
Abstract
A hybrid atomic force microscopy (AFM)-optical fluorescence microscopy is a powerful tool for investigating cellular morphologies and events. However, the slow data acquisition rates of the conventional AFM unit of the hybrid system limit the visualization of structural changes during cellular events. Therefore, high-speed AFM units equipped with an optical/fluorescence detection device have been a long-standing wish. Here we describe the implementation of high-speed AFM coupled with an optical fluorescence microscope. This was accomplished by developing a tip-scanning system, instead of a sample-scanning system, which operates on an inverted optical microscope. This novel device enabled the acquisition of high-speed AFM images of morphological changes in individual cells. Using this instrument, we conducted structural studies of living HeLa and 3T3 fibroblast cell surfaces. The improved time resolution allowed us to image dynamic cellular events.
Similar content being viewed by others

Introduction
Since its invention in 1986 by Binnig1 and introduction to the field of biological sciences in 1988 by Hansma2, atomic force microscopy (AFM) has evolved into several multi-functional tools for biological research and has become indispensable in the post-genomic era. The operational range of AFM imaging is well suited for characterizing structures at the molecular and cellular levels. The sensitivity of the interaction force measurement between biomolecules provides a piconewton level of detection. Furthermore, the ability to function in aqueous environments has made AFM ideal for investigating biological samples such as proteins, nucleic acids and even living cells, under physiological conditions3,4,5,6.
The combination of optical and atomic force microscopy has also accelerated the detailed characterization of cellular structures and processes7,8. High-resolution AFM images have been acquired during simultaneous monitoring of either a fluorescent image of labeled cellular components or a high-contrast optical image. By applying these complementary techniques, correlations between the structure and function of living organisms can be obtained9,10. Although the combined system has the potential to resolve structures that fluoresce, the slow data acquisition rates of a conventional AFM system (several seconds to minutes per frame) limit the visualization of the structural changes of biological macromolecules.
Ando's successful development of high-speed AFM was a breakthrough in efforts to increase the data acquisition rate, achieving a rate of >10 frames per second (fps) for a scan area of 240 × 240 nm2 with 100 × 100 pixels11. Various applications of this device have quickly appeared in the last decade12,13. Conformational changes and dynamics of proteins14,15,16,17,18,19,20,21,22, reaction processes of DNA targeting enzymes23,24,25,26,27,28, nucleosome dynamics29,30 and local conformational changes of DNA strands31,32 have been addressed by the new high-speed AFM. It is noteworthy that presently, both the ‘sub-second time frame’ and ‘nanometer scale’ single molecule observations of functional biological macromolecules cannot be achieved by other techniques.
Currently available high-speed AFM units are based on a sample-scan system, in which the sample is mounted on a piezo scanner that can move the sample in the z-direction for maintaining a constant force and the x-y directions for scanning the sample. One drawback of the high-speed AFM system is its small sample stage (Φ ~ 2 mm × 3 mm at most), which is a prerequisite for a high resonant frequency of the scanner. This limits the size of the sample that can be mounted on the stage. For example, a microscope slide or a cover slip on which cells are cultured cannot be placed on such a small stage. To further develop the technique, we have built a novel tip-scan type of high-speed AFM unit that can be set up on the sample stage of an inverted optical microscope. With the help of the optical microscope, the AFM cantilever can be positioned on a specific area of cell surfaces. In this article, we describe the basic configuration of this newly constructed device and its application to the imaging of cellular events.
Results
Biologically applicable high-speed AFM requires the following:
-
i
Small cantilevers (10 μm long, 2 μm wide and <0.2 μm thick) with fc > 300 kHz in water, k < 0.2 N/m and Q ~ 2 in water.
-
ii
XYZ scanner unit whose resonant frequencies are higher than 10 kHz for the x direction (fast-scanning direction) and 100 kHz for the z direction.
-
iii
Optical beam deflection (OBD) sensor that is capable of detecting the movement of the laser beam reflected back from the cantilever surface, with a spot size of 2–4 μm in diameter.
The sample-scan type high-speed AFM is now well established by Ando's group and commercially available. However, the development of a tip-scan type of high-speed AFM has been difficult. In tip-scan AFM, the scanning motion of the cantilever has to be directly regulated by the scanner and, simultaneously, the laser beam has to remain focused on the back of the cantilever during the x-y scanning motion of the tip. Thus, development of a high-speed tip-scan system requires direct linkage of the OBD sensor and scanner, which satisfies the above criteria ii and iii. Development and current performance of key devices are described below.
Cantilevers and probes
The resonant frequency fc and the spring constant k of cantilevers are in general given by


where E, ρ, l, w and t are the Young's modulus, density, length, width and thickness of the cantilever, respectively. For quick scanning, a high fc, normally higher than 1 MHz, is required. To achieve this, a large t and a small l are preferred. On the other hand, to minimize damage to a sample during imaging, a cantilever with softer mechanical properties, or a small k, preferably less than 0.2 N/m, is required. As seen in equations (1) and (2), those requirements have a trade-off relationship. To practically fulfill both requirements, we employed a small silicon nitride (Si3N4) cantilever having dimensions of 9 × 2 × 0.1 (l × w × t) μm3 (BL-AC10DS, Olympus, Tokyo, Japan). The cantilever had a spring constant (k) of 0.1 N/m with a resonant frequency (fc) of 1.5 MHz in air and 300–600 kHz in water and a quality factor Q ~ 2 in water. The cantilever had a bird beak-like tip at its free end and carbon nanofiber can be grown thereon as an option (BL-AC10FS, Olympus, Tokyo, Japan)33,34,35. Although the lifetime of a tip depends strongly on the sample to be imaged, from our experience, approximately 100–200 continuous images of a cell membrane can be obtained at a scanning rate of 0.1–0.2 fps with the same tip.
High-speed scanner
The scanner we developed for high-speed tip-scan AFM comprises multi-layered piezoelectric bodies. The XYZ-scanner assembly is shown in Fig. 1. The Y-scanner is actuated by a single piezo, while the X-scanner consists of two piezos and actuated by their push-pull motion (the reason for this is described below) (Fig. 1a). These two X-piezos and the Y-piezo are connected to a movable table through flexures. The flexures are flexible enough to be displaced but rigid in the directions perpendicular to the displacement axis.

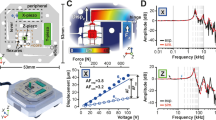
Assembly of the XYZ-scanner.
(a) The top view when viewed at the scanner from +z to −z. (b) The side view when viewed from +x to −x.
The Z-scanner (Fig. 1b) mounted on the movable table comprises two Z-piezo actuators placed in opposition to one another. A cantilever holder is attached to one of the Z-piezos while the other end has a dummy holder for balance. The cantilever is held with a tilt angle of 12 degrees from the x-y plane. The cantilever is elongated in the direction parallel to the y-axis (slow-scan axis). The objective lens for the OBD sensor is mounted on the movable table to focus the laser beam onto the back of the cantilever. A lightweight single convex lens with Φ = 4.5 mm was used to avoid lowering the resonant frequency too much. We achieved an OBD detection sensitivity of 200 mV/nm using the above-mentioned small cantilever. The deflection noise density at approximately the resonant frequency was less than 210 fm/(Hz)1/2 in water with a laser power of 0.8 mW (Supplementary Fig. S1 online).
The constructed XYZ-scanner has an asymmetric configuration about the x-axis while the objective lens and Z-scanner are placed along the y-axis. The quick displacement of the movable table along the x-axis therefore causes mechanical vibrations. To minimize these vibrations, a push-pull motion system was applied to the x-axis because the system can decrease the stress on the movable table. On the other hand, the movable table has a symmetric configuration about the y-axis. Furthermore, in our system, the scanning rate along the y-axis (slow-scan axis) is approximately 100–200 times slower than that along the x-axis (fast-scan axis). Thus, we assumed that the y-component of the stress loaded on the movable table is negligible. For this reason, as for the Y-scanner, we omitted the push-pull system and prioritized the minimization and simplification of the head unit by employing a single piezo system. The signals fed into the X- and Y-scanners are shown in Supplementary Fig. S2 online. The maximum scan size of the XYZ-scanner is 4.48 × 3.36 × 0.60 (X × Y × Z) μm3. The resonant frequencies of the X-, Y- and Z-scanners are 15 kHz, 15 kHz and 150 kHz, respectively. Although this scanner achieved a scan rate of up to 10 fps under fluid, we set a scan rate of 0.1–0.2 fps in this report to obtain acceptable images for relatively large areas of cell surfaces.
Deflection detection system
Our high-speed tip-scan AFM unit contains an OBD sensor (Fig. 2a). On the small cantilever, a 640 nm laser beam is focused passing through a collimator lens, polarized beam splitter, quarter-wave plate, condenser lens and transparent glass. The light is reflected back through the opposite pathway and reflected onto the split photodiode. We used an objective lens with N.A. 0.4 to make the spot size 2–4 μm in diameter.

(a) Schematic layout of a high-speed AFM head unit. (b) Block diagram for the feedback loop. (c, d) Overall view of a high-speed AFM head unit mounted on a standard inverted optical microscope (OLYMPUS IX71, Olympus, Tokyo, Japan). (e) Bottom view of the head unit.
The laser focusing position relative to the cantilever is monitored by the optical microscope and aligned by moving the position of the laser diode against the collimator lens (Supplementary Fig. S3 online) so that the intensity of light reflected back from the cantilever is maximized.
Feedback loop
The tip-sample interactions are detected in amplitude modulation mode (AM-mode) and regulated by a conventional proportional-integral derivative (PID) controller (Fig. 2b). The deflection signal is sent to a RMS-DC (Root Mean Squared value-a Direct Current) and converted to an amplitude signal. The detected cantilever oscillation amplitude is compared with a set point amplitude. The difference between them (error signal) is input to the PID controller, whose output is then fed to a Z-piezo driver. The Z-scanner is finally moved in the z-direction by the output of the Z-piezo driver to make the error signal zero.
Optical imaging system
The AFM unit is designed to be set up on the sample stage of an inverted optical microscope (OLYMPUS IX71, Olympus, Tokyo, Japan) as shown in Fig. 2. The AFM unit can be operated without affecting the optical imaging of the target sample. A white LED oriented with a tilt angle of 12 degrees from the x-y plane was mounted on the bottom surface of the AFM unit for viewing the cantilever (Fig. 2a and 2e). The location of the cantilever can be observed with this LED illumination, which also helps us to position the cantilever onto a target sample. Fluorescent imaging can be achieved by properly filtering the excitation light. HeLa cells stably expressing GFP-fused protein were monitored using this system. The location of the cantilever can be determined more easily and precisely by combining the white LED and filtered excitation light (Fig. 3).

Positioning of cantilever on targeted cell.
(a) Fluorescence image of live HeLa cells stably expressing GFP-actinin-4. (b) Position of cantilever (arrow) observed by white LED on the same area as (a). (c) The relative position of the cantilever to the fluorescently labeled cell can be easily and precisely determined by combining the white LED and filtered excitation light. Scale bars: 10 μm.
Cantilever engagement
Approach and release of the cantilever are performed by actuating the approach motor (Fig. 2a). Before starting the approach, the scanner unit is released at a sufficient distance from the sample stage to avoid any impact. Usually, the scanner unit is released until the cantilever reaches 100–200 μm above the sample surface. The focus of the optical microscope is first set at approximately 10 μm above the sample surface. Then, a coarse approach is performed at an approach velocity of up to 3000 nm/s while carefully watching the relative position of the cantilever and the sample surface on the optical microscope display. Once the shape of the cantilever is clearly seen on the display (at this moment, the cantilever is approximately 10 μm above the sample surface), the approach is stopped (Supplementary Fig. S4a and S4b online). This coarse approach takes approximately 30–60 s. The cantilever is then excited by applying the sinusoidal AC voltage to start the scanning. Subsequently, a slow approach is executed at an approach velocity of 100–200 nm/s until the probe contacts the sample and the AFM image becomes visible (Supplementary Fig. S4c–e online). This process takes an additional 30–60 s.
High-speed imaging of living cell edge protrusion
The coordinated polymerization of actin filaments at the periphery of the cell regulates the dynamics of the cell membrane and organizes numerous cellular events such as cell migration, morphogenesis, endocytosis and phagocytosis. Although many fluorescence approaches have been employed to investigate such events in living cells, observing the dynamics of cell morphology with sufficient time and spatial resolution has been difficult. We aimed to capture the dynamic behavior of the living HeLa cell surface with our combined imaging system.
The location of the cantilever was determined by optical microscopy over the cell and a series of images were recorded for approximately 4 min at a scanning rate of 0.2 fps (Fig. 4a, See also Supplementary Movie S1 online). The images showed protrusions of filamentous structures at the edge of HeLa cells. The elongation event was quantified by plotting the tip of the protrusions at each time point (Fig. 4b and 4c). The protrusion generated at 20 s migrated at rates between 16 and 112 nm per 5 s until 110 s. Intriguingly, accelerated elongation was seen from 110 s and the rate at 130 s was estimated to be 272 nm per 5 s (Fig. 4c). This membrane protrusion is likely operated by filopodial extensions, considering previous reports describing the morphology and growth rate of filopodia36,37, which were similar to our observations.

Dynamics of living HeLa cell surface revealed by high-speed AFM.
(a) High-speed AFM images showed dynamic features of the cell surface reflecting the motion of filopodia. Images were recorded at a scan rate of 0.2 fps. Image size: 4480 nm × 3360 nm. Scale bar: 1 μm. For the complete movie, see Supplementary Movie S1 online. (b) Trajectory of the tip of the protrusion (yellow arrowhead in the image at 20 s) in 23 sequential images (20–130 s) was overlaid on the sepia colored image at 130 s (blue line). Dashed gray lines represent the shape of the cell at 20 s. (c) Changes of the elongation speed of the protrusion (red) and the cumulative migration distance (blue). The elongation speed at each time point was determined by measuring the distance between the tip of the protrusion in the current frame and that in the previous frame.
Direct observation of membrane surface dynamics
Exocytosis is the process by which a cell releases the vesicle content upon the fusion of secretory vesicles with the plasma membrane, which causes a morphological change in the cell surface. AFM imaging studies focusing on such exocytic processes have revealed depression-like structures that are related to the formation of fusion pores in pancreatic acinar cells38,39 and chromaffin cells40. The exocytic release or endocytic uptake of macromolecular complexes occurs on a time scale of seconds to minutes38,41. Therefore, exo/endo-cytosis-related morphological changes within such a time scale have remained unattainable by conventional AFM.
We applied our high-speed AFM to capture morphological events in NIH-3T3 fibroblast cells. Figure 5 shows a series of images of a 3T3 cell surface obtained by live scanning in culture medium (see also Supplementary Movie S2 online). Network structures were observed throughout the scanning, possibly reflecting cytoskeletal organization beneath the cell membrane. In addition, several areas of the surface formed transient invaginations (70 s in Fig. 5a), which may correspond to the vesicle fusion process of exocytosis. The diameter of the invaginations obtained by six individual observations ranged from 69 to 192 nm and the mean size was 104 ± 33 nm. This size was comparable to previously reported structures on the surface of chromaffin cells39 approximately 120 nm in size and those on pancreatic β-cells38 100–180 nm in diameter. Intriguingly, immediately after this invagination step, the swelling structures were observed at the same area, probably indicating that the secretion step occurred (Fig. 5b). After this series of events, the membrane recovered to the original flat shape. The entire process occurred within 140 s, including apparent invagination for ~40 s and the following swelling process for ~30 s. Statistical analysis of six individual events revealed that the time scale of the event ranged between 30 and 140 s (mean 75 ± 43 s), which agrees well with the previously reported time scale38,41.

Dynamics of living fibroblast cell surface revealed by high-speed AFM.
(a) AFM images of the 3T3 cell surface reflecting the morphological events. In addition to the events highlighted by arrowheads, membrane dynamics such as network structures are also captured. Scanning rate: 0.1 fps. Scale bar: 1 μm. For the complete movie, see Supplementary Movie S2 online. (b) Continuous surface plotting images of area within dashed rectangles shown in (a). Scale bar: 0.5 μm. Transition from depression to swelling of the cell surface can be seen as simultaneous three-dimensional AFM images.
Discussion
AFM has been applied to image many kinds of cells for morphological investigations as well as electrophysiological and biochemical studies. However, observing plasma membrane dynamics in living cells by conventional AFM has been difficult primarily due to the limited time resolution. Therefore, a single line scan method, in which an AFM probe repeatedly scans a single line to record z-height changes as a function of time, has been applied to capture the height/volume changes of a cell with sufficient time resolution42,43. Although this method is capable of capturing fast movements of single molecule dynamics, it lacks three-dimensional morphological information. The tip-scan type of high-speed AFM introduced here has opened a new phase of nano/mesoscale analysis of living cell surfaces. The topography and the dynamic features of live cell surfaces such as filopodia elongation and exo/endo-cytosis related events were successfully imaged in physiologically relevant buffer conditions. By combining an inverted optical microscope with our high-speed AFM system, the position of the cantilever over the sample surface can be monitored and optical and high-speed AFM images of the focused areas of interest can be overlaid. Thus, we believe that the present system will offer new ways of addressing novel aspects of cellular events, such as membrane heterogeneity, spatial organization, conformational changes and dynamics of membrane-embedded proteins on living cell surfaces. It is noteworthy that the high-speed AFM unit and fluorescent microscopy system can work independently. Thus, both microscopy systems can be easily modified to accommodate additional techniques. One of the fascinating directions of further development is the combination of the high-speed tip-scan system with FluidFM44 by which one could use the tip to stimulate selected cell domains with soluble agents and image their response. Fluorescent-based techniques, such as fluorescence recovery after photo-bleaching (FRAP) and Förster resonance energy transfer (FRET) could be implemented. Multi-color fluorescence imaging also will be possible by adding appropriate light sources and optical filters.
Current innovations in optical microscopy have shown dramatically how the conventional diffraction limit has been overcome45,46,47 with the emergence of various “single-molecule fluorescence” and “super-resolution” techniques48,49,50,51. Surface and interfacial events such as cell adhesion, exo- and endocytosis are now explored at single-molecule resolution52,53. The translocation or rotational motion of a fluorescent spot emitted from a fluorophore attached to the molecules of interest can also be analyzed54,55. Considering that the observed behavior of the fluorescent spot does not always provide direct information on how the labeled molecule actually behaves, the next advancement of the technology will perhaps come from the integration of high-speed AFM and single-molecule fluorescence microscopy. It is hoped that both the structural changes in individual biomolecules and their nano/mesoscale dynamics will be directly correlated.
Methods
Cell culture
HeLa/GFP cells (Cell Biolabs, Inc., San Diego, CA), HeLa S3 cells and NIH-3T3 cells were grown on a poly-L-lysine (PLL)-coated slide glass in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. An expression vector for GFP-actinin-4 was constructed as previously described56. The stably expressing cell line was obtained by screening the transfected cells with G418 (Nacalai Tesque, Tokyo, Japan) and isolating the drug-resistant single-cloned cells.
High-speed AFM/fluorescence images
AFM imaging of the cells was performed in the culture medium. High-speed AFM images were acquired at a scan rate of 0.1–0.2 fps. Each frame was exported to the bmp file (576 × 432 pixels) and analyzed by ImageJ (http://rsbweb.nih.gov/ij/) software. Fluorescence images were acquired at an excitation wavelength of 480 nm and an emission wavelength of 530 nm with a 60× UPlan FLN objective lens with N.A. 0.7 (Olympus, Tokyo, Japan).
References
Binnig, G., Quate, C. F. & Gerber, C. Atomic force microscope. Phys Rev Lett 56, 930–933 (1986).
Hansma, P. K., Elings, V. B., Marti, O. & Bracker, C. E. Scanning tunneling microscopy and atomic force microscopy: application to biology and technology. Science 242, 209–216 (1988).
Oberleithner, H., Brinckmann, E., Giebisch, G. & Geibel, J. Visualizing life on biomembranes by atomic force microscopy. Kidney Int 48, 923–929 (1995).
Leuba, S. H. & Bustamante, C. Analysis of chromatin by scanning force microscopy. Methods Mol Biol 119, 143–160 (1999).
Henderson, R. M. & Oberleithner, H. Pushing, pulling, dragging and vibrating renal epithelia by using atomic force microscopy. Am J Physiol Renal Physiol 278, F689–701 (2000).
Muller, D. J. & Dufrene, Y. F. Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat Nanotechnol 3, 261–269 (2008).
Pietrasanta, L. I., Schaper, A. & Jovin, T. M. Imaging subcellular structures of rat mammary carcinoma cells by scanning force microscopy. J Cell Sci 107(Pt 9), 2427–2437 (1994).
Tamura, K., Mizutani, T., Haga, H. & Kawabata, K. Nano-mechanical properties of living cells expressing constitutively active RhoA effectors. Biochem Biophys Res Commun 403, 363–367 (2010).
Henderson, E., Haydon, P. G. & Sakaguchi, D. S. Actin filament dynamics in living glial cells imaged by atomic force microscopy. Science 257, 1944–1946 (1992).
Rotsch, C. & Radmacher, M. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J 78, 520–535 (2000).
Ando, T. et al. A high-speed atomic force microscope for studying biological macromolecules. Proc Natl Acad Sci U S A 98, 12468–12472 (2001).
Ando, T. et al. High-speed AFM and nano-visualization of biomolecular processes. Pflug Arch Eur J Phy 456, 211–225 (2008).
Uchihashi, T. & Ando, T. High-speed atomic force microscopy and biomolecular processes. Methods Mol Biol 736, 285–300 (2011).
Yokokawa, M. et al. Fast-scanning atomic force microscopy reveals the ATP/ADP-dependent conformational changes of GroEL. EMBO J 25, 4567–4576 (2006).
Miyagi, A. et al. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. Chemphyschem 9, 1859–1866 (2008).
Shinozaki, Y. et al. Direct Observation of ATP-Induced Conformational Changes in Single P2X4 Receptors. PLoS Biol 7, e103 (2009).
Kodera, N., Yamamoto, D., Ishikawa, R. & Ando, T. Video imaging of walking myosin V by high-speed atomic force microscopy. Nature 468, 72–76 (2010).
Uchihashi, T., Iino, R., Ando, T. & Noji, H. High-speed atomic force microscopy reveals rotary catalysis of rotorless F1-ATPase. Science 333, 755–758 (2011).
Yokokawa, M. & Takeyasu, K. Motion of the Ca2+-pump captured. FEBS J 278, 3025–3031 (2011).
Igarashi, K. et al. Traffic jams reduce hydrolytic efficiency of cellulase on cellulose surface. Science 333, 1279–1282 (2011).
Colom, A., Casuso, I., Boudier, T. & Scheuring, S. High-speed atomic force microscopy: cooperative adhesion and dynamic equilibrium of junctional microdomain membrane proteins. J Mol Biol 423, 249–256 (2012).
Casuso, I. et al. Characterization of the motion of membrane proteins using high-speed atomic force microscopy. Nature Nanotechnology 7, 525–529 (2012).
Yokokawa, M. et al. Fast-scanning atomic force microscopy reveals the molecular mechanism of DNA cleavage by ApaI endonuclease. IEE Proc Nanobiotechnol 153, 60–66 (2006).
Crampton, N. et al. Fast-scan atomic force microscopy reveals that the type III restriction enzyme EcoP15I is capable of DNA translocation and looping. Proc Natl Acad Sci U S A 104, 12755–12760 (2007).
Gilmore, J. L. et al. Single-molecule dynamics of the DNA-EcoRII protein complexes revealed with high-speed atomic force microscopy. Biochemistry 48, 10492–10498 (2009).
Endo, M., Katsuda, Y., Hidaka, K. & Sugiyama, H. Regulation of DNA Methylation Using Different Tensions of Double Strands Constructed in a Defined DNA Nanostructure. Journal of the American Chemical Society 132, 1592–1597 (2010).
Endo, M., Katsuda, Y., Hidaka, K. & Sugiyama, H. A versatile DNA nanochip for direct analysis of DNA base-excision repair. Angew Chem Int Ed Engl 49, 9412–9416 (2010).
Suzuki, Y. et al. Visual analysis of concerted cleavage by type IIF restriction enzyme SfiI in subsecond time region. Biophys J 101, 2992–2998 (2011).
Suzuki, Y. et al. Molecular dynamics of DNA and nucleosomes in solution studied by fast-scanning atomic force microscopy. Ultramicroscopy 110, 682–688 (2010).
Miyagi, A., Ando, T. & Lyubchenko, Y. L. Dynamics of nucleosomes assessed with time-lapse high-speed atomic force microscopy. Biochemistry 50, 7901–7908 (2011).
Sannohe, Y., Endo, M., Katsuda, Y., Hidaka, K. & Sugiyama, H. Visualization of Dynamic Conformational Switching of the G-Quadruplex in a DNA Nanostructure. Journal of the American Chemical Society 132, 16311–16313 (2010).
Endo, M., Yang, Y. Y., Suzuki, Y., Hidaka, K. & Sugiyama, H. Single-Molecule Visualization of the Hybridization and Dissociation of Photoresponsive Oligonucleotides and Their Reversible Switching Behavior in a DNA Nanostructure. Angewandte Chemie-International Edition 51, 10518–10522 (2012).
Tanemura, M. et al. Direct growth of single carbon nanofiber onto tip of scanning probe microscopy induced by ion irradiation. Japanese Journal of Applied Physics Part 1-Regular Papers Brief Communications & Review Papers 45, 2004–2008 (2006).
Kitazawa, M., Ohta, R., Sugita, Y., Inaba, K. & Tanemura, M. Wafer-scale production of carbon nanofiber probes. Journal of Vacuum Science & Technology B 27, 975–979 (2009).
Kitazawa, M. et al. High-Resolution Imaging of Plasmid DNA in Liquids in Dynamic Mode Atomic Force Microscopy Using a Carbon Nanofiber Tip. Japanese Journal of Applied Physics 50 (2011).
Mitchison, T. J. & Cramer, L. P. Actin-based cell motility and cell locomotion. Cell 84, 371–379 (1996).
Katoh, K., Hammar, K., Smith, P. J. & Oldenbourg, R. Birefringence imaging directly reveals architectural dynamics of filamentous actin in living growth cones. Molecular biology of the cell 10, 197–210 (1999).
Schneider, S. W., Sritharan, K. C., Geibel, J. P., Oberleithner, H. & Jena, B. P. Surface dynamics in living acinar cells imaged by atomic force microscopy: identification of plasma membrane structures involved in exocytosis. Proc Natl Acad Sci U S A 94, 316–321 (1997).
Cho, S. J., Wakade, A., Pappas, G. D. & Jena, B. P. New structure involved in transient membrane fusion and exocytosis. Annals of the New York Academy of Sciences 971, 254–256 (2002).
Tsai, C. C. et al. Exocytosis of a single bovine adrenal chromaffin cell: the electrical and morphological studies. The journal of physical chemistry. B 112, 9165–9173 (2008).
Klingauf, J., Kavalali, E. T. & Tsien, R. W. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature 394, 581–585 (1998).
Oberleithner, H. et al. Endothelial cell swelling by aldosterone. The Journal of membrane biology 196, 163–172 (2003).
Schneider, S. W. et al. Volume dynamics in migrating epithelial cells measured with atomic force microscopy. Pflugers Archiv : European journal of physiology 439, 297–303 (2000).
Meister, A. et al. FluidFM: combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett 9, 2501–2507 (2009).
Gustafsson, M. G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 198, 82–87 (2000).
Weiss, S. Shattering the diffraction limit of light: a revolution in fluorescence microscopy? Proc Natl Acad Sci U S A 97, 8747–8749 (2000).
Gustafsson, M. G. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A 102, 13081–13086 (2005).
Yildiz, A. & Selvin, P. R. Fluorescence imaging with one nanometer accuracy: application to molecular motors. Acc Chem Res 38, 574–582 (2005).
Toprak, E. et al. Defocused orientation and position imaging (DOPI) of myosin V. Proc Natl Acad Sci U S A 103, 6495–6499 (2006).
Park, H., Toprak, E. & Selvin, P. R. Single-molecule fluorescence to study molecular motors. Q Rev Biophys 40, 87–111 (2007).
Shroff, H. et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A 104, 20308–20313 (2007).
Betzig, E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006).
Manley, S. et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods 5, 155–157 (2008).
Sako, Y. & Yanagida, T. Single-molecule visualization in cell biology. Nat Rev Mol Cell Biol Suppl, SS1–5 (2003).
Sako, Y. Imaging single molecules in living cells for systems biology. Mol Syst Biol 2, 56 (2006).
Kumeta, M., Yoshimura, S. H., Harata, M. & Takeyasu, K. Molecular mechanisms underlying nucleocytoplasmic shuttling of actinin-4. J Cell Sci 123, 1020–1030 (2010).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Virus-Host Cell Competency” (24115003) of The Ministry of Education, Culture, Sports, Science and Technology, Japan and by a Human Frontier Science Program (K.T.). The authors thank Drs. James Hejna and Masahiro Kumeta for reading the manuscript.
Author information
Authors and Affiliations
Contributions
Y.S., N.S. and K.T. conceived and designed the experiments on living cells. N.S., Y.U., A.Y. (Akira Yagi), Y.I., S.I. and K.K. developed the high-speed AFM instrument. Y.S., N.S. and A.Y. (Aiko Yoshida) performed experiments. Y.S. and A.Y. (Aiko Yoshida) analyzed the data. Y.S. and K.T. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Movie S1
Supplementary Information
Supplementary Movie S2.
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Suzuki, Y., Sakai, N., Yoshida, A. et al. High-speed atomic force microscopy combined with inverted optical microscopy for studying cellular events. Sci Rep 3, 2131 (2013). https://doi.org/10.1038/srep02131
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02131
This article is cited by
-
Chemo-mechanical forces modulate the topology dynamics of mesoscale DNA assemblies
Nature Communications (2023)
-
Hybridization of papain molecules and DNA-wrapped single-walled carbon nanotubes evaluated by atomic force microscopy in fluids
Scientific Reports (2023)
-
Technical advances in high-speed atomic force microscopy
Biophysical Reviews (2023)
-
An ultra-wide scanner for large-area high-speed atomic force microscopy with megapixel resolution
Scientific Reports (2021)
-
High-speed force spectroscopy: microsecond force measurements using ultrashort cantilevers
Biophysical Reviews (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
