
Abstract
Sequence alignments form the basis for many comparative and population genomic studies. Alignment tools provide a range of accuracies dependent on the divergence between the sequences and the alignment methods. Despite widespread use, there is no standard method for assessing the accuracy of a dataset and alignment strategy after resequencing. We present a framework and tool for determining the overall accuracies of an input read dataset, alignment and SNP-calling method providing an isolate in that dataset has a corresponding, or closely related reference sequence available. In addition to this tool for comparing False Discovery Rates (FDR), we include a method for determining homozygous and heterozygous positions from an alignment using binomial probabilities for an expected error rate. We benchmark this method against other SNP callers using our FDR method with three fungal genomes, finding that it was able achieve a high level of accuracy. These tools are available at http://cfdr.sourceforge.net/.
Similar content being viewed by others


Introduction
Sources of error within next-generation sequencing (NGS) projects can result in poorly resolved genotypes1,2,3. Without an assessment of the false discovery rate (FDR) for genome or transcriptome projects, misaligned reads and inaccurate base calls can go unnoticed thereby propagating into SNP discovery. From the onset, datasets comprising sequenced fragments (reads) can harbor a range of potential sequencing errors such as PCR amplification bias in Illumina data4, polyclonal errors in SOLiD data5 and homopolymers in 454 data6. Despite a general trend towards higher success rate at correctly deciphering bases in reads, individual runs can still harbor unexpected levels of error from low quality DNA extractions or library preparations.
In addition to read-quality, an arguably more substantial source of error arises during read-alignment7, the process whereby sequenced reads are mapped to a closely related reference genome. This is the first and most fundamental analysis undertaken once the DNA sequence has been produced8. Alignments are often preferable to de novo assemblies due to increased speed and reduced memory requirements. Given a low read-depth or highly heterogeneous sequence, alignments may also recover more genetic data than assembling without a reference due to current limitations in assembly algorithms9. As is true for de novo assemblies, the accuracy of alignments varies considerably depending on the software and its parameters used10, the type and size of the dataset and the amount of erroneous base calls. Further, alignments are also affected by the genetic distance between reference and newly sequenced genomes.
Most alignment tools score the placement of a read based on the uniqueness of its match. For example, BWA11 and Bowtie12 are frequently used alignment tools that are based on indexing a reference sequence using a Burrows-Wheeler transformation (BWT)13. These tools can align in base (Illumina, 454, Ion Torrent, PacBio) or color space (SOLiD) using a combination of base and alignment mapping quality scores to determine the correct positions of reads and assigning a genotype, whilst ignoring reads containing low quality base calls or low mapping scores. The alignment tool SHRiMP14 is tailored to the platform specific biases in the color-space format of ABI SOLiD reads, using a different set of algorithms to resolve consecutive single nucleotide polymorphisms (SNPs) or miscalled bases, which ‘base-space’ aligners would unsuccessfully determine. Many other examples of short-read alignment tools such as Maq15, SOAP16 and Zoom17 are available and may be preferential given specific experimental requirements such as speed, accuracy or additional features18.
Pre-processing reads prior to alignment by removing low quality reads or 3′ ends is a common initial step to improve alignment accuracy. Sequencing errors can also be detected and removed during the pre-processing of reads using software such as Quake19 or EDAR20, which search the datasets for subsequences that occur in low frequency and therefore likely to be due to sequencing error. Some experiments may also only consider unique matches to the reference sequence. The software Seal21 was recently developed to evaluate alignment tools using simulated reads by testing them for correctly aligned reads, error rates and run-times. However, simulated data will not always correctly recreate the types of systematic biases that may be present within an actual dataset.
More recently, data post-alignment processing is being performed to filter erroneous sites or increase resolution of insertions and deletions (indels). For example, the Genome Analysis Toolkit (GATK)22, which was used by the 1000 Genome Project Consortium found that a local realignment around indels could reduce misaligned reads and nearby false positive SNPs. In addition, GATK uses base and variant quality score recalibrations, whereby the quality scores of reads are re-adjusted according to several covariates over known variant sites as well as use expected transition/transversion ratios (Ti/Tv)22. The option of removing duplicate reads arising from a common progenitor DNA molecule have also been applied as both pre-11 and post-processing steps22, ensuring greater accuracy of subsequent SNP-calling.
Some alignment tools also have inbuilt genotyping such as Maq15, which is able to call SNPs based on a Bayesian statistical model using an expected rate of heterozygotes within the genome and a dependency coefficient. However, most SNP-callers are used post-alignment, such as the UnifiedGenotyper of GATK22, or the Sam/Bcftools' SNP caller23 that considers a number of factors when determining the presence of a polymorphism such as minimum and maximum number of mismatches or filtering by Phred quality scores. Most SNP-callers consider homozygous and heterozygous (bi-allelic) sequences, whilst others such as Bcftools do not properly handle multi-allelic variants and only take the strongest non-reference allele. Many other SNP-callers have been developed, which may be tailored to data-types or expected levels of variation. The number of possible tools and their rate of development make benchmarking an issue that needs to be frequently readdressed.
Increasingly, panels of resequenced isolates include a reference strain for which an assembled genome is already available, in order to determine suitable depth of read coverage for the other isolates and to refine the alignment and SNP-calling parameters to reveal acceptable levels of discrepancies. However, this approach still does not address divergence between the comparison strain and the rates of true positives that will later be called from the alignments. Furthermore, heterozygosity in diploid organisms is still difficult to verify, with few methods developed that do not rely on verification with previously identified sites. Although there have been numerous comparisons of DNA sequence assembly tools for length and accuracy of the contigs, running time, ability to resolve repetitive genomic elements24,25,26 and numerous methods have been proposed for reducing sequencing errors27,5,6 there is currently no standard method for assessing the accuracy of correctly identifying mutations from an alignment and SNP-calling strategy.
Here, we introduce a series of Perl scripts that can reveal and compare the false discovery rates (FDR) for a given NGS dataset used for alignment and SNP calling, requiring only an available reference genome sequence that is closely related to the sequenced strain. Comparing the FDR of methods of alignment and SNP-calling simultaneously can reveal the best combination of tools. In addition to providing a method for determining FDR, we have developed our own tool for calling polymorphisms post-alignment based on cumulative binomial expectations for the number of reads agreeing with a polymorphism, given a depth and an expected error rate. These expectations are stored in lookup tables and used along with the Samtools mpileup format23 as an input. The method is able to identify homozygous and heterozygous mutations with appropriate accuracy. We have benchmarked this method (Binomial SNP Caller from Pileup; BiSCaP) and others, using the scripts for assessing FDR.
Results
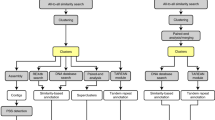
Using fungal NGS datasets and genomes from three separate phyla: Saccharomyces cerevisiae S288C (Ascomycota), Batrachochytrium dendrobatidis (Bd) JEL423 (Chytridiomycota) and Puccinia triticina race 1 isolate 1-1 (Basidiomycota), we assessed the accuracy of the alignment program BWA v0.5.98 with Samtools piped to Bcftools20 by comparing their false discovery rate (cFDR) on 1nt/Kb test SNPs within the coding sequence (CDS) of their corresponding reference genome (workflow shown in Figure 1). We found that these tools resulted in a highly variable accuracy rate for both homozygous SNPs and heterozygous positions dependent on the input datasets, even after normalising the datasets for read length, depth of reads in the alignment and by only considering mutations that were found within the CDS regions (Fig. 2A).

A flow diagram showing the steps to verify false discovery rate and call polymorphic sites using cFDR and BiSCaP.

False discovery rates for variants were ascertained using cFDR for three fungal NGS datasets.
(A) Dataset-specific error rates were identified for both homozygous SNPs and heterozygous positions after alignment with BWA and calling SNPs using SAM/BCFtools with default settings, which persisted after trimming to 30 mers, aligning random 10× deep subsets of aligned reads and only considering SNPs that fell over CDS regions thereby reducing genome size as a factor. (B) Combinations of alignments and SNP-calling methods resulted in different accuracies from the Bd JEL423 30 mer NGS dataset. Experimenting with a variety of methods can therefore reveal the most suitable method for a given dataset based on these metrics of accuracy. Sam/Bcftools only takes the strongest non-reference allele so was not included in the assessment of heterozygous accuracy. Parameters include the percent cut-off for inclusion as a SNP or heterozygous base and minimum depth (MD).
Pre-processing input datasets was briefly assessed for ability to improve the accuracy of downstream SNP calls. Specifically, trimming 3′ ends from all Bd JEL423 reads increased the number of reads that were aligned using BWA nearly 5 fold from 2.2 million to 10.4 million, in turn, enabling > 4 times the number of true positives SNPs and heterozygous positions to be called by Sam/Bcftools. Furthermore, read-trimming decreased the percent of false positives SNPs from 15.6% to 5.9% and false positive heterozygous positions from 14.4% to 3.4%, demonstrating how much error can and was present within this NGS dataset, often at greater frequencies towards the 3′ ends of reads. Trimming the reads only achieved an improvement in the number of true positive and false positives with the Bd JEL423 reads and conversely reduced both the number of false and true positives in the P. triticina and S. cerevisiae datasets. The variation between each of these datasets demonstrates the importance of assessing quality control as a preliminary step for resequencing projects.
To demonstrate how alignment and SNP-calling varied, we compared combinations of methods and parameters on the Bd JEL423 SOLiD 30-mer dataset (Fig. 2B). Owing primarily to the high levels of sequencing errors, even after removing low quality 3′ ends, none of the tested methods called > 73% true positives SNPs or < 5% false positive SNPs. The alignment program SHRiMP, which is specifically designed for the ‘color-space’ reads of SOLiD sequencing aligned 60% of the Bd reads and 70% after trimming to 30 mers, compared with BWA that aligned just 9% full length and 41% 30 mers. Despite the differences in the output alignment depth between the two programs, BWA (with the Bd 30 mers) resulted in a greater number of true positives and approximately equal number of false positives than SHRiMP called using either read length and any of the SNP-calling methods we tested. This comparison shows that BWA is a more appropriate tool for this particular dataset. The comparably low false discovery rate and number of reads aligned by BWA on the SOLiD Bd dataset was not found to the same extent in the Illumina Sc and Pt datasets, where > 59% full-length reads and > 68% trimmed reads were aligned to their reference sequence. For heterozygous base-calls, GATK2 had the greatest accuracy of any of the methods tested with 86.78% true positives and remarkably not a single false positive.
A comparison of the FDR for SNPs achieved by the alignment program BWA and the SNP calling method presented here (BiSCaP) using default settings on the Bd 30-mer dataset revealed 6582/12458 (52.83%) true positives and 494 (6.98%) false positive SNPs that were covered ≥ minimum required depth (MD). This result is a considerably preferable outcome compared to using SAM/Bcftools for SNP-calling (as shown in Fig. 2B), whilst the UnifiedGenotyper of GATK performed competatively in terms of specificity at a small expense in sensitivity compared with one of the tested settings of BiSCaP (MD 4). This variation of FDR demonstrates the importance of comparing candidate tools for a given study. Of the false positives called by BiSCaP, 328 were identified when aligning to the non-modified reference genome (351 total) and 321/328 were covered by 100% uniquely aligned reads, suggesting that they may be real genetic changes occurring between the separate batches of isolate Bd JEL423, or genuine mistakes in the reference genome.
The remaining 45.83% of modified positions in Bd that were not identified, consisted of 98.5% that were uncovered by the minimum depth, 1.47% called as heterozygous or ambiguous if haploid setting is used and a single incorrectly called homozygous SNP. Of the 84 bases that were incorrectly identified as heterozygous, 83 sites correctly called the modified base but also inferred the presence of the reference allele' and one consisted of the reference base and a deletion. None of these false positives were identified without first modifying the reference for cFDR, which may suggest they arise from misaligned reads. These results demonstrate that by far the greatest impact on alignment/SNP-calling error on the Bd 13.4× deep dataset arises from lack of coverage. Surprisingly however, the S. cerevisiae 67.9× deep dataset (Fig. 1A) also revealed a similar situation where 1920/1929 of the false negatives were due to below required depth compared to the remaining 9 that were incorrectly called as heterozygous positions.
To explain this pattern of enrichment for uncovered polymorphic sites (Bd and S. cerevisiae had 98.4% and >99.9% CDS covered respectively, compared to 45.8% and 15.7% of the introduced mutations within the CDS), we compared the distances between the introduced SNPs (Fig. 3A). Firstly, we found that Sc and Pt tended to have more closely associated introduced mutations (both had 19% occurring within 20 nt of each other) than Bd (only 2% within 20 nt). In terms of number of contigs and genes, number and average length of exons and length of coding and introns sequence, Bd is situated between Pt and Sc. However, a clear difference is found in the number of genes selected by IRMS for introducing mutations in: 953, 1976 and 5701 for Sc, Pt and Bd respectively, which could be caused by one or more computational or biological differences such as differing numbers of genes specified with overlapping exons or splice variants specified in the feature file, which IRMS excludes. In any case, the difference in the number of modified genes likely explains the difference in distance between mutations. Despite this, false negatives were predominately more closely associated in Bd and Pt, whilst false negatives in Sc appeared to not be so clearly correlated with distance from other mutations.

Erroneous base calls (homozygous SNPs) from BiSCaP and percent cutoff methods were compared for proximity and depth of read coverage using full dataset 30 mer Bd, Sc and Pt reads.
(A) False negatives were predominantly more closely associated for all 3 datasets (SNPs called with BiSCaP). False negatives were almost entirely caused by lack of coverage in each of these datasets demonstrating the most divergent part of each of the genomes is the most poorly resolved. (B) Homozygous errors were more frequently called over lower depth regions using strict cut-off methods than with BiSCaP.
We also looked at minimum depth cut-off points for different numbers of reads agreeing or disagreeing for a particular base, a parameter often taken into account by mutation callers, but compounded by variable depths over each of base in the genome. To examine this issue, we looked at the depth of read coverage over false positives and false negatives called using the percent cut-off methods and BiSCaP, which uses the depth-dependent cut-offs (Fig. 3B). As expected, homozygous erroneous base calls using either BiSCaP or percent cut-off methods were more frequently called over lower read depth positions in the alignment. BiSCaP achieved fewer false positive/negatives over the lowest read depths for Bd and Sc and also over bases covered by more than 5 reads. The dataset Pt had fewest false base calls with the 90% cut-off method, which again highlights that different algorithms are suitable for different datasets and only after testing them for FDR could you be certain of the success rate, or determine the most appropriate method.
Discussion
The rapid rate of increase in large-scale population studies using genome resequencing for SNP detection necessitates the development of improved tools to assess the quality of resequencing projects. Here we describe the development and efficacy of two such tools. We have tested the comparison of false discovery rate (cFDR) and Binomial SNP Caller from pileup (BiSCaP) scripts on sequence data from fungal genomes from three separate phyla: Saccharomyces cerevisiae S288C (Ascomycota), Batrachochytrium dendrobatidis (Bd) JEL423 (Chytridiomycota) and Puccinia triticina race 1 isolate 1-1 (Basidiomycota). These fungal genomes were chosen to represent a range of genome sizes and structures in terms of introns numbers, repeat richness and sequence heterogeneity. These methods can be applied to any resequencing study regardless of taxon. A large number of similar alignment, SNP-calling tools or pre-processing methods could have also been tested using these FDR scripts in addition to those we have tested here (BWA11, SHRiMP14, GATK22, SAMTools23).
To identify the factors involved in the FDR variation between the three datasets composed of equal alignment depth, length and modified genetic distance to its reference sequence, we extracted the positions corresponding to true and false positives. A large amount of variation between datasets, alignments and SNP calling accuracy was identified using these tools and was used to identify the most suitable combination of methods to accurately detect variants. Similar approaches to the generalised tools we present here have already been used by a number of NGS projects28,29 and facilitated by the release of these packages, a wider range of projects could also make use of either, or both cFDR and BiSCaP. Each combination of methods performed differently on the Bd JEL423 genome with one simulated divergence rate and from this test we could decide a single set of methods and parameters that performed optimumally. However, FDR validation and SNP-calling should be readdressed for every new dataset. For example, GATK would be most suitably benchmarked against a dataset for which a training set of known variation is available. However, even without variant quality score recalibration, GATK performed well on the fungal JEL423 genome, in particuar over bi-allelic heterozygotes.
Alternative methods for assessing homozygous variants that rely largely on simulated reads21 or cross checking databases of polymorphisms30 to determine FDR would have less power than our method in terms of realistic read and alignment error, or relying on resources that harbour their own sources of error. The alignment tool Maq15 includes a command for introducing mutations into a reference sequence, with the intent of assessing alignment accuracy from simulating reads. Our method is able to make use of and assess real read data, which can harbor any number of platform or non-platform specific errors that may influence ability to call SNPs, in contrast to simulated reads or use of databases. This feature makes our method the only currently available technique to simultaneously assess the quality of data generated and the quality of methods used to analyze those data.
BiSCaP has been designed for variant-calling across haploid, diploid or triploid sequences, with corresponding binomial probabilities provided. In its current form, BiSCaP takes longer than the other assessed SNP-callers to complete: roughly one hour on a desktop computer on a modestly sized genome and dataset (the 13.4× Bd dataset to the 23 Mb genome), which is likely to persist until scripts are converted into a lower level programming language. The FDR method is also able to verify heterozygous alleles called by either GATK or BiSCaP using simulated reads and the cFDR scripts finish running within a few minutes using the ouput from either of these (and SAMTools) SNP-calling tools. BiSCaP is able to call polymorphisms from standard input and output formats, making it a versatile tool for projects utalising these formats. We have not assessed how quality scores could be used to improve accuracy of those SNP-calls although both GATK and BiSCaP are able to filter potential SNPs based on these scores, so could be incorporated into the analysis.
Each of the methods presented here rely on a reference genome strain that is both high quality in terms of accurately assembled and with correct base calls and has few discrepancies to the consensus (resequenced) isolate. For example, more distantly related isolates (reference and consensus) will result in a greater number of ‘false positives’ called by cFDR. Furthermore, if the reference sequence is poorly resolved (missing sequence or low quality or repetitive areas), the method may identify genuine polymorphisms that will also be considered false positives. This limitation can be partially resolved using a separate quality control measure for those false positives using either quality scores or called without first modifying the reference.
We found the ideas for and implementation for SNP-calling based on cumulative binomial probabilities a suitable method for determining polymorphisms from an alignment. We tested both of these methods on three unique fungal pathogens, each of which are thought to be predominantly diploid and therefore had both homozygous and heterozygous polymorphic positions called, which we found homozygous polymorphisms using the cFDR scripts to have a high level of accuracy. Either or both of these tools could be used with any other sequenced panel of diploid or haploid isolates to gauge the accuracy of alignment and SNP-calling.
Methods
The genome sequence and feature files for Saccharomyces cerevisiae S288C were downloaded from the SGD on 31.3.11 (http://www.yeastgenome.org/). Genomes of Puccinia triticina race 1 isolate 1-1 and Batrachochytrium dendrobatidis (Bd) JEL423 were downloaded from the Broad Institute (http://www.broadinstitute.org/). Illumina reads were obtained from the Short Read Archive under accessions SRR003681 and SRA009871 for S. cerevisiae S288C and P. triticina respectively. We previously resequenced the genome of Bd JEL42329 using SOLiD, which is available for download in the Short Read Archive under accession SRA030504. Genome sequences were modified by randomly choosing and modifying 1 nt/Kb within the coding sequence (CDS) using a script that is part of the toolset (Introduce Random Mutation into Sequence; IRMS.pl).
SRA files were converted to FASTQ and aligned to their modified reference genome sequences using BWA v0.5.911 with default parameters and SHRiMP v211 with an 80% identity threshold for read alignment. Pileups were made using SAMTools v0.1.1823 and polymorphisms called using the mpileup command piped to Bcftools v0.1.17-dev and filtered using vcfutils.pl with default parameters. In order to assess heterozygous variants, we randomly chose and modified 1 nt/Kb within the CDS using the “HET” setting of IRMS.pl, which first generates a duplicate (homologous) genome. We then simulated single-end reads from these modified sequences to the same depth as the ‘real’ data using simLibrary and simNGS (http://www.ebi.ac.uk/goldman-srv/simNGS/) using the default runfile (s_3_4x), which describes how “noise and cluster intensities are distributed in a real run of an Illumina machine” and aligned those reads to the non-modified reference genomes.
The Genome Analysis Toolkit (GATK) v2.1-922 was assessed according to the "Best Practice Variant Detection with the GATK v4, for release 2.0" detailed on the Broad Institute website. Briefly, Picard Tools v1.68 (http://picard.sourceforge.net) was first used for marking duplicates. Indel-realignment was performed using the GATK2 RealignerTargetCreator and IndelRealigner tools. Next, the UnifiedGenotyper was used to output raw variants that were used for base quality score recalibration (BaseRecalibrator and PrintReads). The UnifiedGenotyper was then assessed using default parameters on the new BAM file. Without a training dataset, variant recalibration is still considered experimental, so this step was left out for each of the three fungal genomes. Percent cut-offs with variable minimum read-depths and using the Binomial SNP caller from Pileup (BiSCaP) v0.11 (presented here) were also used to call polymorphisms. Each alignment/SNP-calling combination was assessed for accuracy using the Comparison of False Discovery Rate script (cFDR).
BiSCaP is based on the binomial expectations for the number of reads agreeing with a reference base over a given locus. These expectations allow for polymorphisms to be called with different levels of leniency for sequencing errors dependent on the depth of read coverage and for heterozygous positions to be called without a bias for the read depth. Briefly, an expected alignment and base calling error rate (e.g. 0.1 or 0.01) is used to generate a list of binomial probabilities p for sequencing and aligning k number of correct bases, given a read depth of n (number of trials) or f (k; n, p). The probability a base is homozygous (h1) can be considered as P (1 – error rate). In a diploid, the probability of a heterozygous base (h2) can be considered as P ((h1/2) + ((error rate * 0.5)), where half the sequencing or alignment ‘errors’ are now specifying the two correct bases. Equally, a heterozygous allele in a triploid sequence (h3) can be considered as either P ((h1/3) + ((error rate * 0.25)) or P ((h1/3) * 2) + ((error rate * 0. 5)). From these, a cumulative h1 probability for the lower tail can be calculated (ch1) in addition to the minimum values found from the cumulative h2 or h3 upper tail and the cumulative h2 or h3 lower tail (ch2 and ch3 respectively). Binomial values are generated by the script GBiD.pl (Generate Binomial Distributions) and stored in a lookup table. Pre-calculated tables for error rates of 0.1 and 0.01 up to a read depth of five hundred are provided in the current version. BiSCaP then uses one such look up table to infer the most probable consensus nucleotides from the alignment.
Briefly, the algorithm for determining the consensus sequence is to tally each of the four possible aligned bases, each of which needs to be ≥ the minimum read depth to be considered a consensus allele. The most common base is considered homozygous where ch1 ≥ error rate and ch1 > ch2. The most common and 2nd most common base are considered heterozygous where ch2 ≥ error rate and ch2 > ch1 for both bases. A triploid heterozygous site is considered when the ch3 for each of the three most common bases ≥ error rate. Indels are treated separately but using the same criteria and sub routine.
BiSCaP by default provides details of polymorphic sites in Variant Call Format (VCF)31 and can also output pileup lines into separate files based on the identified mutation-type. Other optional parameters include different lookup table based on error rate, minimum read depth, ploidy, stringency for heterozygous SNP calling and a Phred quality score filter. If the read depth is greater than the lookup table depth (default 500 for error rates 0.1 and 0.01), reads up to the maximum lookup table depth can be used to determine the genotype (default), or printed to a separate file named ‘outside-distribution’. The cFDR script considers Percent True Positive (TP) homozygous SNPs as ((N° TP hom. SNPs/N° Introduced mutations) × 100) and the Percent False Positive (FP) homozygous SNPs as ((N° FP hom. SNPs/(N° TP and FP hom. SNPs)) × 100).
References
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Kiezun, A. et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet., 44, 623–630 (2012).
Aird, D. et al. Analyzing and minimizing PCR amplification bias in Illumina Sequencing libraries. Genome Biol. 12, R18 (2011).
Sasson, A. & Michael1, T. P. Filtering error from SOLiD Output. Bioinformatics 26, 849–850 (2010).
Huse, S. M., Huber, J. A., Morrison, H. G., Sogin, M. L. & Welch, D. M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 8, R143 (2007).
Landan, G. & Graur, D. Characterization of pairwise and multiple sequence alignment errors. Gene 441, 141–7 (2009).
Flicek, P. & Birney, E. Sense from sequence reads: methods for alignment and assembly. Nature Methods 6, S6-12 (2009).
Alkan, C., Sajjadian, S. & Eichler, E. E. Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65 (2011).
Frith, M. C., Hamada, M. & Horton, P. Parameters for accurate genome alignment. BMC Bioinformatics 2010, 11, 80 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25 (2009).
Burrows, M. & Wheeler, D. J. A block-sorting lossless data compression algorithm. Technical report 124. Palo Alto, CA: Digital Equipment Corporation (1994).
Rumble, S. M. et al. SHRiMP: Accurate Mapping of Short Color-space Reads. PLoS Computational Biology 5 (2009).
Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–8 (2008).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–7 (2009).
Lin, H., Zhang, Z., Zhang, M. Q., Ma, B. & Li, M. ZOOM! Zillions of oligos mapped. Bioinformatics 24, 2431–7 (2008).
Li, H. & Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics 11, 473–483 (2010).
Kelley, D. R., Schatz, M. C. & Salzberg, S. L. Quake: quality-aware detection and correction of sequencing errors. Genome Biology 11, R116 (2010).
Zhao, X. et al. EDAR: an efficient error detection and removal algorithm for next generation sequencing data. J. Comput. Biol. 17, 1549–60 (2010).
Ruffalo, M., LaFramboise, T. & Koyutürk, M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 27, 2790–6 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009).
Farrer, R. A., Kemen, E., Jones, J. D. & Studholme, D. J. De novo assembly of the Pseudomonas syringae pv. Syringae B728a genome using Illumina/Solexa short sequence reads. FEMS Microbiol. Lett 1, 103–111 (2009).
Miller, M. J. & Powell, J. I. A quantitative comparison of DNA sequence assembly programs. J. Comput. Biol. 1, 257–69 (1994).
Zhang, W. et al. A Practical Comparison of De Novo Genome Assembly Software Tools for Next-Generation Sequencing Technologies. PLoS One 6 (2011).
Salmela, L. Correction of sequencing errors in a mixed set of reads. Bioinformatics 26, 1284–90 (2011).
Raffaele, S. et al. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science. 330, 1540–3 (2010).
Farrer, R. A. et al. Multiple emergence of genetically diverse amphibian-infecting chytrids include a globalised hypervirulent lineage. Proc. Natl. Acad. Sci. U.S.A. 108, 18732–6 (2011).
Musumeci, L. et al. Single nucleotide differences (SNDs) in the dbSNP database may lead to errors in genotyping and haplotyping studies. Human mutation 31, 67–73 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–8 (2011).
Acknowledgements
Funding: R.A.F. was funded by the Natural Environment Research Council (NERC). D.A.H. and M.C.F. were supported by the Wellcome Trust. No additional external funding received for this study.
Author information
Authors and Affiliations
Contributions
R.A.F., D.A.H. and M.C.F. wrote the main manuscript text. R.A.F. prepared the figures. R.A.F., D.M. and D.J.S. conceived of the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Farrer, R., Henk, D., MacLean, D. et al. Using False Discovery Rates to Benchmark SNP-callers in next-generation sequencing projects. Sci Rep 3, 1512 (2013). https://doi.org/10.1038/srep01512
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01512
This article is cited by
-
HaplotypeTools: a toolkit for accurately identifying recombination and recombinant genotypes
BMC Bioinformatics (2021)
-
Next-Generation Sequencing and Its Impacts on Entomological Research in Ecology and Evolution
Neotropical Entomology (2021)
-
Comparison of the Chinese bamboo partridge and red Junglefowl genome sequences highlights the importance of demography in genome evolution
BMC Genomics (2018)
-
Genomic innovations linked to infection strategies across emerging pathogenic chytrid fungi
Nature Communications (2017)
-
Cascade: an RNA-seq visualization tool for cancer genomics
BMC Genomics (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
