
Abstract
In this article we propose a Fast Optimal Global Sequence Alignment Algorithm, FOGSAA, which aligns a pair of nucleotide/protein sequences faster than any optimal global alignment method including the widely used Needleman-Wunsch (NW) algorithm. FOGSAA is applicable for all types of sequences, with any scoring scheme and with or without affine gap penalty. Compared to NW, FOGSAA achieves a time gain of (70–90)% for highly similar nucleotide sequences (> 80% similarity) and (54–70)% for sequences having (30–80)% similarity. For other sequences, it terminates with an approximate score. For protein sequences, the average time gain is between (25–40)%. Compared to three heuristic global alignment methods, the quality of alignment is improved by about 23%–53%. FOGSAA is, in general, suitable for aligning any two sequences defined over a finite alphabet set, where the quality of the global alignment is of supreme importance.
Similar content being viewed by others

Introduction
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA or protein to identify their degree of similarity that may be important in identifying functional, structural or evolutionary relationships between them. If two sequences in an alignment share a common ancestor, mismatches can be interpreted as point mutations and gaps as indels, introduced in one or both lineages in the time since they diverged from one another. In sequence alignment of proteins, the degree of similarity between amino acids occupying a particular position in the sequence can be interpreted as a rough measure of how conserved a particular region is. The alignment algorithms are generally of two types: Local Alignment Methods such as1,2,3,4, are designed to search locally similar segments between two sequences while in Global Alignment Methods the overall similarity is mapped out.
Global alignment algorithms like5,6 are found useful as biological sequences from related organisms satisfy some ordering assumption. For example, the human and mouse genome share a conserved region up to 8 Megabases in length7. The fundamental contribution of global alignment described in8 was the widely adopted one for optimal alignment of sequences. But this algorithm is very expensive with respect to time and space, proportional to the product of the length of two sequences and hence is not suitable for long sequences. Then GAP39 was proposed with improved sensitivity and was suitable for comparing sequences with intermittent similarities. But as the underlying principle is based on dynamic programming, the computing time is still proportional to the product of the sequence lengths. Beside this, the optimality of the alignment, as performed by GAP3, is highly sensitive to the parameter values given in the program. There are some heuristic based fast alignment programs also, like ACANA10, AVID11, ClustalW12, BLASTZ13, NUMmer14, LAGAN15 etc. However these often compromise on the quality of the alignment.
In this article we propose a Fast Optimal global sequence alignment that overcomes the shortcomings of the existing methods and provides the optimal alignment of sequences without any parameter tuning. FOGSAA gives exactly the same result as that provided by the Needleman-Wunsch method (NW)8, but in much less time. The Result Section shows that among the three optimal global alignment programs (NW, GAP3, FOGSAA), FOGSAA is the fastest. With respect to the heuristic alignment methods mentioned earlier, FOGSAA provides an improvement of alignment scores of about 22.8% on simulated benchmark data16 and 53% on real human-mouse ortholog sequences over these methods. FOGSAA also outperforms GAP39 on the overall quality of the alignment. Not only for the gene sequences, it can do equally well for protein sequences with or without affine gap penalty. In such cases FOGSAA takes the match and mismatch scores from the substitution matrices like BLOSUM62, PAM etc. and the gap penalties including the gap_open and gap_extension scores can have any value as specified by the user. Experimental results show that for protein sequences FOGSAA achieves a time gain of (25% – 40%).
The algorithm, FOGSAA, is basically a branch and bound approach of global pairwise sequence alignment. It works by building a branch and bound tree where each root-to-leaf path represents a possible way to align the given pair of sequences. FOGSAA starts the branch expansion in a greedy way taking the symbols from the input sequences (protein or nucleotide) and continues till the end of the path. If at an intermediate point, some other branch is found more promising than the current one, then it is started for expansion. The procedure is repeated until no other branch is found better. Finally it returns the optimal alignment along with the optimal score, by traversing the optimal path. During expansion, if a path is found no longer promising, it is pruned to save unnecessary computation. However, if less than 30% similarity of the input sequences is detected, then the algorithm is terminated with an approximate score which is equal to the best score obtained so far. Although FOGSAA can give the accurate optimal alignment for any sequence pair even if they are less than 30% similar, it may not be worthwhile to spend the resources for aligning such dissimilar sequences. Therefore, we terminate FOGSAA in these cases. The threshold of 30% was chosen based on the intuition, though it can be changed if required. The pruning strategy and the way of computing an approximate score for highly dissimilar sequences are described in the Method Section. The workflow of FOGSAA is depicted in Fig. 1. Some relevant definitions and the theoretical formulation of the algorithm are provided below.

FOGSAA workflow : The basic working principle of FOGSAA is descried through flow-chart.
Let the two sequences of length m and n, respectively, be the following:

Let P1 and P2 be the pointers to symbols in S1 and S2 respectively, having initial values P1 = 0 and P2 = 0. FOGSAA computes the optimal alignment between S1 and S2 by finding the optimal branch in the corresponding branch and bound tree. Each node of this tree shows the alignment between one pair of symbols pointed by (P1,P2) which is (0,0) for the root node. A node has four components:
-
(P1,P2) value pair
-
The Type of alignment:

-
PrS (Defined later)
-
(Tmax,Tmin) (Defined later).

Then, a path from the root to a node ((P1, P2); Type; PrS; (Tmin, Tmax)) represents an alignment of a1a2…aP1 and b1b2…bP2 with the last pair of characters being aligned as Type. The PrS and (Tmin, Tmax) give the Present Score and Fitness Score values respectively after alignment of a1a2…aP1 and b1b2…bP2. These are defined later. In this way, a path from the root to a leaf node i.e., a complete path represents one possible way to align the given pair of sequences. Therefore, a branch and bound tree is a search tree which searches for an optimal alignment path while using an objective score to bound the search space. Starting from the root of this tree, FOGSAA proceeds in one of the following three ways:
-
Advance both the pointers P1 and P2, i.e., align a1 with b1, which can lead to either a match or mismatch.
-
Move the pointer P1 keeping P2 fixed which will introduce a gap in S2, i.e., a1 will be paired with a gap.
-
Move the pointer P2 keeping P1 fixed, thereby introducing a gap in S1.
Hence the root can have three children (a1,b1), (a1,–) or (–,b1), where the first child indicates a match or mismatch, the second child indicates a gap in S2 and the third one indicates a gap in S1. The corresponding P1 and P2 values of these three children would be (1, 1), (1, 0) and (0, 1) respectively. Likewise every node of this branch and bound tree can have at most three children (as shown in Fig. S1 in Supplementary). Among them only the best one will be expanded according to the ‘Fitness Score’ (see Definitions below), while the others are inserted in a hashed priority queue ordered by their scores. These nodes might be expanded later on if they come in the top of the priority queue.
This approach of selecting the fittest child based on the ‘Fitness Score’ continues till the end of the branch. One branch, from the root to a leaf, gives one alignment. Then the algorithm proceeds to the next branch in search for a better alignment. This branch always starts from a node which has the highest Fitness Score and is on the top of the priority queue. All the decisions i.e., whether we should go for a next branch or not, which should be that next branch and how far a branch should be expanded, are taken according to the above mentioned score. If at any point FOGSAA detects that the score of the top most node of the priority queue indicates that the two sequences have less than 30% similarity, then it terminates with the best alignment and corresponding score that has been obtained so far. In other cases, FOGSAA terminates with an optimal alignment of the sequences based on the given values of match, mismatch and gap scores. The gap score can also include affine gap penalties17 with Gap-open (Go) and Gap-extension (Ge) costs. In such cases, the total Gap cost of length L would be (Go + L × Ge). Similarly, for protein sequence alignments, FOGSAA can use any substitution matrix, with or without affine gap penalties.
The working principle of FOGSAA is based on two strategies: i) Select the best child in the current branch. ii) Start next branching from a node showing highest potential. The potential of any node, say X, is computed using Fitness Score. If its potential is greater than other siblings, then X will be expanded in the tree. The Fitness Score is the summation of two other scores, the Present Score and Future Score. The Present Score is the sum of all match/mismatch/gap scores that have been encountered so far in the current branch starting from root to the node X, while the Future Score is an estimated score value that can result when the remaining parts of the sequences would be aligned. These scores are defined below.
Let the given pair of sequences be

where |S1| = m and |S2| = n. If the current node is at position (P1, P2) i.e., P1 symbols from S1 and P2 symbols of S2 have been checked and (i1, j1),(i2, j2), … ,(ik, jk) are the k nodes that are expanded so far in the current branch where, ik = P1 and jk = P2, then the Present Score, denoted by PrS, is defined as:

The addition of scores for each node, from root to the current node of the current branch, gives the Present Score. Here,

Where M = Match Score, Ms = Mismatch Score and G = Gap Penalty.
The Future Score reflects the scenario from the node X to the leaf of the current branch. Unlike Present Score, the Future Score is not known at this moment. It will attain its maximum value when there are all matches in the path X to the leaf. On the other hand, all mismatches will lead to its minimum value of the optimal alignment. There can be any other alignment worse than this, but it is surely not the optimal one. Note that there will be at least as many number of gaps as the difference of lengths of the two strings. If the current node is at (P1, P2), then the Present Score includes the alignment of the symbols a1…aP1 of S1 and b1…bP2 of S2. For the remaining portion, i.e., for aP1 + 1…am of sequence S1 and bP2 + 1…bn of S2, we have to compute the minimum and maximum scores. In the Future Score, without loss of generality, it may be stated that there must be at least |(m − P1) − (n − P2)| gaps. For the remaining part, at best there may be (m − P1) matches and at worst (m − P1) mismatches.
If the two sequences to be aligned are a1…am and b1…bn and the present node is at position (P1, P2), then the two components Fmin and Fmax of Future Score, for the subsequences aP1+1…am and bP2+1…bn, are defined as:


where, x1 = (n − P2) and x2 = (m − P1).
Note that for amino-acid sequences, M and Ms take values from a substitution matrix, which is by default BLOSUM 62.
The Fitness Score of a node, based on which the potential of a branch is evaluated, is the sum of the Present Score (PrS) and the Future Score. Fitness Score, having two components denoted by Tmin and Tmax, is defined as follows:


The entire method for the selection of the best child depending on these scores, which finally results in the optimal alignment, is summarized in Algorithm 1. The nodes are inserted in the priority queue based on their Tmax values i.e., the node having the highest value of Tmax will be on the top.
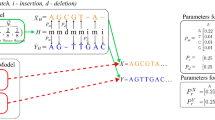
An example of Fitness Score calculation using Algorithm 1 from the partially computed FOGSAA tree is shown in Fig. 2. It uses +1 and −1 for the Match and Mismatch scores respectively and −2 as the Gap penalty (without using affine gap).

FOGSAA tree: partially computed for the sequences S1 = ACGGTTGC and S2 = AGCGTC, having m = |S1| = 8 and n = |S2| = 6.
Each node is annotated with (P1, P2) on the left and [Tmax, Tmin] on the right. The label of a node indicates the symbol pairs that are being aligned.
The root starts with P1 = 0 and P2 = 0 and since no alignment has been made so far, the Present Score, PrS = 0. Here m = 8 and n = 6, thus in the Future Score there will be at least (m − n) = 2 gaps. The best case would be if the remaining 6, as min(m, n) = 6, are all matches yielding Fmax = 6 * 1 + 2 * (−2) = 2. Similarly, the worst scenario would be if these 6 are all mismatches, giving Fmin = 6 * (−1) + 2 * (−2) = −10. Therefore, Tmin = P + Fmin = 0 + (−10) = −10 and Tmax = P + Fmax = 0 + 2 = 2. This [Tmax, Tmin] = [2, −10] value pair is shown in the top-right side of the root node in Fig. 2. Now, from the root there are three possible moves (1, 1), (1, 0) or (0, 1). For the first one PrS = 1, as it is a match. Here the Future Score is computed for length x1 = (m − P1) = 7 and x2 = (n − P2) = 5 of sequences S1 and S2 respectively. So, Fmax = 5 + 2 * (−2) = 1 and Fmin = 5 * (−1) + 2 * (−2) = −9. Finally, Tmin = P + Fmin = 1 + (−9) = −8 and Tmax = P + Fmax = 1 + 1 = 2. Similarly, it can be shown that, node (1,1) has higher Tmax value than the other two children (1,0) and (0,1). Hence this node is expanded in Fig. 2. The algorithm continues in this way. A detailed illustration of FOGSAA can be found in the Supplementary Figures2,3,4,5,6.
Note that although the example provided here is for a specific scoring scheme without affine gap penalty, FOGSAA is able to handle any scoring scheme including substitution matrices for protein sequences and also affine gap penalty. In the case of affine gap penalty, the G of Eq. 2 will be computed as follows:

where Go and Ge stand for Gap-open and Gap-extension penalties respectively.
In case of calculating Fmin, we have to consider the worst case where each gap is a new gap. That means all the gaps are scattered separately and the cost of each gap would be (Go + Ge). In contrast, for Fmax we can take the best case scenario in which all the gaps are clubbed together and there is only one gap open penalty. Therefore, the Eq. 3 and Eq. 4 can be extended as follows to include affine gap penalty.


Note that, Eq. 5 and Eq. 6 remain unchanged, though the computation of PrS and (Fmin, Fmax) are modified as described above.
Results
FOGSAA is basically a branch and bound algorithm which starts its branch expansion by greedy selection of nodes based on some specific score value. Branch and bound techniques can take exponential time in the worst case. However, the average complexity of branch and bound method is significantly lower18. It has already been shown that the average case analysis of branch and bound problem has polynomial complexity19. Here in FOGSAA, if the two input sequences are of length m and n respectively, then there cannot be more than m × n nodes in the branch and bound tree. Therefore, the worst case running time of FOGSAA is bounded by O(m × n), though, on an average, it is much lower. The best case, when FOGSAA finds the optimal alignment just after expanding the first branch, has complexity O(m + n), equal to the maximum length of a branch. This is why FOGSAA achieves a large time gain in comparison to NW, whose complexity is O(m × n) for all the cases -best, average and worst. Note that the alignment quality of FOGSAA and NW are exactly the same.
We have divided the results into two categories: 1) Running time comparison between three optimal global alignment programs, NW, GAP3 and FOGSAA; 2) Comparative study of alignment quality between FOGSAA, NW and GAP3 and three heuristic methods ACANA, AVID and ClustalW.
Running time comparison
To assess the performance of FOGSAA, we have compared its running time with those of two other optimal alignment programs, NW and GAP3, on 178 real DNA sequences collected from NCBI GenBank (Code, test data and results are available at http://www.isical.ac.in/~bioinfo_miu/FOGSAA.htm). These DNA sequences are then divided into three classes based on their similarity: i) greater than 80% similar, ii) 30% – 80% similar and iii) less than 30% similar. Fig. 3 and Fig. 4 show the performance of all the three methods for sequences having > 80% similarity and 30% – 80% similarity respectively, when they are run on Intel(R) Core(TM) i7 CPU @ 2.93 GHz machine with 4 GB RAM with the scoring scheme as M = 1, Ms = −1 and G = −2. As can be clearly seen from the graphs, FOGSAA comprehensively outperforms NW as well as GAP3 in every case for sequences upto 6000 bp.

Time comparison between Needleman-Wunsch, GAP3 and FOGSAA for sequences having > 80% similarity.

Time comparison between Needleman-Wunsch, GAP3 and FOGSAA for sequences having (30 – 80)% similarity.
If FOGSAA encounters a situation when the most promising node of the branch and bound tree (or, the first entry in the priority queue) shows less than 30% similarity, then it terminates with an approximate alignment. A detailed description of pruning strategy and approximate score can be found in the Method Section. Table 1 shows the behavior of FOGSAA in comparison to NW and GAP3 for real gene sequences of less than 30% similarity. As can be seen from the table, the optimal alignment score, whenever available, is negative reflecting the low similarity of the sequences. And the approximate score as given by FOGSAA, is very close to the optimal one. In certain cases where the input sequences are very long and dissimilar, then most of the times NW and GAP3 fail. However, FOGSAA is able to provide at least a good approximate score as shown in the last row of Table 1.
FOGSAA performs significantly well even for different scoring schemes with or without affine gap penalties. This is reflected in Table 2. Here results are shown for 5 different scoring schemes with and without affine gap penalty. Each scheme is tested on nearly 100 pairs of sequences having length upto approximately 10,000 bp, which have been collected from NCBI GenBank. These sequences are of varied similarity as they are picked arbitrarily. As can be seen from the table, FOGSAA performs consistently better over all the scoring schemes and produces an average time gain of 82%. Here, it is also found that on an average 64% of the total possible nodes are pruned.
As mentioned earlier, FOGSAA is equally applicable for protein sequences with any substitution matrix, both with and without affine gap penalties. Here we provide the results for BLOSUM62. Fig. 5 summarizes the performance of FOGSAA as compared with NW for 100 pairs of amino-acid sequences with affine gap penalty. These sequences are also selected arbitrarily from NCBI. Here we have used −90 and −25 as the Gap-open and Gap-extension penalties respectively. From the histogram plot as shown in this figure, it is evident that FOGSAA provides a high time gain for a very large number of times. Here time gain is computed as (TimeNW − TimeFOGSAA)/TimeNW. Similar results using different scoring functions with or without affine gap penalty can be found in the Supplementary Tables (S3–S6).

Time gain of FOGSAA over Needleman-Wunsch for protein sequence alignment with affine gap penalty.
Result on alignment quality
FOGSAA is not only a faster alignment tool, it also provides the best or optimal alignment of the input sequences (having > 30% similarity). FOGSAA is sometimes slower than some fast heuristic based alignment approaches. However, the quality of alignment of these faster methods often degrades and is far from the optimal alignment. In this section, we provide results pertaining to the alignment quality. Table S1 of Supplementary, shows the comparison between FOGSAA, ACANA, AVID, ClustalW and GAP3 for the benchmark gene sequences16. The mean and median of the alignment scores are provided. Greater the alignment score, better is the alignment quality.
As can be seen from Table S1 in Supplementary, FOGSAA shows the highest mean as well as median scores among all the methods. As expected, the corresponding values for NW are the same since both of them have exactly the same alignment quality. GAP3 has the property of removing some base pairs, when they are found not potential for alignment. That is why GAP3 provides good alignment for sequences having intermittent similarity. But the performance often degrades for the overall alignment as reflected by the negative mean in the table. Here we have tuned the parameters of GAP3 in such a way that no base pairs are removed, otherwise it will be difficult to make the comparison as the sequence length would get reduced. GAP3 provides the optimal alignment only in certain cases, but not always as verified through personal communication with the authors.
Table 3 shows the result of a comparative study on real sequences containing human-mouse orthologs. Here FOGSAA is compared only with ACANA, as it is one of the more recent methods, on 25 pairs of real ortholog sequences. The detailed count of matches (M), Mismatches (MS) and Gaps (G) are given for both the methods. It is evident from Table 3 that in general, FOGSAA provides better alignment than ACANA for all the sequences with more matches (M) and introducing lesser gaps (G). It is therefore apparent from the results that FOGSAA provides a good balance of running time and alignment quality. Some more results are included in the Supplementary Table S2 based on alignment quality for 94 pairs of real gene sequences.
Discussion
Obtaining high quality sequence alignment while minimizing the running time is a challenge in bioinformatics. Though several efforts have already been made in this regard, the problem is not totally solved. When existing optimal alignment programs were found too slow, faster heuristics were developed. However these faster solutions compromised on the quality of alignment being better suited for sequences with short regions of high similarity. Not only that, the difficulty also lies in the selection of the alignment output because almost no two alignment programs (other than the optimal ones) give the same result for the same input sequences.
In this article we report on the development of FOGSAA that provides optimal global alignment of a pair of sequences while being remarkably fast. The results reported in this article demonstrate that FOGSAA is effective for nucleotide sequences as well as amino acid sequences, given any scoring scheme. It can also handle affine gap penalty. Compared to the optimal NW algorithm, FOGSAA is faster by 70%–90% for sequences having high similarity, while providing the same optimal score. Compared to some heuristic alignment methods, FOGSAA provides much improved alignment with higher number of matches and smaller number of mismatches and gaps. We believe that FOGSAA is of high significance with applications covering a large number of areas in Computational Biology, as pairwise alignment is a fundamental process in sequence analysis. Most often, it is the first step in any biological analysis, which is used to identify evolutionary relationship between some novel sequences to existing ones. Use of FOGSAA can also significantly reduce the time requirement of database searches, with no reduction in the accuracy of alignment. Evidently, accuracy of the alignment affects the downstream processing tasks. Highly accurate alignments will help to uncover subtle signals embedded in the sequences, that might otherwise be missed or overlooked.
In future we want to demonstrate the application of FOGSAA for analysis of Next Generation Sequencing data set20. We believe that the underlying technique of FOGSAA can also bring significant advancement in multiple sequence alignment methods. This is an important direction in future research. Although the effectiveness of FOGSAA is demonstrated for nucleotide and protein sequences, it is equally applicable in other domains, such as web-clustering, where the quality of alignment is of great concern.
Methods
Being a branch and bound method, FOGSAA starts its branch expansion from the root node, selecting the best child at each step and inserting the other children in the priority queue according to the values of Tmax, using separate chained hashing technique. Hashing is a specialized technique for storing data which ensures constant time search operation in ideal scenario. In this scheme, the data are placed in a specific cell of the hash table depending on its hashed value, which is Tmax here. Collision occurs if two or more data have the same hashed value. Separate chaining is one of the most popular collision resolution techniques where the data that has the same hashed value are placed in a chain of linked nodes. That means, all the nodes in a particular chain will always have the same Tmax value and they are ordered by their corresponding values of Tmin. The largest difference between Tmax and Tmin value provides the theoretical bound on the number of possible hashed values. This node selection procedure continues till the end of the first path (root-to-leaf path) which provides an initial alignment of the sequences. Now, FOGSAA has to check whether there is a chance of obtaining a better alignment. Note that the Tmax value of a node is the best possible score that might be obtained by aligning along one of the branches starting from it. If the Tmax value of the top node of the priority queue is greater than the best alignment score that is obtained so far, then there is a possibility of improving the alignment. Therefore, FOGSAA starts a new branch expansion from the corresponding node. In the middle of a branch expansion, if it comes to a node having the same (P1, P2) value as one of the existing nodes, which has already been expanded in a better way producing better PrS score, then the current branch is pruned. The process of selecting a new branch from the top node continues until the Tmax value of top node falls below the best alignment score achieved till now. Then, FOGSAA reports the optimum alignment along with the score and the algorithm terminates.
If the best possible score i.e., the Tmax value of the top node of priority queue indicates less than 30% similarity of the input sequences, then rather than searching for the actual optimal score, FOGSAA terminates with an approximate score which is the score of the best alignment path (root-to-leaf) that has been obtained so far. The detailed method is described in Algorithm 1.
In the remaining part of this section, we provide some technical insights into the working principle of FOGSAA.
Lemma 1. Let a node X in FOGSAA tree have Fitness Score [Tmax, Tmin], then the score of its child will be [Tmax, Tmin + (M − Ms)] if it makes a match, where M and Ms are the match and mismatch scores respectively.
Proof. For the parent node X, let (Tmax)parent = PrSparent + (Fmax)parent, where PrS denotes the Present Score (PrS) and (Fmax)parent = x × match_scores + y × gap_penalties and (Fmin)parent = x × mismatch_scores + y × gap_penalties. Where x is the number of matches in the best case and number of mismatches in the worst and y is the number of gaps introduced. However, for the child: PrSchild = PrSparent + M, as it has already made a match. Thus the future part is reduced by length one, i.e., there can be (x − 1) matches/mismatches but the gap penalties remain the same as it is proportional to the length difference of the two sequences. So, (Fmax)child = (x − 1) × match_scores + y × gap_penalties, (Fmin)child = (x − 1) × mismatch_scores + y × gap_penalties. Thus (Fmin)child = (Fmin)parent − Ms, as one mismatch is reduced and (Fmax)child = (Fmax)parent − M because the child can have one less match than that of the parent. Therefore,

and

Lemma 2. Let a node X in FOGSAA tree have Fitness Score [Tmax, Tmin], then the score of its child will be [Tmax + (Ms − M), Tmin] if it makes a mismatch.
Proof. For the parent node X, let (Tmax)parent = PrSparent + (Fmax)parent and (Fmax)parent = x × match_scores + y × gap_penalties and (Fmin)parent = x × mismatch_scores + y × gap_penalties. However, for the child: PrSchild = PrSparent + Ms, as it has already made a mismatch. Thus the future part is reduced by length one, i.e., there can be (x − 1) matches/mismatches but the gap penalties remain the same, as it is proportional to the length difference of the two sequences. So, (Fmax)child = (x − 1) × match_scores + y × gap_penalties, (Fmin)child = (x − 1) × mismatch_scores + y × gap_penalties. Thus (Fmin)child = (Fmin)parent − M s, as one mismatch is reduced and (Fmax)child = (Fmax)parent − M because the child can have one less match than that of the parent. Therefore,

and

Lemma 3. Let a node X in FOGSAA tree have Fitness Score [Tmax, Tmin], then the score of its child will be [Tmax, Tmin] or [Tmax + (2 × G − M), Tmin + (2 × G − Ms)], if it inserts a gap.
Proof. For the parent node X, let (Tmax)parent = PrSparent + (Fmax)parent and (Fmax)parent = x × match_scores + y × gap_penalties, (Fmin)parent = x × mismatch_scores + y × gap_penalties. However, for the child: PrSchild = PrSparent + G, where G is the gap penalty. However the gap can be inserted in any of the two sequences.
Case 1: If the gap is introduced in the shorter sequence then it makes no change, as this gap is due to the length difference of the two sequences and it is already counted within ‘y gap penalties' in the parent node. The only change is that the gap has become ‘present’ now leaving y − 1 gaps in the future. So, (Fmax)child = x × match_scores + (y − 1) × gap_penalties, (Fmin)child = x × mismatch_scores + (y − 1) × gap_penalties. Thus (Fmin)child = (Fmin)parent − G and (Fmax)child = (Fmax)parent − G as one gap is reduced . Therefore,

and

Case 2: If the gap is introduced in the longer sequence then it is an extra gap which will always cause an insertion of another gap at some position of the shorter sequence. Hence in the future there can be (x − 1) matches/mismatches and (y + 1) gaps. So, (Fmax)child = (x − 1) × match_scores + (y + 1) × gap_penalties, (Fmin)child = (x − 1) × mismatch_scores + (y + 1) × gap_penalties. Thus (Fmin)child = (Fmin)parent − Ms + G = (Fmin)parent + (G − Ms) and (Fmax)child = (Fmax)parent − M + Gp = (Fmax)parent + (G − M). Therefore,

and

Lemma 4. The branches that are pruned by FOGSAA (Algorithm 1) will never give the optimal alignment solution.
Proof. The two reasons for which a branch is pruned according to Algorithm 1 are specified in the lines 12 and 17 respectively.
Case 1: If the current node (say, X) of the branch has a Present Score which is smaller than the Present Score of an existing node (say, Y) having the same P1 and P2 value pair, then the current branch is pruned (Line 12 of Algorithm 1), where the P1 and P2 represents the position in the string S1 and S2 respectively. As the P1, P2 values of the nodes X and Y are same, both of them will have the same successors. Therefore, the remaining part of the alignment, for both the nodes, will be the same. Let the score of this remaining part be S. So, the actual score of the full alignment of the branch containing X is (PrS)X + S and similarly, the actual score of the entire branch having the node Y would be (PrS)Y + S. As (PrS)X ≤ (PrS)Y, the branch containing X node cannot give better alignment than the branch having node Y. Therefore, if this branch of node X is pruned, it will not affect the optimal solution.
Case 2: If the Tmax value of the current node (say, Z) of a branch is less than the optimal score which has been obtained so far (say, along branch B1), then this branch is pruned [Line 17 of Algorithm 1]. Note that, the Tmax value of a node is the best possible score that might be obtained by aligning along one of the branches starting from it. So, a node cannot achieve an alignment having score better than Tmax. Therefore, even if we expand the branch containing node Z, it cannot ever produce an alignment better than B1. Hence, the branch which is pruned here will never give the optimal alignment, as at least one better solution has already been found.
Corollary 1. Let a node X in FOGSAA tree have three children X1, X2, X3, then the child having a match or a gap in the shorter sequence, is always the best child according to the Fitness Score(Tmax).
Proof. Let the node X have (P1, P2) = (i, j), then its children X1, X2, X3 will have values (i + 1, j + 1), (i + 1, j), (i, j + 1) respectively. The node X1 can have either a match or a mismatch depending upon the symbol at that position of the two sequences. But X2 and X3 will always have a gap. If X has Fitness Score value [Tmax, Tmin], then according to Lemma 1 and 2, X1 will have [Tmax, Tmin + (M − Ms)] if it's a match and [Tmax + (Ms − M), Tmin] otherwise. X2 and X3 will have Fitness Score [Tmax, Tmin] or [Tmax + (2 × G − M), Tmin + (2 × G − Ms)], for the two different cases as specified in Lemma 3. As M > 0, Ms < 0, G < 0 and usually G < Ms, it is obvious that the child with a match or a gap in the shorter sequence has the highest Tmax value and hence it is most promising.
Proof of Correctness of FOGSAA
Given a pair of input sequences that have more than 30% similarity, the alignment score provided by FOGSAA is optimal for the given scoring scheme.
We will prove this by the method of contradiction. Let us consider the following two cases:
Case 1: Without affine gap: Let us assume that the alignment reported by FOGSAA is not optimal. Say B is the branch corresponding to the non-optimal alignment provided by FOGSAA on termination. Also assume that there is another branch  which leads to the optimal alignment. Let X and
which leads to the optimal alignment. Let X and  be the terminal (leaf) nodes of the branches B and
be the terminal (leaf) nodes of the branches B and  respectively. At a leaf, there is no Future Score, hence
respectively. At a leaf, there is no Future Score, hence  and (PrS)X = (Tmax)X. Since
and (PrS)X = (Tmax)X. Since  is the leaf on the optimal branch
is the leaf on the optimal branch  while X is the leaf on the non-optimal branch B, so
while X is the leaf on the non-optimal branch B, so  . Obviously the Tmax values of the ancestors of
. Obviously the Tmax values of the ancestors of  is greater than or equal to
is greater than or equal to  , since while the ancestors overestimate the Tmax values,the value at the leaf reflects the actual alignment score [The scores of a branch become accurate as the Algorithm 1 moves down through it and makes the modification of scores as specified in the lines 15,16 of Algorithm 1]. Consequently, the Tmax values of the ancestors of
, since while the ancestors overestimate the Tmax values,the value at the leaf reflects the actual alignment score [The scores of a branch become accurate as the Algorithm 1 moves down through it and makes the modification of scores as specified in the lines 15,16 of Algorithm 1]. Consequently, the Tmax values of the ancestors of  are all greater than (PrS)X as
are all greater than (PrS)X as  . Moreover, as FOGSAA has not expanded the branch
. Moreover, as FOGSAA has not expanded the branch  , as per out assumption, at least one of the ancestor nodes of
, as per out assumption, at least one of the ancestor nodes of  are still there in the priority queue because Algorithm 1 inserts the current node in the priority queue according to the Tmax values of its best child (Line no.9 of Algorithm 1). That means, the top node of the priority queue has a Tmax value which is greater than (PrS)X. But there cannot be any such node because FOGSAA stops only when the Tmax value of top node of the priority queue becomes smaller than the PrS value of its best branch i.e., the optimal score obtained so far (See the loop termination condition of Algorithm 1, line 25). Hence our initial assumption that FOGSAA terminates with a non-optimal alignment, is wrong. Therefore, if FOGSAA has terminated with an alignment along branch B, then there can be no branch
are still there in the priority queue because Algorithm 1 inserts the current node in the priority queue according to the Tmax values of its best child (Line no.9 of Algorithm 1). That means, the top node of the priority queue has a Tmax value which is greater than (PrS)X. But there cannot be any such node because FOGSAA stops only when the Tmax value of top node of the priority queue becomes smaller than the PrS value of its best branch i.e., the optimal score obtained so far (See the loop termination condition of Algorithm 1, line 25). Hence our initial assumption that FOGSAA terminates with a non-optimal alignment, is wrong. Therefore, if FOGSAA has terminated with an alignment along branch B, then there can be no branch  providing better score than B.
providing better score than B.
Case 2: With affine gap: When the scoring scheme includes affine gap penalty, then also the branch expansion and termination strategy of FOGSAA remains the same. Only the way Tmax is being calculated, is different. Here also Tmax shows the best possible score, but the gap penalties are computed using the formula (Go + L × Ge) where L is the gap length. As the inherent technique remains same, it can be shown in the same way that there cannot be any other branch producing better alignment than the one provided by FOGSAA.
Thus, FOGSAA is correct and always outputs the optimal alignment.
Proof of termination of FOGSAA
In the best case, FOGSAA terminates after the expansion of the first branch if the Tmax value of the top node of the priority queue becomes smaller than the best alignment score obtained so far. Otherwise, it starts expanding a new branch from the top node. This process continues until either Tmax value of top node falls below the optimal score obtained so far, or the queue becomes empty i.e., all possible paths have been checked. If the given sequences are of length m and n, then there can be no more than m × n nodes. Again, each node can be pushed into the queue only once. Therefore, even if FOGSAA checks all the nodes of the queue, it will terminate in O(m × n) time, which is finite. Hence, FOGSAA terminates within a finite amount of time.
Proof of Completeness of FOGSAA
It is quite obvious that FOGSAA is applicable for any sequence over a finite alphabet. Just the scoring matrix for this alphabet needs to be defined. This justifies the completeness property of FOGSAA.
Algorithm 1: FOGSAA
Input: A pair of DNA or protein sequence S1 and S2.
Output: Optimal alignment of given sequence pair having ≥ 30% similarity. Otherwise it terminates with an approximate alignment and score.
Data Structure:
c[i][j]: The node of FOGSAA tree having P1 = i and P2 = j.
Priority queue: Stores the nodes of FOGSAA tree for future expansion based on their Fitness Score, using separate-chained hashing. (See the Method section for a discussion).
|S1| = m and |S2| = n, P1 = 0, P2 = 0, c[0, 0]. PrS = 0, optimal = c[0, 0]. Tmin
if m ≠ 0 AND n ≠ 0 then
repeat
while P1 ≤ (m − 1) OR P2 ≤ (n − 1) do
Select the best child from the remaining children according to the Tmax.
Let the corresponding P1, P2 values of the selected child be x and y respectively.
if any child of the current node remains to be expanded then
insert the current node in the priority queue according to the Tmax score of the next better child.
end if
if child_node.PrS ≤ c[x, y].PrS then
Prune the current branch, as it has already been traversed in a better way.
else
c[child_node] ← new_score
P1 ← x, P2 ← y
if child_node.Tmax ≤ optimal then
Prune the current branch. The Tmax of a node shows the maximum score that the branch can achieve and if this max value is smaller than the optimal branch score obtained so far, then it can not ever lead to the optimal solution.
end if
end if
end while
if c[P1, P2].Tmax ≥ optimal then
optimal = c[P1, P2].Tmax and set the current path as the optimal one.
end if
pick the top most node from the priority queue and update new Tmax.
if The top most node has Tmax such that it cannot have more than 30% similarity then
end the process and report approximate score.
end if
until optimal ≥ new Tmax
end if
References
Smith, T. F. & Waterman, M. S. Identification of Common Molecular Subsequences. J. Mol. Biol. 147, 195–197 (1981).
Pearson, W. R. & Lipman, D. Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA 85, 2444–2448 (1988).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biology 215, 403–410 (1990).
Huang, X. & Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math 12, 337–357 (1991).
Huang, X. On global sequence alignment. Comput Appl Biosci 10(3), 227–235 (1994).
Chenna, R. et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acid Research 31(13), 3497–3500 (2003).
Mural, R. et al. A comparison of whole genome shotgun-derived mouse chromosome 16 and the human genome. Science 296, 1667–1671 (2002).
Needleman, S. B. & Wunsch, C. D. A. general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453 (1970).
Huang, X. & Chao, K. A. generalized global alignment algorithm. Bioinformatics 19, 228–233 (2003).
Huang, W., Umbach, D. M. & Li, L. Accurate anchoring alignment of divergent sequences. Bioinformatics 22, 29–34 (2006).
Bray, N., Dubchak, I. & Pachter, L. AVID : A Global Alignment Program. Genome Res. 13, 97–102 (2003).
Thompson, J. D. et al. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Schwartz, S. et al. Human-mouse alignment with BLASTZ. Genome Research 13, 103–107 (2003).
Delcher, A. L. et al. Fast algorithms for large scale genome alignmentand comparison. Nucleic Acid Res. 30, 2478–2483 (2002).
Brudno, M. et al. LAGAN and Multi-LAGAN: efficient tools for large scale multiple alignment of genomic DNA. Genome Res. 13, 721–731 (2003).
Pollard, D., Bergman, C., Stoye, J., Celniker, S. & Eisen, M. Benchmarking tools for the alignment of functional noncoding DNA. BMC Bioinformatics 5(1) (2004).
Gotoh, O. An improved algorithm for matching biological sequences. Journal of Molecular Biology 162(3), 705–708 (1990).
Thakoor, N. & Devarajan, V. Computation Complexity of Branch-and-Bound Model Selection. IEEE 12th International Conference on Computer Vision (ICCV) (2009).
Zhang, W. & Korf, R. E. An average case analysis of Branch and Bound with applications :Summary of results. AAAI-92 Proceedings (1992).
Rizk, G. & Lavenier, D. GASSST: global alignment short sequence search tool. Bioinformatics 26, 2534–2540 (2010).
Acknowledgements
Prof. Sanghamitra Bandyopadhyay acknowledges the Swarnajayanti Fellowship scheme of Department of Science and Technology, Government of India (No.DST/SJF/ET-02/2006-07).
Author information
Authors and Affiliations
Contributions
A.C. developed the idea, carried out the work, wrote the main text, prepared the Figures 1–5 and the software tool for FOGSAA. S.B. conceived of the study, planned the work, provided laboratory facilities and wrote the manuscript. Both authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary of FOGSAA
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chakraborty, A., Bandyopadhyay, S. FOGSAA: Fast Optimal Global Sequence Alignment Algorithm. Sci Rep 3, 1746 (2013). https://doi.org/10.1038/srep01746
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01746
This article is cited by
-
A review of alignment based similarity measures for web usage mining
Artificial Intelligence Review (2020)
-
MPSAGA: a matrix-based pair-wise sequence alignment algorithm for global alignment with position based sequence representation
Sādhanā (2019)
-
Protein Sequence Comparison Based on Physicochemical Properties and the Position-Feature Energy Matrix
Scientific Reports (2017)
-
The identification of abrasive grains in the decohesion process by acoustic emission signal patterns
The International Journal of Advanced Manufacturing Technology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
