
Abstract
With modern scanning probe microscopes, it is possible to manipulate surface structures even at the atomic level. However, manipulation of nanoscale objects such as clusters is often more relevant and also more challenging due to the complicated interactions between the surface, cluster and apparatus. We demonstrate the manipulation of nanometer scale gold clusters on the NaCl(001) surface with a non-contact atomic force microscope and show that the movement of clusters is in certain cases constrained to specific crystallographic directions. First principles calculations explain this kinetic anisotropy as the result of the cluster attaching to surface defects: cation vacancies allow the clusters to bond in such a way that they only move in one direction. Constraining the movement of clusters could be exploited in the construction of nanostructures or nanomechanical devices and the manipulation signatures may also be used for identifying cluster-defect complexes.
Similar content being viewed by others
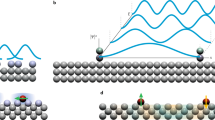

Introduction
Controlling structures at the atomic level is the ultimate challenge in nanofabrication - absolute control over the positions of individual atoms, molecules, or clusters on substrates allows for nearly complete freedom in material composition or in the fabrication of nanodevices. Although arranging structures atom-by-atom is attractive from the perspective of fundamental interest in surface physics and chemistry, for many applications manipulation of larger units is more relevant. With respect to metallic clusters, interest stems from catalysis where clusters promote many catalytic reactions1,2,3, but also from nanotechnology where clusters can be used as building blocks for nanoelectronic devices4. In tribology, clusters can even serve as units for probing the friction at the nanoscale5,6. In all these domains, manipulating clusters as single objects enables the systematic study of the impact of the specific and voluntarily chosen adsorption site on the physical, mechanical, electronic and chemical properties of a single cluster7. While manipulation of atoms was achieved already in 1990 with scanning tunneling microscope (STM)8, atomic manipulation with non-contact atomic force microscopy (nc-AFM)9 has been a much more recent development10. In particular, manipulation on insulating surfaces remains particularly challenging11,12, yet insulators are a crucial class of materials in nanoelectronics and nanocatalysis.
Similar to STM13, two methods are used to manipulate clusters by nc-AFM: the first method consists of moving the cluster by pushing during a spectroscopy step (Fig. 1 (a)). Here, the AFM tip is brought down vertically near the cluster until at some point the repulsive interaction between the tip and the cluster is strong enough to make the cluster move. In the second method the tip is scanned above the cluster (Fig. 1 (b)), preferentially in the constant height AFM mode14,15,16. At a specific distance, as the tip approaches the cluster laterally, a force between both moves the cluster. For simplicity, we will call the former method kicking and the latter sliding.

Principles used in the manipulation of clusters.
(a) the cluster is moved by kicking during a spectroscopy step; (b) moving the cluster by sliding during scanning.
This work investigates methods for directing the manipulation of a nanocluster beyond the constraint of the tip's scanning direction. In particular we demonstrate the possibility of using intrinsic defects on the surface to mediate the diffusive behaviour of the clusters. We have chosen the NaCl(001) surface as a model insulator substrate firstly because it can be easily prepared and examined by nc-AFM even at the atomic scale17, but also because the characteristic defects at it's surface are well-established18. Furthermore, NaCl and other alkali halide surfaces have been key in breakthrough nc-AFM studies in atomic19,20,21 and charge identification18,22, as well as providing prototype substrates for the study of adsorbed molecules23,24,25, self-assembly26, friction27,28 and local electronic structure29,30.
Results
Manipulation on terraces
When single neutral gold atoms are deposited on perfect NaCl(001) surfaces, the gold atoms diffuse on the terraces due to their high mobility at room temperature31,32,33 until they reach a step-edge, where they aggregate into clusters1,34. At room temperature and low gold coverage, the steps are decorated by gold clusters whereas on the flat terraces very few clusters can be found because of the lack of surface defects that would act as nucleation sites (see further discussion in the Supplementary information). The image in Fig. 2 (a) exemplifies a typical decoration of steps (along 〈100〉 equivalent directions) by gold clusters. The clusters have a mean diameter of 5 nm (~2500 atoms), which was estimated from the flux of incoming gold during the deposition.

Manipulation of a Au cluster (green) on NaCl(001) along the [110] and [100] directions (nominal Au thickness: 0.3 mono-layers, flux: 4.1 × 1012 atoms/cm2/s).
(a) The surface region before the manipulation (density: 0.9 × 1011 clusters/cm2, cluster-cluster mean distance at steps: ~ 20 nm). (b) – (d) The cluster before (b), after the first (c) and after the second (d) manipulation experiment. (e) During the first manipulation, a part of the cluster could be still imaged until the position labeled by the arrow where the cluster suddenly started to escape behind the tip (backheel). (f) During the second manipulation step, the cluster was moved by sliding along the [110] direction. (g) Schematic of the two manipulation steps. (a) 250 × 250 nm2, Δf = −22 Hz, (b, c, d) 65 × 136 nm2, Δf ≈ −22; −18; −20 Hz, (e) 40 × 40 nm2, Δf = −48 Hz, (f) 30 × 26 nm2, Δf = −69 Hz, All images: f0 = 275 kHz, vScan = 10 Hz (e: 24 Hz, f: 33 Hz), k = 28 N/m, App = 15 nm.
In this first series of manipulation experiments, we focus on one of the rare clusters on the terrace (highlighted in green in Fig. 2) and consider its manipulation characteristics. The cluster was first imaged with a high magnification in the constant height mode (Fig. 2 (b)). In the figures, the slow scanning direction of the AFM tip is always from the bottom to the top. The same cluster was imaged several times while gradually bringing the tip closer to the surface. At a specific tip-surface distance the cluster suddenly disappeared during imaging (arrow in Fig. 2 (e)) by escaping behind the tip to the region that had just been imaged to the position shown in Fig. 2 (c). This kind of backheel led to a 16 nm long movement of the cluster along the [100] direction. In a second manipulation step the same cluster was imaged again and the tip-surface distance was further reduced. Now, the cluster could be moved by sliding in front of the tip along the [110] direction (Fig. 2 (f)). Because the cluster is moved in front of the tip from line to line, it appears with an elongated contrast, reflecting its movement. The new position of the cluster, which is 11 nm further away from the old one, can be seen in Fig. 2 (d).
Manipulation from step-edges
In Fig. 3, similar clusters of about 2000 atoms decorate intersecting 〈110〉 and 〈100〉 steps on the NaCl(001) surface. One of the clusters is highlighted (I in Fig. 3 (a), green color) at the 〈110〉 step-edge. After recording a series of high resolution images at this site, the last image of this series shows that the cluster was moved 12 ± 1 nm from the step (to position II in Fig. 3 (b)). We continued to move the same cluster, now on the terrace, by sliding about 15 ± 1 nm along the 〈110〉 direction (Fig. 3 (c)), which has been verified in a following image with a larger scale (Fig. 3 (d)). We could move the cluster also by kicking with a vertically approaching tip in a spectroscopy step. Figure 3 (e) shows that in this case the cluster could also be moved by 16 ± 1 nm from its initial position. Altogether, the cluster was moved by the tip dozens of times during the whole manipulation process. Regardless of the method of manipulation, the events always resulted in cluster movement along equivalent 〈110〉 directions on the surface (see path of cluster in Fig. 3 (f)). This extremely anisotropic movement is in clear contrast to the cluster found on the terrace, which could easily be moved in different directions.

Experimental constant height images presenting several manipulation steps of Au clusters on NaCl(001) (nominal thickness: 0.64 mono-layers, flux: 1.0 × 1013 atoms/cm2/s).
(a) Decoration of NaCl steps by Au clusters (cluster-cluster distance: 8.4 nm, cluster density: 4.6 × 1011 clusters/nm2). (b) A single cluster (green) is separated from the step during a scan with the AFM. The dashed circle represents the position of the manipulated cluster in the previous image. (c) Controlled manipulation of the cluster by sliding. (d) The result after the movement. (e) Controlled manipulation of the cluster by kicking in a spectroscopy step. (f) Schematic of the manipulation steps. (a) 88 × 88 nm2, Δf = −52 Hz, (b, d, e) 54 × 51 nm2, Δf ≈ −54 Hz, (c) 20 × 20 nm2, Δf = −120 Hz, All images: f0 = 247 kHz, k = 32 N/m, vScan ≈ 15 Hz, App = 14 nm.
Calculated manipulation kinetics
In order to understand the movement of clusters during manipulation and pinpoint the origin of the experimentally observed differences, we sample the possible pathways for moving 20 and 100 atom gold clusters (Fig. 4 (a)) using density functional calculations. With the smaller Au20 cluster we perform extensive analysis on the adsorption configurations and diffusion pathways while the larger Au100 cluster is used in selected cases to assess scaling with respect to cluster size.

Calculated diffusion barriers for various cluster-vacancy structures in eV.
(a) The Au20 and Au100 clusters used in calculations. (b) Au20 without defect (c) Au20 on a Cl vacancy (d) Au20 on a Na vacancy (e) Au100 on a Na vacancy (vacancy at the energetically favoured edge site). (f) The 〈110〉 directions are set to run horizontally and vertically in the figures. Positions of Na and Cl vacancies (VNa and VCl) are denoted by dark and light squares, respectively.
The Au20 cluster is found to adsorb on the perfect surface (adsorption energy 0.5 eV) and the energy barriers for moving the cluster in any given direction are very low, about 0.2 eV (Fig. 4 (b)). At room temperature, such a diffusion barrier corresponds to a Boltzmann factor of 10−4 and thus such a small cluster should move spontaneously due to thermal energy. Furthermore, the barriers for moving the cluster in either 〈100〉 or 〈110〉 directions are practically the same and thus no direction should be preferred.
The calculated high mobility on the ideal terrace strongly implies the presence of a defect under the cluster in Fig. 2 — else the cluster would have diffused to a step during growth. Hence, we also consider the role of defects in cluster adsorption and diffusion. The calculations show that in the presence of surface vacancies, or F-centres, the cluster adsorbs even more strongly than on the perfect surface, as the adsorption energies are 2.1, 2.5 and 1.1 eV for adsorption at neutral Cl, Na and double vacancies, respectively. (Defect charge state influence is discussed in the Supplementary information.) Also the diffusion energy barriers are much higher when defects are present, as discussed below, meaning it is much more difficult to move the cluster and this verifies that defects can act as anchors for the clusters.
In the presence of a Cl vacancy, the barriers for moving the cluster are 0.8 – 1.1 eV and the anisotropy is relatively weak (Fig. 4 (c)). A cluster bonded to a Na vacancy, on the other hand, prefers a [110] direction where the barrier is calculated to be 0.7 eV. In comparison, the 〈100〉 barriers are 0.9 eV and in the perpendicular [ ] direction the barrier is 1.2 eV (Fig. 4 (d)). Similar calculations with a 100 atom gold cluster show that this is still the case even when the clusters grow. Figure 4 (e) shows the calculated barriers for the Au100 + Na vacancy complex. When the vacancy is located near the cluster edge, in the energetically favored position, the anisotropy is even stronger than for the small cluster (barriers 0.5 – 1.1 eV).
] direction the barrier is 1.2 eV (Fig. 4 (d)). Similar calculations with a 100 atom gold cluster show that this is still the case even when the clusters grow. Figure 4 (e) shows the calculated barriers for the Au100 + Na vacancy complex. When the vacancy is located near the cluster edge, in the energetically favored position, the anisotropy is even stronger than for the small cluster (barriers 0.5 – 1.1 eV).
Discussion
The charged Na+ and Cl− vacancies (latter are F+-centres) are expected to be mobile on the NaCl(001) surface and diffuse to steps during annealing35,36. Recent nc-AFM and Kelvin Probe Force Microscopy (KPFM) studies have further shown that Na+ vacancies are the dominant defect at step-edges and kinks18,37, which is due to the Debye-Frenkel layer formation on alkali halide (001) surfaces38,39. This suggests that a cluster taken from a step, as shown in Fig. 3, is with a high probability anchored by a Na+ vacancy. The experimentally observed kinetic anisotropy of these clusters can then be explained as the characteristic behaviour of this particular cluster-defect complex, as revealed by the simulations. The third type of possible defect on NaCl(001) is the F0-center (Cl− vacancy + electron, i.e., a Cl0 vacancy) and these defects are expected to be stable on the terrace (calculated diffusion barrier for the defect is 0.9 eV). Therefore, they are also the most probable defect on which clusters found on terraces are adsorbed (as in Fig. 2). From this consideration, the theoretically predicted small diffusion anisotropy for a cluster adsorbed on a Cl vacancy agrees with the experimentally found isotropic movement of the cluster on the terrace.
The reason why the defects influence the movement of clusters differently can be understood by examining the calculated structures of the bonded cluster-vacancy pairs. In Fig. 5 (a) and (b), the differential charge densities are shown for clusters in the presence of Cl and Na vacancies, respectively. This density represents the change in electronic charge upon bringing the cluster and the surface together40. In the presence of a Cl vacancy, the Au cluster bonds almost exclusively at one of its corners, attaching to a Cl ion next to the vacancy. In fact, the cluster tilts 10° as the bonded corner is attracted to the Cl ion. Because of this, the cluster prefers to move by pivoting around the bonded corner, as schematically shown in Fig. 5 (c) (although the preference is not strong, as shown by the almost isotropic barriers in Fig. 4 (c)). Simultaneously, the Cl vacancy jumps to a neighboring site, allowing the cluster to bond to another corner. Combining these rotations leads to movement in the 〈100〉 direction, but since the cluster is constantly reorienting itself, on average none of the 〈100〉 surface directions is preferred and the cluster may in fact move in any direction by taking a zig-zag path.

Differential charge densities ρAu+NaCl − ρAu − ρNaCl plotted at isosurfaces −0.01 eÅ−3 (grey) and 0.01 eÅ−3 (orange) for Au20 on a Cl vacancy (a) and on a Na vacancy (b).
The [110] direction runs into the image. Schematic representation of the preferred diffusion modes for the Au20 on a Cl vacancy (c) and a Na vacancy (d), with the [110] direction of the NaCl running vertically as shown in (e). Differential charge density for Au100 on a Na vacancy (f) at the same isosurfaces as in (a) and (b). Rendered using VMD57.
On the other hand, if the cluster attaches to a Na vacancy, it does not bond at a corner, but along an edge which orients itself with an entire row of Cl ions on the surface. This is seen in Fig. 5 (b). Again, the cluster is seen to slightly tilt (5.1°) towards the active edge to facilitate the bonding. For such a configuration, the energetically favored mode of movement of the cluster is sliding along the row of Cl ions with which it has bonded, so that the active edge always stays on the Cl row. This is represented in Fig. 5 (d). Since this movement follows a single row of Cl ions, the cluster moves in a [110] direction. Notably, since the cluster cannot move perpendicularly to the Cl row without detaching or rotating first, a much more difficult task than sliding in the preferred direction, this configuration results in truly anisotropic movement. Exactly the same kind of attaching to a Cl row occurs with a larger cluster as shown in Fig. 5 (f), demonstrating that this behaviour does not depend on the cluster size.
There is a discrepancy between the cluster sizes in the presented theory and experiments (100 and 1000 atom clusters, respectively). Although such a difference in the number of atoms only means a linear size difference by a factor of two, difference in size at this scale may lead to changes in the cluster morphology. On ionic surfaces like NaCl(001) or MgO(001), gold clusters expose either their (001) or (111) facets to form rectangular or triangular shapes, respectively and the shape may depend on the size and growth conditions44,45. However, as our simulations predict the anisotropic movement for the cluster–vacancy complex to be due to bonding of low-coordinated gold atoms at the cluster edge with Cl ions on the substrate near a Na vacancy, the anisotropy should not depend on the size and exact shape of the cluster. Instead, the behaviour is a characteristic feature of this kind of cluster–defect pair. We therefore expect the same characteristics also for the larger clusters, as seen in the experiments.
To summarize, we experimentally demonstrate nanomanipulation of gold clusters roughly of the size of 2000 atoms (5 nm diameter) as single units on the terraces of the insulating NaCl(001) surface using nc-AFM. From first principles calculations we show that stable clusters on the terrace are expected to be adsorbed at defects, which always accompany a cluster during its movement on the surface. Since the NaCl(001) surface only has vacancies as potential defect candidates at the surface, the manipulation properties are dominated by the type of vacancy: clusters on cation vacancies exhibit an anisotropic movement along the 〈110〉 surface directions whereas clusters on anion vacancies do not. Especially with respect to the cation vacancies, which are known to be located at the step edges, theory perfectly agrees with experiments since clusters that are detached from steps by the tip move indeed along 〈110〉 surface directions.
In general, this work demonstrates both experimentally and theoretically that even on smooth surfaces cluster-defect pairs can have extremely anisotropic movement patterns and that this anisotropy can be strong enough to be the dominant factor controlling their manipulation. Such constrained movement can be exploited when building structures through nanomanipulation or even in the design of nanomechanical machines. Also conversely, observing specific anisotropy patterns could be used for distinguishing the types of defects on which clusters are adsorbed. Of course, this is not necessarily a one-to-one mapping: in a system where different kinds of defects exist, it is possible that different cluster-defect complexes have very similar manipulation patterns. Still, examining the cluster movement may allow one to distinguish the most likely defect types, especially if the intrinsic occurrence probabilities of defects are known.
Aside from providing an important characterization of defects, the ability to control and create defect-cluster complexes offers great potential for future studies in catalysis where, for instance, the cluster reactivity must be studied as a function of adsorption site. Such manipulation experiments can be combined with a control over the defects, which can be created by e.g. the AFM tip followed by imaging and spectroscopy identification, or by doping or irradiation of the substrate material such that known defects appear on the surface. When manipulation can be used for an overall characterization of the defect-cluster system, it will be a powerful tool for linking observed properties with the atomic and electronic structure of the defect-cluster complex.
Methods
Sample preparation
The undoped NaCl crystals are grown in an argon atmosphere by the Czochralski growth method in our laboratory at the CINaM41. Before surface preparation, crystals and sample holders are cleaned by annealing (~250°C) in an oven located inside the main UHV chamber42. After cooling, the (001) surfaces are prepared by cleavage of the crystals along the (001) plane at room temperature in the UHV chamber42. The crystals are then annealed a second time in the same oven at ~ 150°C for a few hours to remove surface charges43. The preparation of a clean NaCl(001) surface by cleavage and following annealing, the deposition of gold onto such a NaCl(001) surface and following AFM experiments are done in one UHV chamber, which is equipped with a sample transfer system.
Gold coverages were estimated by measuring the flux of a beam of neutral gold atoms from a Knudsen cell using a quartz crystal microbalance. The pressure during the deposition is in the lower 10−9 mbar range. The nc-AFM images were not used for coverage estimation due to the strong tip-cluster convolution, which is of importance when the clusters have a size of only a few nanometers15,16. Especially for the small clusters studied in this work, their size (diameter ~ 5 nm) is smaller than the tip apex, the diameter of which is typically ≤ 20 nm for new unused tips46. In this case, the shape of the tip apex is basically imaged at each cluster site, which becomes immediately evident when the apex changes upon tip changes during imaging. The convolution therefore overestimates the cluster size and induces large errors in any kind of estimates where the cluster size is a parameter.
AFM and manipulation protocols
Frequency-modulated noncontact AFM (nc-AFM) and Kelvin probe force microscopy (KPFM) experiments are performed in the low 10−10 mbar pressure range and at room temperature with an Omicron RT-AFM/STM. Conducting silicon cantilevers are used (Nanosensors, p-Si, 0.015 Ω.cm, 247 and 275 kHz resonance frequency, 32 (247 kHz tip) and 28 N/m (275 kHz tip) spring constant). Noncontact AFM primarily measures the change of frequency (detuning Δf) of the cantilever oscillation that is due to the interaction of the attached sensing tip with the surface17,47. In standard constant Δf operation mode, a high feedback gain of the distance regulation loop is used such that variations in Δf from a pre-set value (Δfpre-set) are nullified by regulating the tip-surface distance (topography images). For imaging the surface in a quasi-constant height mode, which was the scanning mode used for this work, the gain is drastically lowered such that the tip follows only the mean inclination of the surface. In this mode, the tip-surface interaction is stored in the Δf image. When scanning above clusters the tip is closest above the top facet of the clusters so that the tip-surface interaction is strongest (dark contrast ≡ stronger interaction ≡ more negative detuning Δf). More details about scanning modes with respect to clusters can be found in Refs. 15, 16. In order to measure the detuning, a demodulator from NanoSurf is used (EasyPLL). All images presented in this work were conducted in the constant height mode of the AFM. Images were acquired with the Omicron SCALA system and analyzed with the Gwyddion software48.
Two methods are used to manipulate the clusters by nc-AFM. The first method consists of moving the cluster by pushing during a spectroscopy step (Fig. 1 (a)). The scanning is switched off, the tip is positioned in a specific spot located on or very close to the cluster and the tip-sample distance is finally decreased monotonously. At a specific tip-cluster distance, at which the tip apex is very close to the cluster, the cluster moves on the surface due to a sufficiently large lateral force between the foremost atoms of the tip and the cluster. In the second method the tip is scanned preferentially in the constant height mode14,15,16 above the cluster (Fig. 1 (b)). From image to image, the tip-surface distance is reduced by choosing more negative detuning values Δf. At a specific distance, the tip comes from the side and is sufficiently close to the cluster such that again a lateral force between both moves the cluster. This produces an elongated contrast of the cluster, as can be seen in Fig. 2(f) and Fig. 3(c). From this specific contrast it can be concluded that the cluster slides by some small distance in front of the tip - the tip images almost always the same part of the cluster such that the same contrast is reproduced in each scanning line.
Calculations
Calculations were performed using spin polarized density functional theory (DFT), as implemented in the periodic plane wave code vasp49,50. Generalized gradient functionals (PBE)51 and the projector augmented wave method (PAW)49,52 were applied. An energy cutoff of 300 eV and a 2 × 2 × 1 k-point mesh were found to converge all key parameters of the system. The NaCl (001) surface was modeled using slabs three ionic layers thick with the bottom layer frozen, separated by 2.5 nm of vacuum. Energy barriers were determined using the climbing image nudged elastic band method53, where typically three or five chain images were sufficient. For simplicity, we ignore the effect of the AFM tip and concentrate on examining the energy landscape as felt by the clusters on the NaCl (001) surface.
Dispersion forces were not explicitly included in the calculations and although they may influence adsorption energies, their effect on the diffusion barriers is expected to be small54,55. In order to verify that this is the case, we calculated as a benchmark a series of diffusion pathways using both PBE and the vdW-DF functionals56, the latter implementing van der Waals forces. We especially used the vdW-DF functional to re-examine the diffusion pathways predicted by PBE to have the lowest and highest barrier energies for the Au20 cluster and the different defects. In all cases, the functional including dispersion forces yielded energy barriers close to that given by PBE, 0.0 – 0.2 eV lower and the hierarchy of energies was the same for both functionals.
Lateral forces needed to move the clusters were also estimated from the calculated trajectories. In general, in the preferred pathways where the energy barriers were found to be the lowest, also the lateral force on the cluster was low, in the range of 0.1 – 0.2 eV/Å. On the high barrier trajectories also the lateral forces were higher, about 0.2 – 0.3 eV/Å. That is, a direct correlation was seen between the energy barriers and lateral forces. We focus on the energy barriers in the discussion, since these are more accurately determined in the calculations.
The calculated results were obtained for a charge neutral system unless stated otherwise. This means that to create a Na vacancy, we remove a Na atom from the lattice leaving behind effectively a Na+ vacancy and a missing electron. Similarly, a Cl vacancy is effectively a Cl− vacancy with an additional electron. In the case of the anion vacancy, an electron remains localized in the vacancy forming an F0-centre. This is the expected form of defects at the surface, particularly with the abundance of electron sources (i.e. metal) in the system. However, it is also possible to change the number of electrons in the calculations to simulate Na+ and Cl− vacancies. These systems are not charge neutral, but the excess is compensated by a background charge. Extensive tests were performed to study the influence of the charge state of the defects on the results. These calculations are discussed in the Supplementary information.
20 and 100 atom gold clusters were used in the simulations, constructed in the shape of a tetrahedral pyramid or a truncated pyramid, respectively, as shown in Fig. 4 (a). Much larger systems are not feasible at this level of accuracy. Gold clusters prefer to grow in (100) epitaxy but also in (111) epitaxy with a triangular or hexagonal shape1,44,45. For consistency, we focus on small Au20 and Au100 clusters in (111) epitaxy in the calculations. For Au100, our calculations show the (111) faceted truncated tetrahedron structure to be energetically preferred to the best (100) epitaxial geometry we could find by 2.5 eV.
References
Henry, C. R. Surface studies of supported model catalysts. Surf. Sci. Rep. 31, 231–325 (1998).
Libuda, J. & Fruend, H.-J. Molecular beam experiments on model catalysts. Surf. Sci. Rep. 57, 157–298 (2005).
Heiz, U. & Landman, U. Nanocatalysis Springer-Verlag, Berlin (2007).
Adams, D. M. et al. Charge transfer on the nanoscale: Current status. J. Phys. Chem. B 107, 6668–6697 (2003).
Dietzel, D. et al. Frictional Duality Observed during Nanoparticle Sliding. Phys. Rev. Lett. 101, 125505 (2008).
Guerra, R., Tartaglino, U., Vanossi, A. & Tosatti, E. Ballistic nanofriction. Nat. Materials 9, 634–637 (2010).
Mougin, K. et al. Manipulation of gold nanoparticles: Influence of surface chemistry, temperature and environment (vacuum versus ambient atmosphere). Langmuir 24, 1577–1581 (2008).
Eigler, D. & Schweizer, E. Positioning single atoms with a scanning tunneling microscope. Nature 344, 524–526 (1990).
Giessibl, F. Advances in atomic force microscopy. Rev. Mod. Phys. 75, 949–983 (2003).
Custance, O., Perez, R. & Morita, S. Atomic force microscopy as a tool for atom manipulation. Nat. Nanotechnol. 4, 803–810 (2009).
Bouju, X., Joachim, C. & Girard, C. Moving gold atoms with an atomic-force-microscope tip: A study of dimer and trimer formation on NaCl(100). Phys. Rev. B 50, 7893–7902 (1994).
Trevethan, T., Watkins, M., Kantorovich, L. N. & Shluger, A. Controlled manipulation of atoms in insulating surfaces with the virtual atomic force microscope. Phys. Rev. Lett. 98, 028101 (2007).
Bartels, L., Meyer, G. & Rieder, K. H. Basic steps of lateral manipulation of single atoms and diatomic clusters with a scanning tunneling microscope tip. Phys. Rev. Lett. 79, 679–700 (1997).
Barth, C., Foster, A. S., Reichling, M. & Shluger, A. L. Contrast formation in atomic resolution scanning force microscopy on CaF2(111): experiment and theory. J. Phys.: Condens. Matter 13, 2061 (2001).
Barth, C., Pakarinen, O. H., Foster, A. S. & Henry, C. R. Imaging nanoclusters in the constant height mode of the dynamic SFM. Nanotechnology 17, S128 (2006).
Pakarinen, O. H., Barth, C., Foster, A. S. & Henry, C. R. Imaging the real shape of nanoclusters in scanning force microscopy. J. Appl. Phys. 103, 054313 (2008).
Barth, C., Foster, A. S., Henry, C. R. & Shluger, A. L. Recent trends in surface characterization and chemistry with high-resolution scanning force methods. Adv. Mater. 23, 477–501 (2011).
Barth, C. & Henry, C. R. Surface double layer on (001) surfaces of alkali halide crystals: A scanning force microscopy study. Phys. Rev. Lett. 98, 136804 (2007).
Hoffmann, R., Kantorovich, L., Baratoff, A., Hug, H. & Guntherodt, H. Sublattice identification in scanning force microscopy on alkali halide surfaces. Phys. Rev. Lett. 92, 146103 (2004).
Foster, A. S., Barth, C. & Henry, C. R. Chemical identification of ions in doped NaCl by scanning force microscopy. Phys. Rev. Lett. 102, 256103 (2009).
Teobaldi, G. et al. Chemical resolution at ionic crystal surfaces using dynamic atomic force microscopy with metallic tips. Phys. Rev. Lett. 106, 216102 (2011).
Gross, L. et al. Measuring the charge state of an adatom with noncontact atomic force microscopy. Science 324, 1428–1431 (2009).
Nony, L. et al. Observation of individual molecules trapped on a nanostructured insulator. Nano Lett 4, 2185–2189 (2004).
Mohn, F. et al. Reversible bond formation in a gold-atom-organic-molecule complex as a molecular switch. Phys. Rev. Lett. 105, 266102 (2010).
Lämmle, K. et al. Unambiguous determination of the adsorption geometry of a metal-organic complex on a bulk insulator. Nano Lett. 10, 2965–2971 (2010).
Dienel, T., Loppacher, C., Mannsfeld, S. C. B., Forker, R. & Fritz, T. Growth-mode-induced narrowing of optical spectra of an organic adlayer. Adv. Mater. 20, 959–963 (2008).
Steiner, P. et al. Atomic-scale friction on stepped surfaces of ionic crystals. Phys. Rev. Lett. 106, 186104 (2011).
Steiner, P., Roth, R., Gnecco, E., Baratoff, A. & Meyer, E. Angular dependence of static and kinetic friction on alkali halide surfaces. Phys. Rev. B 82, 205417 (2010).
Nony, L., Foster, A. S., Bocquet, F. & Loppacher, C. Understanding the atomic-scale contrast in Kelvin probe force microscopy. Phys. Rev. Lett. 103, 036802 (2009).
Glatzel, T. et al. Determination of effective tip geometries in Kelvin probe force microscopy on thin insulating films on metals. Nanotechnology 20, 264016 (2009).
Harding, J., Stoneham, A. & Venables, J. Predicting nucleation and growth processes: Atomistic modeling of metal atoms on ionic substrates. Phys. Rev. B 57, 6715–6719 (1998).
Hakala, M. H., Pakarinen, O. H. & Foster, A. S. First-principles study of adsorption, diffusion and charge stability of metal adatoms on alkali halide surfaces. Phys. Rev. B 78, 045418 (2008).
McKenna, K., Trevethan, T. & Shluger, A. Interplay between adsorbate diffusion and electron tunneling at an insulating surface. Phys. Rev. B 82, 085427 (2010).
Bethge, H. Electron microscopic studies of surface structures and some relations to surface phenomena. Surf. Sci. 3, 33–41 (1964).
Pakarinen, O., Barth, C., Foster, A. S., Nieminen, R. & Henry, C. High-resolution scanning force microscopy of gold nanoclusters on the KBr (001) surface. Phys. Rev. B 73, 235428 (2006).
Foster, A. S., Trevethan, T. & Shluger, A. L. Structure and diffusion of intrinsic defects, adsorbed hydrogen and water molecules at the surface of alkali-earth fluorides calculated using density functional theory. Phys. Rev. B 80, 115421 (2009).
Egberts, P., Filleter, T. & Bennewitz, R. A kelvin probe force microscopy of charged indentation-induced dislocation structures in KBr. Nanotechnology 20, 264005 (2009).
Kliewer, K. & Koehler, J. Space Charge in Ionic Crystals. I. General Approach with Application to NaCl. Phys. Rev. 140, A1226–A1240 (1965).
Kliewer, K. Space Charge in Ionic Crystals. II. The Electron Affinity and Impurity Accumulation. Phys. Rev. 140, A1241–A1246 (1965).
Molina, L. M. & Hammer, B. Theoretical study of CO oxidation on Au nanoparticles supported by MgO(100). Phys. Rev. B 69, 155424 (2004).
Grange, G. Formation of the suzuki phase on the solid-liquid interface of czochralski grown crystals. Surf. Sci. 105, 265 (1981).
Barth, C., Claeys, C. & Henry, C. R. Surface preparation of hard ionic crystals by ultrahigh vacuum cleavage. Rev. Sci. Instrum. 76, 083907 (2005).
Barth, C. & Henry, C. R. Kelvin probe force microscopy on surfaces of UHV cleaved ionic crystals. Nanotechnology 17, S155–S161 (2006).
Henry, C. R. Morphology of supported nanoparticles. Prog. Surf. Sci. 80, 92–116 (2005).
Matthews, J. W. & Grunbaum, E. The structure of gold films grown in ultra-high vacuum on sodium chloride substrates. Phil. Mag. 11, 1233–1244 (1965).
Nanosensors Neuchatel (Switzerland) http://www.nanosensors.com (accessed Jan 11th 2013).
Morita, S., Wiesendanger, R. & Meyer, E. Noncontact Atomic Force Microscopy Springer, Berlin (2002).
Gwyddion http://gwyddion.net (accessed Jan 11th 2013).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Henkelman, G., Uberuaga, B. & Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Pakarinen, O. H. et al. Role of van der Waals forces in the adsorption and diffusion of organic molecules on an insulating surface. Phys. Rev. B 80, 085401 (2009).
Mura, M., Gulans, A., Thonhauser, T. & Kantorovich, L. Role of van der Waals interaction in forming molecule-metal junctions: flat organic molecules on the Au(111) surface. Phys. Chem. Chem. Phys. 12, 4759 (2010).
Gulans, A., Puska, M. & Nieminen, R. Linear-scaling self-consistent implementation of the van der Waals density functional. Phys. Rev. B 79, 201105 (2009).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33 (1996).
Acknowledgements
We acknowledge financial support through the FANAS project NOMCIS. CB and GC highly acknowledge stimulating discussions with C. R. Henry. TH and ASF acknowledge support by the Academy of Finland through its Centres of Excellence Program (project no. 251748), as well as the computational resources offered by CSC, Finland.
Author information
Authors and Affiliations
Contributions
All authors contributed to writing the article. CB and GC designed and performed experiments and analysis. TH and ASF designed the computational study. TH performed the calculations and analysis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Defect mediated manipulation of nanoclusters on an insulator - supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hynninen, T., Cabailh, G., Foster, A. et al. Defect mediated manipulation of nanoclusters on an insulator. Sci Rep 3, 1270 (2013). https://doi.org/10.1038/srep01270
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01270
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
