sec pulsed ultrasound application. Thus, the time-averaged shear stress during the whole ultrasound stimulation was much less. It has been reported that sustained shear stress above a value of 1–2.5 Pa can affect behaviors of NIH/3T3 fibroblasts40; however cells examined in such studies have been exposed to a sustained shear stress for >8 hr.
A time-resolved measurement of actual bubble expansion and contraction at the ultrasound frequency of 1.25 MHz and with the ultrasound pulse of 50 ms is technically challenging and requires a frame rate above several Mframes per sec. Nonetheless, we recorded high-resolution images of microbubbles during the entire ultrasound application duration (10 sec) at a frame rate of 20 kframes per sec to assess changes of bubble size. We observed that microbubbles gradually reduced their size during ultrasound application ( Fig. 3C ), likely owing to disruption of bubble lipid shell during repeated bubble expansion and contraction, causing gas diffusion out of bubble28. A reduction of microbubble size resulted in a decrease of acoustic radiation force acting on the bubble, leading to a shortened bubble displacement driven by later pulses as shown in Fig. 3B . The total or accumulative forward displacement of a microbubble during the entire 10-sec ultrasound application period appeared to be positively correlated with the overall cytoskeleton contractile force increase during the 30-min observation period ( Fig. 3D ) (R2 = 0.72 and n = 13), suggesting the important role of the physical displacement of integrin-mediated cell adhesion in regulating cytoskeleton contractile force response. The broad distribution of accumulative displacement of microbubbles was attributable to the bubble initial size distribution (and thus different acoustic radiation forces acting on bubbles) as well as inevitable variations in RGD-integrin binding and their intracellular connection to actin cytoskeleton.
Both shear stress (due to ultrasound-driven microbubble expansion and contraction) and microbubble displacement (due to acoustic radiation force) might have contributed to reactive cytoskeleton contractile force change. The relative dominance of microbubble displacement over their expansion and contraction can be adjusted by tuning ultrasound parameters such as ultrasound frequency, acoustic pressure and pulse duration. Additional future work is needed to determine precisely the relative role of these two biophysical factors in ultrasonic bubble excitation-mediated cytoskeleton contractile force enhancement.
Discussion
Our study demonstrates that ultrasound excitation of targeted microbubbles provides a robust strategy for biomechanical stimulation of mechano-sensitive and -responsive mammalian cells. The strong contractile response of NIH/3T3 fibroblasts as well as hMSCs to ultrasound excited microbubbles suggests that our ultrasound tweezing cytometry technique may have a great utility in regulating cellular behaviors.
Cells can sense mechanical stimuli and convert them into downstream intracellular biochemical signals and such mechanotransduction process has profound impact in many physiological and pathological contexts. For example, endothelial cells respond to blood flow shear stress by regulating actin cytoskeleton structure4. Cytoskeleton contractility has also been implicated as a non-destructive live-cell predictor for hMSC differentiation34. Thus, the ultrasound tweezing cytometry methodology reported here can be effectively applied, in principle, to regulate biomechanical and cellular responses of different cell types.
Like magnetic and optical tweezers, our acoustic tweezing cytometry can generate controllable localized mechanical forces to single live mammalian cells. However, acoustic tweezing cytometry has several unique advantages. Unlike microbeads used in magnetic or optical tweezers, microbubbles provide a versatile multifunctional platform that can enable multifaceted applications. Microbubbles have been established as an effective imaging contrast agent for ultrasound imaging41,42,43 and they are also routinely used as drug carrier44,45. Thus, the acoustic tweezing cytometry reported here has the potential for in vivo imaging guided manipulation of cellular contractility and thus downstream cellular effects. In addition, ultrasound excitation of targeted microbubbles is capable of intracellular delivery of drugs in a spatiotemporally controlled fashion, as we have previously demonstrated28. Thus, besides direct mechanical perturbation of cells, sonoporation can be seamlessly implemented and integrated in the same platform with acoustic tweezing cytometry to deliver membrane-impermeable drugs into cells for additional biochemical modulations of intracellular signaling. Many types of mammalian cells, either adherent in a monolayer or embedded in a three-dimensional matrix, can be treated simultaneously by ultrasound stimulation that can readily excite all microbubbles within the ultrasound field, providing a high-throughput cytometric assay on a large number of single cells with a subcellular precision. Further, by focusing ultrasound field to a local region or spatially patterning microbubbles46, a selected group of cells within a monolayer or other multicellular configurations can be targeted for biomechanical manipulations, thus enabling investigation of intercellular biomechanical interactions.
In summary, we demonstrated here for the first time that ultrasound excitation of targeted microbubbles could enable controlled application of subcellular biomechanical stimuli to live single cells. Drug inhibition experiments demonstrated that intact actin cytoskeleton, myosin activity and RhoA/ROCK signaling were all required for force sensing and transmission in the acoustic tweezing cytometry system. We envision that the acoustic tweezing cytometry employing targeted microbubbles as a biocompatible and multifunctional agent can be further developed in the future as a powerful cell mechanics and mechanobiology tool to regulate cell function, gene expression and stem cell differentiations both in vitro and in vivo.
Methods
Fabrication and surface functionalization of PDMS microposts
The PDMS micropost array was fabricated using a protocol described previously47. Briefly, a silicon micropost array master was fabricated by standard photolithography and deep reactive ion etching (DRIE). The height of silicon micropost was modulated by controlling DRIE etch time. The silicon master was silanized with (tridecafluoro-1, 1, 2, 2-tetrahydrooctyl)-1-trichlorosilane (United Chemical Technologies, Bristol, PA) for 4 hrs under vacuum before 1:10 (w/w, curing agent/base monomer) ratio PDMS prepolymer (Sylgard 184, Dow-Corning, Midland, MI) was poured over the silicon master and cured at 110°C for 20 min. Negative PDMS molds were then peeled off the silicon master, oxidized with oxygen plasma for 1 min (200 mTorr; Plasma Prep II, West Chester, PA) and silanized with (tridecafluoro-1, 1, 2, 2-tetrahydrooctyl)-1-trichlorosilane for 24 hrs. To generate final PDMS micropost arrays, 1:10 (w/w, curing agent/base monomer) ratio PDMS prepolymer was poured over the negative PDMS mold and degassed under vacuum for 10 min. A clean 25 mm × 25 mm cover glass was then placed on top of the negative mold and the whole assembly was cured at 110°C for 40 hrs. The final PDMS micropost array was then peeled off the negative PDMS mold. When peeling induced collapse of PDMS microposts, we regenerated the array by sonication in 100% ethanol for 30 sec followed by dry-release with liquid CO2 using a critical point dryer (Samdri®-PVT-3D, Tousimis, Rockville, MD). The cover glass holding the PDMS micropost array was finally attached to a 35 mm dish with a 20 mm hole at the dish center (MatTek dish; Glass Bottom Dishes, MatTek, Ashland, MA).
To promote cell adhesion, top surface of the PDMS micropost array was functionalized with fibronectin (Sigma, St. Louis, MO) using microcontact printing. Briefly, a flat PDMS stamp made from 1:30 (w/w, curing agent/base monomer) ratio PDMS prepolymer was soaked in a fibronectin solution (50 μg mL−1) in distilled water for 1 hr at room temperature. The PDMS stamp was then thoroughly rinsed with distilled water and blown dry with a stream of nitrogen. In parallel, the PDMS micropost array was treated with a UV-ozone cleaner (Jelight, Irvine, CA) for 7 min for surface activation. The PDMS stamp was placed in conformal contact with the PDMS micropost array for 10 sec to transfer fibronectin from the stamp to the top surface of the PDMS micropost array. The PDMS micropost array was then labeled with 5 μg mL−1 Δ9-DiI (Invitrogen, Grand Island, NY) in distilled water for 1 hr. To prevent non-specific protein absorption to non-functionalized surface of the PDMS micropost array, the PDMS micropost array was incubated in 0.1%–1% (w/v) Pluronics F127 NF (BASF, Sigma, St. Louis, MO) in distilled water for 30 min at room temperature.
Cell culture
NIH/3T3 mouse fibroblasts (ATCC, Manassas, VA) were maintained at 37°C and 5% CO2 in a growth medium consisting of low-glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% bovine serum (Invitrogen), 1% L-glutamine and 1% penicillin/streptomycin. Human mesenchymal stem cells (Lonza, Walkersville, MD) were maintained at 37°C and 5% CO2 in the MSCGM mesenchymal stem cell growth medium (Lonza). Cells were harvested 8–16 hrs before experiments and seeded as single cells on the PDMS micropost array in the MatTek dish at a density of 3,000 cells cm−2.
Targeted lipid microbubbles and their attachment to cells
Targestar™-SA lipid microbubbles from Targeson (San Diego, CA) were used in this study. Microbubbles (with a mean diameter of 2.3 ± 0.2 μm; n = 39) were first mixed with biotinylated Arg-Gly-Asp (RGD) peptides (Peptides International, Louisville, KY) for 20 min at room temperature, with a molar ratio of 5:1 for microbubbles (5 × 108 mL−1) and RGD (0.01 mg mL−1). Following a recommended procedure from the manufacturer, immediately after removal of culture media in the dish where cells were seeded on the PDMS microposts, 20 μL of the microbubble-RGD mixture was added into the dish (see Supplementary Fig. S1 ). The dish was then flipped upside down for 10 min to allow bubbles to float up towards cells to facilitate binding of microbubbles to the cells. Unbound bubbles were removed by some gentle wash with culture media. The concentration of microbubbles used in this study ensured that for most single cells, only one bubble was attached to cell membrane.
Ultrasound application
An ultrasound transducer (Advanced Devices, Wakefield, MA) driven by a function generator (Agilent Technologies 33250A, Palo Alto, CA) and a 75 W power amplifier (Amplifier Research, Souderton, PA, USA) was used to generate ultrasound pulses to excite microbubbles. The ultrasound transducer was positioned at a 45° angle relative to the horizontal MatTek dish, in order to avoid formation of standing wave in the dish and to permit unobstructed microscopic imaging. The transducer activation surface was submerged in the medium 7 mm (Rayleigh distance) away from cells. To further minimize formation of standing waves in the MatTek dish, a chirp technique48 for ultrasound application was applied. Each ultrasound pulse was produced by sweeping or chirping the driving frequency around the center frequency, i.e. 1.1–1.4 MHz. The acoustic pressure amplitude of the chirped pulses was further calibrated in a free field using a needle hydrophone (HNR-0500, ONDA, CA, USA).
Videomicroscopy and ultrasound driven microbubble activities
Images of microbubbles excited by ultrasound exposures were acquired using a high-speed camera (FASTCAM SA1, Photron, San Diego, CA; with the frame rate up to 20 Kframes s−1) and a water immersion 60× objective (1.0 NA; Nikon, Melville, NY, USA) mounted on an inverted fluorescence microscope (Eclipse Ti-U; Nikon). Ultrasound driven bubble activities such their translational movement and change in bubble radius were quantified using commercial image processing software (FASTCAM Viewer 3, Photron). Specifically, bubble displacement was quantified by manually locating the bubble center and tracking its position in each frame. Bubble radius was determined by counting pixels within a manually-defined boundary for the bubble in each frame.
Acoustic radiation force acting on microbubbles
A first-order estimate of the primary acoustic radiation force F acting on microbubbles by an ultrasound field at a resonance frequency of ω0 was obtained from  49, where PAis the acoustic pressure, R0 is the bubble radius, δtotis the total damping constant (δtot = 0.16), ρ0 is the medium density (1000 kg m−3) and c is the sound speed in media (1500 m s−1). It should be noted that this estimation did not consider the effect of the lipid shell on bubble resonant frequency.
49, where PAis the acoustic pressure, R0 is the bubble radius, δtotis the total damping constant (δtot = 0.16), ρ0 is the medium density (1000 kg m−3) and c is the sound speed in media (1500 m s−1). It should be noted that this estimation did not consider the effect of the lipid shell on bubble resonant frequency.
Flow velocity and shear stress generated by ultrasound excitation of microbubbles
Ultrasound driven microbubble expansion and contraction generates microstreaming or flow field near the bubble50,51. We applied the micron-resolution Particle Image Velocimetry (micro-PIV) to measure flow velocity near a bubble, by using 0.5 μm diameter polystyrene particles (Thermo Scientific, CA, USA) as flow tracers. Specifically, tracer particles at a stock solution (1.46 × 1011 particles mL−1) were diluted first in deionized water with a 1:2 (v/v) ratio. 2 μL of diluted tracer solution was then added to culture media right before ultrasound applications for cells seeded on the PDMS micropost array in the MetTak dish. Positions of tracer particles near a bubble during ultrasound application were continuously recorded using the high-speed camera at a frame rate of 20 Kframes s−1. Velocities of polystyrene particles and thus the flow field near the bubble were obtained by processing the image sequence using Image J (US National Institutes of Health, Bethesda, MD). Shear stress τ exerted on cell membrane due to microstreaming of fluid was then calculated as τ = μΔv/Δy, where μ is the medium viscosity (7.26 × 10−4 Pa sec), Δv is the flow velocity difference between bubble equator plane and cell membrane and Δy is treated equal to the bubble radius.
Fluorescence imaging
An air-cooled CCD camera (Photometrics QuantEM, Tucson, AZ, USA) was used for fluorescence imaging. Calcium imaging was performed using a method described previously52. Briefly, cells seeded on the PDMS micropost array were first incubated with 10 μM Fura2-AM (Invitrogen) and 0.05% (w/v) Pluronic F-127 (Invitrogen) in culture media for 60 min at 37°C and 5% CO2. Fluorescence emission intensities of Fura2 at 510 nm from cells excited at wavelengths of 340 nm and 380 nm were acquired to generate ratiometric images inferring to intracellular free calcium concentrations.
Changes in cell membrane permeability were assessed by detecting intracellular fluorescence intensity of nucleic acid intercalating agent propidium iodide (PI) (Sigma Aldrich, St. Louis, MO). PI was added into culture media (final PI concentration of 100 μM) right before ultrasound application. Cells were excited at 539 nm and emission was detected at 610 nm to detect the presence of PI inside cells.
To assess cell viability, calcein AM (Invitrogen) was added into culture media 30 min after ultrasound treatments at a final working concentration of 1 μM. In live cells, non-fluorescent calcein AM is converted to green fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterases.
Quantification of cytoskeleton contractile forces using PDMS microposts
Fluorescent images showing the top surface of PDMS microposts were obtained from fluorescence microscopy (Eclipse Ti-U; Nikon) and processed by a custom-developed MATLAB program (MathWorks, Natick, MA; details of the program was described previously47) to quantify deflections of PDMS microposts from their unbent, unloaded positions, which were then converted to horizontal contractile forces by multiplying with the nominal spring constant K of the PDMS micropost.
To study spatial distribution of subcellular contractile force changes, we divided cells into two groups based on the relative location of microbubbles on the cells. In the first group, each cell had a bubble attached to a position in the cell peripheral region, defined as the outer 1/3 radial position from the cell centroid. The other group included cells with bubbles attached at locations in the more interior region. Subcellular maps of contractile force changes before and 30 min after ultrasound applications were generated for all individual cells in each group and these maps were spatially aligned such that microbubbles were aligned along with the nearest cell boundary. All the force change maps for each group of cells were summed up to demonstrate show the overall subcellular contractile force changes based on the microbubble location.
Drug inhibition assays
Cells seeded on the PDMS micropost array in the MatTek dish were treated with Cytochalasin D (40 μM; Invitrogen), Blebbistatin (100 μM; Sigma) and Y-27632 (10 μM; Enzo Life Sciences, Farmingdale, NY) in culture media for 30 min before applying ultrasound to activate lipid microbubbles attached to cells.
References
Discher, D. E., Mooney, D. J. & Zandstra, P. W. Growth factors, matrices and forces combine and control stem cells. Science. 324, 1673–1677 (2009).
Sun, Y., Chen, C. S. & Fu, J. Forcing stem cells to behave: A biophysical perspective of the cellular microenvironment. Annu Rev Biophys. 41, 519–542 (2012).
Mann, J. M., Lam, R. H., Weng, S., Sun, Y. & Fu, J. A silicone-based stretchable micropost array membrane for monitoring live-cell subcellular cytoskeletal response. Lab Chip. 12, 731–740 (2012).
Lam, R. H. W., Sun, Y. B., Chen, W. Q. & Fu, J. P. Elastomeric microposts integrated into microfluidics for flow-mediated endothelial mechanotransduction analysis. Lab Chip. 12, 1865–1873 (2012).
Svoboda, K., Schmidt, C. F., Schnapp, B. J. & Block, S. M. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 365, 721–727 (1993).
Smith, S. B., Finzi, L. & Bustamante, C. Direct mechanical measurements of the elasticity of single DNA-molecules by using magnetic beads. Science. 258, 1122–1126 (1992).
Strick, T. R., Allemand, J. F., Bensimon, D., Bensimon, A. & Croquette, V. The elasticity of a single supercoiled DNA molecule. Science. 271, 1835–1837 (1996).
Sheetz, M. P. Laser tweezers in cell biology. Introduction. Methods in cell biology vol. 55, 1998, pp xi–xii.
Huang, H., Kamm, R. D. & Lee, R. T. Cell mechanics and mechanotransduction: Pathways, probes and physiology. Am J Physiol. 287, C1–11 (2004).
Tanase, M., Biais, N. & Sheetz, M. Magnetic tweezers in cell biology. In: Wang Y.,, Discher D. E., eds. (eds). Methods in cell biology vol. 83, Academic Press, 2007, pp 473–493.
Chen, J. X., Fabry, B., Schiffrin, E. L. & Wang, N. Twisting integrin receptors increases endothelin-1 gene expression in endothelial cells. Am J Physiol. 280, C1475–C1484 (2001).
Chowdhury, F., Na, S., Li, D., Poh, Y. C., Tanaka, T. S., Wang, F. et al. Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat Mater. 9, 82–88 (2010).
O'Brien, W. D., Jr Ultrasound-biophysics mechanisms. Prog Biophys Mol Bio. 93, 212–255 (2007).
Wu, J. & Nyborg, W. L. Ultrasound, cavitation bubbles and their interaction with cells. Adv Drug Deliver Rev. 60, 1103–1116 (2008).
Miller, D. L. Overview of experimental studies of biological effects of medical ultrasound caused by gas body activation and inertial cavitation. Prog Biophys Mol Bio. 93, 314–330 (2007).
Dayton, P. A., Morgan, K. E., Klibanov, A. L. S., Brandenburger, G., Nightingale, K. R. & Ferrara, K. W. A preliminary evaluation of the effects of primary and secondary radiation forces on acoustic contrast agents. IEEE Trans Ultrason Ferroelectr Freq Control. 44, 1264–1277 (1997).
Dayton, P. A., Morgan, K. E., Klibanov, A. L., Brandenburger, G. H. & Ferrara, K. W. Optical and acoustical observations of the effects of ultrasound on contrast agents. IEEE Trans Ultrason Ferroelectr Freq Control. 46, 220–232 (1999).
Zhou, Y., Yang, K., Cui, J., Ye, J. Y. & Deng, C. X. Controlled permeation of cell membrane by single bubble acoustic cavitation. J Control Release. 157, 103–111 (2012).
Kitzman, D. W., Goldman, M. E., Gillam, L. D., Cohen, J. L., Aurigemma, G. P. & Gottdiener, J. S. Efficacy and safety of the novel ultrasound contrast agent perflutren (definity) in patients with suboptimal baseline left ventricular echocardiographic images. Am J Cardiol. 86, 669–674 (2000).
Wei, K., Mulvagh, S. L., Carson, L., Davidoff, R., Gabriel, R., Grimm, R. A. et al. The safety of definity and optison for ultrasound image enhancement: A retrospective analysis of 78,383 administered contrast doses. J Am Soc Echocardiogr. 21, 1202–1206 (2008).
Ellegala, D. B., Leong-Poi, H., Carpenter, J. E., Klibanov, A. L., Kaul, S., Shaffrey, M. E. et al. Imaging tumor angiogenesis with contrast ultrasound and microbubbles targeted to alpha(v)beta3. Circulation. 108, 336–341 (2003).
Takalkar, A. M., Klibanov, A. L., Rychak, J. J., Lindner, J. R. & Ley, K. Binding and detachment dynamics of microbubbles targeted to p-selectin under controlled shear flow. J Control Release. 96, 473–482 (2004).
Klibanov, A. L. Preparation of targeted microbubbles: Ultrasound contrast agents for molecular imaging. Med Biol Eng Comput. 47, 875–882 (2009).
Kaufmann, B. A., Sanders, J. M., Davis, C., Xie, A., Aldred, P., Sarembock, I. J. et al. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 116, 276–284 (2007).
Winter, P. M., Morawski, A. M., Caruthers, S. D., Fuhrhop, R. W., Zhang, H., Williams, T. A. et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with alpha(v)beta3-integrin-targeted nanoparticles. Circulation. 108, 2270–2274 (2003).
Leong-Poi, H. Molecular imaging using contrast-enhanced ultrasound: Evaluation of angiogenesis and cell therapy. Cardiovasc Res. 84, 190–200 (2009).
Anderson, C. R., Hu, X., Zhang, H., Tlaxca, J., Decleves, A. E., Houghtaling, R. et al. Ultrasound molecular imaging of tumor angiogenesis with an integrin targeted microbubble contrast agent. Invest Radiol. 46, 215–224 (2011).
Fan, Z., Liu, H., Mayer, M. & Deng, C. X. Spatiotemporally controlled single cell sonoporation. Proc Natl Acad Sci U S A. 109, 16486–16491 (2012).
Kooiman, K., Foppen-Harteveld, M., van der Steen, A. F. W. & de Jong, N. Sonoporation of endothelial cells by vibrating targeted microbubbles. J Control Release. 154, 35–41 (2011).
van Wamel, A., Kooiman, K., Harteveld, M., Emmer, M., ten Cate, F. J., Versluis, M. et al. Vibrating microbubbles poking individual cells: Drug transfer into cells via sonoporation. J Control Release. 112, 149–155 (2006).
Marmottant, P. & Hilgenfeldt, S. Controlled vesicle deformation and lysis by single oscillating bubbles. Nature. 423, 153 (2003).
Prentice, P., Cuschieri, A., Dholakia, K., Prausnitz, M. & Campbell, P. Membrane disruption by optically controlled microbubble cavitation. Nat Phys. 1, 107 (2005).
Postema, M., van Wamel, A., Lancee, C. T. & de Jong, N. Ultrasound-induced encapsulated microbubble phenomena. Ultrasound Med Biol. 30, 827–840 (2004).
Fu, J. P., Wang, Y. K., Yang, M. T., Desai, R. A., Yu, X. A., Liu, Z. J. et al. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Meth. 7, 733–736 (2010).
Ingber, D. E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 20, 811–827 (2006).
Riento, K. & Ridley, A. J. Rocks: Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 4, 446–456 (2003).
Rooney, J. A. Shear as a mechanism for sonically induced biological effects. J Acoust Soc Am. 52, 1718–1724 (1972).
Wu, J. Shear stress in cells generated by ultrasound. Prog Biophys Mol Bio. 93, 363–373 (2007).
Marmottant, P. & Hilgenfeldt, S. Controlled vesicle deformation and lysis by single oscillating bubbles. Nature. 423, 153–156 (2003).
Ji, J. Y., Jing, H. & Diamond, S. L. Shear stress causes nuclear localization of endothelial glucocorticoid receptor and expression from the gre promoter. Circ Res. 92, 279–285 (2003).
Gramiak, R. & Shah, P. M. Echocardiography of the aortic root. Invest Radiol. 3, 356–366 (1968).
Goldberg, B. B., Liu, J.-B. & Forsberg, F. Ultrasound contrast agents: A review. Ultrasound Med Biol. 20, 319–333 (1994).
Kaul, S. Myocardial contrast echocardiography: A 25-year retrospective. Circulation. 118, 291–308 (2008).
Lentacker, I., De Smedt, S. C., Demeester, J. & Sanders, N. N. Microbubbles which bind and protect DNA against nucleases. J Control Release. 116, e73–75 (2006).
Lum, A. F., Borden, M. A., Dayton, P. A., Kruse, D. E., Simon, S. I. & Ferrara, K. W. Ultrasound radiation force enables targeted deposition of model drug carriers loaded on microbubbles. J Control Release. 111, 128–134 (2006).
Frampton, J. P., Fan, Z., Simon, A., Chen, D., Deng, C. X. & Takayama, S. Aqueous two-phase system patterning of microbubbles: Localized induction of apoptosis in sonoporated cells. Adv Fun Mater In press (2013).
Yang, M. T., Fu, J., Wang, Y.-K., Desai, R. A. & Chen, C. S. Assaying stem cell mechanobiology on microfabricated elastomeric substrates with geometrically modulated rigidity. Nat Protoc. 6, 187–213 (2011).
Erpelding, T. N., Hollman, K. W. & O'Donnell, M. Bubble-based acoustic radiation force using chirp insonation to reduce standing wave effects. Ultrasound Med Biol. 33, 263–269 (2007).
Dayton, P. A., Morgan, K. E., Klibanov, A. L., Brandenburger, G., Nightingale, K. R. & Ferrara, K. W. A preliminary evaluation of the effects of primary and secondary radiation forces on acoustic contrast agents. IEEE Trans Ultrason Ferroelectr Freq Control. 44, 1264–1277 (1997).
Gormley, G. & Wu, J. R. Observation of acoustic streaming near albunex (r) spheres. J Acoust Soc Am. 104, 3115–3118 (1998).
Marmottant, P. & Hilgenfeldt, S. A bubble-driven microfluidic transport element for bioengineering. Proc Natl Acad Sci USA. 101, 9523–9527 (2004).
Fan, Z., Kumon, R. E., Park, J. & Deng, C. X. Intracellular delivery and calcium transients generated in sonoporation facilitated by microbubbles. J Control Release. 142, 31–39 (2010).
Acknowledgements
This work is supported in part by the National Science Foundation (CMMI 1129611 and CBET 1149401 to J. F.), the American Heart Association (Scientist Development Grant, 12SDG12180025 to J. F.), the National Institutes of Health (CA116592 to C. X. D.), departments of Mechanical Engineering and Biomedical Engineering of the University of Michigan. The Lurie Nanofabrication Facility at the University of Michigan, a member of the National Nanotechnology Infrastructure Network (NNIN) funded by the National Science Foundation, is acknowledged for support in microfabrication.
Author information
Authors and Affiliations
Contributions
C.X.D. and J.F. developed initial concept. Z.F., Y.S., D.C., C.X.D. and J.F. designed experiments. Z.F., Y.S., D.C. and W.C. performed experiments and analyzed data. D.T. analyzed data. Z.F., Y.S., D.C., C.X.D. and J.F. co-wrote the manuscript. C.X.D. and J.F. supervised the study. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
upplementary Information
Supplementary Information
Supplementary Video
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Fan, Z., Sun, Y., Di Chen et al. Acoustic tweezing cytometry for live-cell subcellular modulation of intracellular cytoskeleton contractility. Sci Rep 3, 2176 (2013). https://doi.org/10.1038/srep02176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02176
This article is cited by
-
Trends in mechanobiology guided tissue engineering and tools to study cell-substrate interactions: a brief review
Biomaterials Research (2023)
-
Rapid responses of human pluripotent stem cells to cyclic mechanical strains applied to integrin by acoustic tweezing cytometry
Scientific Reports (2023)
-
Microskeletal stiffness promotes aortic aneurysm by sustaining pathological vascular smooth muscle cell mechanosensation via Piezo1
Nature Communications (2022)
-
Acoustic tweezing cytometry for mechanical phenotyping of macrophages and mechanopharmaceutical cytotripsy
Scientific Reports (2019)
-
Probing Single-Cell Mechanical Allostasis Using Ultrasound Tweezers
Cellular and Molecular Bioengineering (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
