
Abstract
The interaction of nanoparticles (NPs) with living organisms has become a focus of public and scientific debate due to their potential wide applications in biomedicine, but also because of unwanted side effects. Here, we show that superparamagnetic iron oxide NPs (SPIONs) with different surface coatings can differentially affect signal transduction pathways. Using isogenic pairs of breast and colon derived cell lines we found that the stimulation of ERK and AKT signaling pathways by SPIONs is selectively dependent on the cell type and SPION type. In general, cells with Ras mutations respond better than their non-mutant counterparts. Small negatively charged SPIONs (snSPIONs) activated ERK to a similar extent as epidermal growth factor (EGF) and used the same upstream signaling components including activation of the EGF receptor. Importantly, snSPIONs stimulated the proliferation of Ras transformed breast epithelial cells as efficiently as EGF suggesting that NPs can mimic physiological growth factors.
Similar content being viewed by others


Introduction
Cells contain a variety of sensitive receptors on their plasma membranes that activate intracellular signaling pathways, resulting in specific cellular responses to variations in environmental cues1. Membrane-embedded receptors are typically activated by peptide ligands, such as growth factors, cytokines, chemokines or hormones, whose binding activates the respective receptor leading to the assembly of signaling complexes at the cell membrane that mediate specific cellular responses, such as apoptosis, survival, proliferation, differentiation, cell-to-cell communication, contraction, migration and secretion. The EGF receptor is a tyrosine kinase receptor that is overexpressed or activated in many human cancers and has become a prominent target for anti-cancer therapeutics2,3. EGF binding induces receptor autophosphorylation and the binding of signal transducing proteins to the receptor, resulting in the activation of several downstream signaling pathways including the Ras-ERK and Ras-AKT pathways that stimulate cell growth and survival4,5.
Due to their large surface to volume ratio, nanoparticles (NPs) have distinct properties compared with the bulk form of the exact same materials6. These unique properties are now being exploited in biology and biomedicine to probe biological systems and deliver biosensors or drugs7. However, relatively little is known about the interactions of nanoscale objects with living systems, such as cells, per se.
Recent evidence suggests that NPs not only passively interact with cells and cell membranes, but that they can interact with membrane receptors thereby actively and specifically modulating signal transduction pathways8. Both, different NP sizes as well as cell type dependent differences can play a role in determining the biological response. In lung epithelial cells, carbon NPs were shown to interact with and activate the epidermal growth factor receptor (EGFR) and β1-integrins thus inducing cell proliferation of lung epithelial cells9. These effects were mediated through activation of PI3K and Akt. In another study, ultrafine carbon particles were shown to activate EGFR in lung epithelial cells leading in parallel to apoptotic events as well as cell proliferation10. Furthermore, increasing evidence suggests that NPs are not only passively interacting with living cells, but that they can modulate various signal transduction pathways through the production of reactive oxygen species or induction of growth factors and cytokine expression8,11,12,13,14. These effects may depend on the size15 and surface charge of NPs6,8,16.
Here, we have investigated the effects of superparamagnetic iron oxide NPs (SPIONs) with various physicochemical properties on the activation of intracellular signaling pathways downstream of the EGF receptor. SPIONs represent a new class of NPs, which due to their versatility and excellent biocompatibility have found widespread biomedical applications including the targeted delivery of therapeutic agents, imaging, induction of hyperthermia (as they heat up in electromagnetic fields), transfection and cell/biomolecules separation6,17,18. We found that small negatively charged SPIONs (snSPIONs) could activate EGF receptor induced signaling independently of ROS production. Importantly, snSPIONs stimulated the proliferation of Ras transformed breast epithelial cells as efficiently as EGF suggesting that NPs can mimic physiological growth factors.
Results
Synthesis of SPIONs
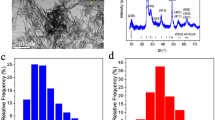
To test the hypothesis that NPs with well-defined sizes and surfaces can affect cellular signaling pathways, highly uniform SPIONs with core sizes of 9 and 15 nm and either plain, or dextran based negative or positive surface charges were generated (Fig. 1). Both small- and large-coated SPIONs have very narrow size distribution as determined by transmission electron microscopy (TEM). Dynamic light scattering (DLS) and zeta potential measurements showed that the electrokinetic potential and average sizes of NPs in different solutions are highly dependent on the surface charges and core sizes of NPs (Supplementary Tables S1 and S2). The average DLS sizes of small-SPIONs with different coatings are 18.9 nm, 20.3 nm and 24.6 in water, phosphate buffered saline (PBS) and fetal calf serum (FCS), respectively. The average sizes of large-particles under these conditions are 29.3, 31.2 and 37.5, respectively. The DLS results are in good agreement with TEM data. High-resolution TEM showed that the 9 nm and 15 nm NPs are highly monodisperse (Fig. S1).

Scheme for the ultra-uniform synthesis of monodisperse SPIONs and their coating process.
Iron–oleate precursor was prepared from the reaction of iron chlorides and sodium oleate. The monodisperse SPIONs in various sizes (i.e. 9 and 15 nm) were prepared by thermal decomposition of the metal–oleate precursors in high boiling solvent. The coating processes were performed by ligand exchange method in DMSO. TEM images show the formation of uniform bare- and dextran-coated-SPIONs with two different sizes. (The scale bars are 10 nm and 20 nm, respectively).
SPIONs activate cellular signaling pathways
While most studies focus on long-term effects and cytotoxicity of NPs19, we asked whether these differently sized and modified SPIONs influence short-term signaling events and whether this is related to the oncogenic state of the cells. Therefore, we used two isogenic cell line pairs that differ by the presence of an oncogene. Colon cancer derived HCT116 cells harbor a mutant K-Ras allele that is deleted in Hke3 cells causing reversion of the oncogenic phenotype20. MCF10A cells are derived from normal breast tissue, while their derivative MCF10A-CA1 (CA1) cells were transfected with the oncogenic H-RasG12V mutant and selected for a highly aggressive and metastatic phenotype21 Serum starved HCT116/Hke3 (Figs. 2 and S2) and MCF10A/CA1 (Figs. 3 and S3) were incubated with SPIONs or EGF and assayed for the activation of ERK and AKT, which are main downstream effector pathways of EGF and Ras4,5,22.

Regulation of ERK and AKT signaling by SPIONs in HCT116 and Hke3 cells.
HCT116 and Hke3 cells were incubated with SPIONs or EGF after serum-withdrawal for 18 hours. Activation of ERK (A) and activation of AKT (B) was assessed at the indicated time-points by immunoblotting. Results represent the mean of 3 independent experiments with SD.

Regulation of ERK and AKT signaling by SPIONs in MCF10A/CA1 cells.
MCF10A and CA1 cells were incubated with SPIONs or EGF after serum-withdrawal for 18 hours. Activation of ERK (A) and activation of AKT (B) was assessed at the indicated time-points by immunoblotting. Results represent the mean of 3 independent experiments with SD.
In Hke3 and HCT116 cells EGF activated ERK with similar kinetics peaking at five minutes and decreasing to basal levels within one hour, although the amplitude of activation was higher in HCT116 cell (Fig. 2A). The response to SPIONs was delayed and prolonged with maximal activation between 30 and 60 minutes, again higher in the HCT116 cells. Positively charged small SPIONs had the highest effects with 32% activation (relative to the maximal amplitude induced by EGF) in HCT116 and 50% in Hke3, respectively, although no significant differences between differently sized and charged SPIONs were observed. By contrast, EGF activated AKT to similar extent in both cell lines but with slightly prolonged kinetics in Hke3. The response to SPIONs was low and comparable between cell lines, except that plain small SPIONs activated AKT equally well as EGF (94% of the maximal activation by EGF) at 30 minutes in HCT116 cells (Fig. 2B).
In the breast epithelial lines MCF10A and CA1 EGF induced a transient but more sustained ERK activation than in the colorectal cancer lines and the non-oncogenic MCF10A responded better than the malignant CA1 (Fig. 3A). Interestingly, snSPIONs activated ERK to 50% of the maximal activation by EGF in MCF10A and 86% in CA1 cells, respectively. In CA1 the activation kinetics were very similar to EGF. Large negatively charged SPIONs (lnSPIONs) activated ERK selectively in CA1 cells to 60% of the maximal EGF response and with sustained kinetics. Other SPIONs were ineffective. The activation of AKT by EGF was to equal extent but slightly prolonged in CA1 compared to MCF10A cells (Fig. 3B). SPIONs did not activate AKT in MCF10A, but elicited a sustained activation in CA1 cells with up to 52% of the maximal activation by EGF. Similarly, SPION treatment of NIH3T3 cells and NIH3T3 cells stably transfected with the oncogenic H-Ras mutant (NIH3T3/H-Ras) corroborate these results (Fig. S4). Taken together, these results suggest that SPIONs can activate signaling pathways downstream of Ras with efficiencies and kinetics that in selected cases come close to the physiological growth factor EGF. While NP size did not play much role, the surface charge and the presence of mutant Ras seemed main response determinant.
In order to exclude that the selective regulation of signaling pathways was due to differential cytotoxicity of the various SPIONs or production of reactive oxygen species (ROS), we assessed mitochondrial respiration as an acute indicator of cell viability using MTT tests as well as intracellular ROS levels (Fig. S5). None of the SPION formulations significantly affected viability or ROS levels within the assay times suggesting that the effects on signaling are not due to NP toxicity or ROS production.
Given the above results, we asked whether SPIONs activate signaling pathways similar to physiological ligands. Therefore, several small chemical inhibitors targeting MEK and phosphoinositide-3 kinase (PI3K), the upstream activating kinases of ERK and AKT, were used in combination with SPION treatment (Fig. 4A). As snSPIONs in the MCF10A/CA1 cell system showed the biggest effects, we used these combinations for further evaluation. MCF10A and CA1 cells were serum starved for 18 hours and incubated with inhibitors for one hour before snSPIONs or EGF were added (Fig. 4B,C, Fig. S6). Both MEK inhibitors UO126 and PD184352 completely abolished the activation of ERK by EGF and snSPIONs. They also had a small and opposite effect on AKT activity, slightly enhancing EGF stimulated AKT activation, while diminishing snSPION mediated AKT activation. The PI3K inhibitor LY294002 abrogated AKT activation in response to both EGF and snSPIONs. Interestingly, LY294002 increased EGF stimulated ERK activation in MCF10A, but strongly inhibited snSPION mediated ERK activation in both MCF10A and CA1 cells. These results indicate that snSPIONs activate ERK and AKT like EGF via their canonical upstream kinases, but that snSPIONs affect the crosstalk between ERK and AKT pathways differently than EGF. Cross-regulation of ERK activation by AKT pathways has been reported to be inhibitory23 or stimulating24,25 probably depending on cell types and ligands. snSPION induced ERK activation is strongly AKT dependent, which resembles the scenario that EGF mediated ERK activation requires PI3K for post-peak activity25.

Signaling-specific inhibitors inhibit activation by snSPIONs.
(A) Schematic representation of the small-molecule inhibitors used and (B) the time course. MCF10A and CA1 cells were incubated with SPIONs or EGF after serum-withdrawal for 18 hours and pre-treatment with inhibitors. Activation of ERK (A) and activation of AKT (B) was assessed at the indicated time-points by immunoblotting. Results represent the mean of 3 independent experiments with SD.
SPIONs activate the EGF receptor
Next, we tested if SPIONs act even further upstream of MEK and PI3K by activating the EGF receptor (EGFR). Treatment of CA1 cells with snSPIONs activated the EGFR with similar kinetics as the natural ligand, albeit weaker than EGF (Fig. 5A, Fig. S7). In MCF10A cells snSPIONs caused a similarly strong, but even more sustained activation of the EGFR than EGF. EGF- and snSPION-mediated signaling not only resulted in the transient tyrosine phosphorylation of the receptor but also subsequent downstream substrates (Fig. S8). Furthermore, two different EGFR-specific inhibitors, BIXB1382 and gefitinib, reduced AKT activation and completely abolished ERK activation in response to snSPIONs or EGF (Fig. 5B,C, Fig. S7). Thus, snSPIONs trigger biochemical signaling events comparable to EGF. In order to assess whether snSPIONs can produce similar biological effects we monitored the proliferation of MCF10A and CA1 cells in response to EGF and snSPIONs, (Fig. 5D). Both agents stimulated robust proliferation indicating that SPIONs and EGF have comparative effects, while lnSPION or small plain SPIONs (spSPION) had no effects.

snSPIONs activate the EGF receptor.
(A) Serum starved MCF10A and CA1 cells were incubated with snSPIONs or EGF.Activation of the EGFR was assessed with phosphospecific antibodies. Activation of EGFR by SPIONs can be inhibited by EGFR-specific inhibitors. Serum starved MCF10A and CA1 cells were treated with EGFR inhibitors before stimulation. Activation of ERK (B) and activation of AKT (C) was assessed at the indicated time-points by immunoblotting. (D) Cell numbers of MCF10A and CA1 cells were determined at the indicated timepoints following incubation with snSPIONs, lnSPIONs, spSPIONs or EGF. Results represent the mean of 3 independent experiments with SD. * - p<0.05. ** - p<0.01.
Discussion
SPIONs represent a new class of NPs, which, due to their multi-task ability along with their excellent biocompatibility, have been selected as model NPs for several biomedical applications. These include targeted, e.g. tissue specific, delivery of therapeutic agents; imaging, e.g. magnetic resonance imaging (MRI) contrast enhancement; therapeutic applications such as hyperthermia due to their capability to be heated by an externally applied electromagnetic field; as well as in vitro applications such as the separation of cells and biomolecules6,26,27.
Increasing evidence suggests that NPs are not only passively interacting with living cells and cell membranes, but that they can trigger specific biochemical and biological responses. Many of these effects have been attributed to the production of ROS, mostly mediating stress signaling and cytotoxic processes, while suppressing survival and proliferative pathways, such as ERK and AKT28,29,30. Occasionally, ROS production induced ERK signaling and proliferation31, but it is unclear whether this effect is due to differences in cell types or NPs used in these studies. Our results show that the stimulation of ERK and AKT by SPIONs occurs fast, within minutes and does not involve the production of ROS. In addition, ERK and AKT activation requires EGF receptor activity suggesting that snSPIONs act directly on or through the EGF receptor. To our knowledge such direct receptor mediated effects of NPs on signal transduction pathways have not been observed in previous studies.
While it has been suggested that NPs can activate cell membrane receptors8, the mechanism is currently unclear. NPs may induce receptor aggregation which could promote receptor activation8. Another, not mutually exclusive possibility is the adsorption of peptide ligands from body fluids or serum added to cell culture media that bind to and activate receptors. NPs are known to bind proteins forming a so-called protein corona, which depends on the surface chemistry and size of the NPs7. In our experiments the incubation of cells with NPs was conducted under serum free conditions precluding that NPs may have absorbed EGF receptor ligands from serum. However, as cells can produce and secrete autocrine growth factors, NPs may have bound such autocrine factors during incubation with cells. As it has been shown that SPIONs do not bind EGF32, this possibility is unlikely. However, more detailed studies will be required to address the mechanism of EGF receptor stimulation by SPIONs.
The interaction of NPs with cell membrane receptors, including the EGF receptor, was also described for other NPs and surface receptors. For instance, carbon NPs can induce cell proliferation of lung epithelial cells via interaction and activation with EGFR and β1-integrins9. Using specific inhibitors it was demonstrated that this proliferation is mediated by PI3K and AKT. However, maximal activation by NPs occurred after several hours in comparison to our results, where maximal activation was observed within minutes. An earlier study showed that ultrafine carbon particles can activate the EGF receptor in lung epithelial cells inducing both apoptosis and proliferation at the same time10. While EGF receptor kinase activity was essential for both processes, proliferation required co-stimulation of ERK by β1-integrin activation, whereas the activation of JNK was necessary for apoptosis. These findings suggest that NPs can have very specific and subtle effects on signaling.
Our results extend this observation by showing that cells harboring mutated Ras are selectively sensitive to the stimulatory effects of SPIONs on signal transduction pathways. This effect is dependent on the type of SPION and surface modification as well as cell type specific. For instance, snSPIONs activated ERK strongly in the breast derived but not in colon cancer cell lines and small plain SPIONs selectively activated AKT but not ERK in HCT116 cells. Thus, potentially highly selective and cell type specific effects could be achieved through NPs. However, there is also some caution. NPs used for imaging, drug delivery or other therapeutic purposes could have unwanted side effects due to their effects on signaling pathways. As Ras mutations are common in human cancers22, NPs may have growth stimulatory effects when used in patients with mutant Ras cancers. It will be interesting to learn how we can control these effects through the purposeful design of NPs and their surface modifications.
Methods
Synthesis of Ultra-Uniform SPIONs
Ultra-uniform SPIONs were synthesized with different sizes of 9 and 15 nm using thermal decomposition of iron-oleate33. The iron–oleate complex was prepared by reaction of iron chloride (FeCl3·6H2O, 40 mmol, Aldrich, 98%) and sodium oleate (120 mmol, TCI, 95%). In a representative synthesis of 9 nm particles, 10.8 g of iron chloride and 36.5 g of sodium oleate was dissolved in a mixture solvent composed of 80 ml ethanol, 60 ml distilled water and 140 ml hexane. The resulting solution was kept at 70 °C for four hours. After completion of the reaction the upper organic layer, which contains the iron–oleate complex, was collected and washed three times using 30 ml distilled water in a separatory funnel. Then, hexane was evaporated off resulting in formation of an iron–oleate complex in a waxy solid form. 36 g (40 mmol) of the resulting iron-oleate complex together with 5.7 g of oleic acid (20 mmol, Aldrich, 90%) were dissolved in 200 g of 1-octadecene (Aldrich, 90%) at room temperature. The reaction mixture was heated to 320 °C with a constant heating rate of 3.3 °C min–1 and then kept at that temperature for 30 minutes. When the reaction temperature reached 320°C the initial transparent solution became turbid and brownish black. The resulting solution containing the nanocrystals was then cooled to room temperature and 500 ml of ethanol was added to the solution to precipitate the nanocrystals. Finally, the nanocrystals were separated by centrifugation.
Synthesis of various coating polymers
The carboxylated-dextran was prepared as reported elsewhere34. First, the hydroxyl groups in dextran were oxidized to aldehyde groups by sodium periodate35. Briefly, sodium periodate was dissolved in de-oxygenated DI water and added to dextran (molecular average weight 5000) solution (4 g in 30 mL of de-oxygenated DI water). The obtained solution was homogenized for 2 hours at room temperature followed by dialysis with a 1,000 Da molecular weight cut-off for 4 days. The solution was reacted with potassium cyanide for preparation of cyanohydrins intermediate. Finally, the carboxylic acid group was created on the terminal units of dextran by hydrolysis of the obtained cyanohydrins intermediate. The prepared carboxylated dextran was lyophilized and stored at −80°C. The amino dextran was prepared as reported elsewhere36. Typically, 10 g of dextran were dissolved in 75 mL of de-oxygenated DI water containing 2.5 g sodium hydroxide and 0.2 g sodium borohydride at pH of 11, which was fixed by drop wise addition of 2.5 N NaOH and 2 mL allyl bromide at a temperature of around 50°C. The solution was neutralized by addition of acetic acid followed by incubation at 4°C for 2 hours. The top organic layer was removed, 100 mL of fresh de-oxygenated DI water was added and the solution was dialyzed for 24 hours using a 50,000 Damolecular weight cut-off membrane. In order to prepare amino dextran conjugate, the dialyzed mixture was reacted with 7.5 g of amino alkylthiol compound in 30 mL dimethyl sulfoxide where 0.1 g ammonium persulfate was used as initiator. After 3 hrs, the same volume of the fresh de-oxygenated DI water was added to the reactor followed by pH adjustment using sodiumhydroxide and the product was diluted with 140 mL sodiumacetate buffer (0.02 mol/L, pH 4). To ascertainthe removal of excess materials the obtained amino dextran conjugate was dialyzed using a membrane bag with a 50,000 Da molecular weight cut-off for 24 hours. The dialysed amino dextran conjugate was lyophilized and stored at −80°C.
Coating process
For coat the hydrophobic NPs with plain, carboxylated-dextran, or dextran with amine terminal groups, the ligand exchange method was used37. Briefly, 4 mL of 9 nm and 15 nm SPION solutions (1 mg/ml iron) were mixed with the dextran ligands in 90 mL DMSO at room temperature for 72 hours in a shaking incubator. After completion of the reaction, the SPIONs were collected using magnetic-activated cell sorting (MACS®) system and re-dispersed into 1 mL of DI water. Atomic absorption confirmed that the iron concentration of various samples were 4 mg/ml. These water-soluble SPIONs were completely stable at room temperature without detectable precipitation for several months.
Characterization methods
The morphologies of various SPIONs were analyzed by transmission electron microscopy (TEM) using JEOL-2010 and FEI Tecnai F20 electron microscopes operating at 200 kV. To prepare samples for TEM, a drop of the SPION suspension was placed on a copper grid and dried. Dynamic light scattering (DLS) measurements were performed with a Malvern PCS-4700 instrument equipped with a 256-channel correlator. The 488.0 nm line of a Coherent Innova-70 Ar ion laser was used as the incident beam at 250 mW. The scattering angles, θ, ranged between 40°–140°. The temperature was maintained at 25 °C with an external circulator. Zeta potential determination was performed using a Malvern Zetasizer 3000 HSa. Each measurement was an average of six repetitions of one minute each and repeated five times. Data analysis was performed according to standard procedures and interpreted through a cumulant expansion of the field autocorrelation function to the second order. In order to obtain a distribution of decay rates, a constrained regularization method, CONTIN, was used to invert the experimental data. To determine the variations of sizes and surface charges of negative-, plain- and positive-SPIONs after incubation with fetal calf serum (FCS), the protein-SPIONs solutions were captured in a strong MACS system in a magnetic column. The magnetically fixed NPs were washed with phosphate buffered saline (PBS) to remove unbound or loosely bound serum proteins. Full DLS and zeta potential data of native, PBS, or FCS interacted-particles are presented in TablesS1 & S2.
Cell lines
HCT116 and Hke3 are isogenic human colon cancer cell lines that differ only by the presence or absence of a mutant KRASG13D allele20. They were cultured in standard DMEM containing 10% FCS. From the MCF10 human breast epithelial model system we used non-malignant MCF10A and malignant metastatic MCF10CA1 (CA1) cells, which were derived by transfection with a mutant HRAS transgene and serial passage through mice21. They were cultured in DMEM supplemented with 5%horse serum, 20 ng/ml) epidermal growth factor (EGF), 0.5µg/ml hydrocortisone, 100 ng/ml cholera toxin and 10µg/ml insulin. NIH3T3 is a mouse fibroblast cell line and NIH3T3/H-Ras are derived by stable transfection with a mutant HRAS transgene. They were cultured in standard DMEM containing 10% FCS.
Toxicity evaluation of SPIONs
Cells were cultivated for 24 hours in 96-well plates before incubation with 200µg/ml of different SPIONs for 15 min up to 3 hours. For control cells were incubated without NPs. Cytotoxicity was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, which is a non-radioactive, colorimetric technique. Following incubation of the cells with NPs, the medium was removed and formazan crystals were solubilised by incubation in 150 µL of isopropanol for 20 minutes. The absorbance of each well, which assesses viable cells, was read at 545 nm on a microplate reader (Stat Fax-2100, AWARENESS, Palm City, USA). All experiments were carried out in triplicate. The results were analyzed using the MINITAB software (Minitab Inc., State College, PA). Statistical evaluation was performed by one-way analyses of variance (ANOVA), for which p<0.05 was considered as statistically significant.
ROS evaluation of SPIONs
For the determination of intracellular ROS levels the fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Invitrogen) was used. In the presence of intracellular esterases this nonpolar component is converted into the nonfluorescent polar derivative H2DCF. This intermediate is membrane impermeable and rapidly oxidized to fluorescent 2′,7-dichlorofluorescein (DCF) by ROS. Cells were cultivated for 24 hours in 96-well plates before incubation with 200 µg/ml of NPs for 30 min (50 µM H2DCF-DA was added to the wells). As control, cells were treated with H2O2 a strong inducer of ROS production. The intracellular DCF fluorescence was measured with a plate reader infinite M200 (Tecan) with an excitation wavelength of 485 nm. Emission was recorded at 535 nm. All data were corrected for background fluorescence. ROS production in NP treated cells was adjusted to untreated cell cultures.
Treatment and analysis of cells by immunoblotting
Cells were grown in standard medium before serum withdrawal for 18 hrs. Cells were incubated with SPIONs at a final concentration of 200 µg/ml or with 10 nM EGF. At indicated timepoints cells were harvested, washed with ice-cold PBS and lysed in 10 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl and 0.5% NP40 supplemented with protease and phosphatase inhibitors (Roche Diagnostics). Lysates were cleared of debris by centrifugation (15,000 × g, 10 minutes), separated by SDS-PAGE (10–12%) and analyzed by multistrip Western blotting38. The following antibodies were used: Polyclonal rabbit anti-human mitogen-activated protein (MAP) kinase/extra- cellular signal-regulated kinase (ERK) 1 and ERK2 antibody (Sigma); monoclonal mouse anti-human MAP kinase activated (diphosphorylated ERK1 and ERK2) antibody (Sigma); polyclonal rabbit anti-human AKT antibody (Cell Signalling); polyclonal rabbit anti-human activated Akt (phosphorylated at Ser473) antibody (Cell Signalling); polyclonal rabbit anti-human EGF receptor antibody (Cell Signalling); monoclonal rabbit anti-human activated EGF Receptor (phosphorylated at Tyr1173) antibody (Cell Signalling). Proteins were detected using horseradish peroxidase–conjugated secondary antibodies and enhanced chemiluminescence (ECL). The Chemi Image digital imager (Advanced Molecular Vision) was used for image acquisition and immunoblots were quantified using ImageJ software39. The relative activation of ERK (ppERK) and AKT (pAKT), respectively, was calculated by normalizing the signal by the amounts of total ERK and AKT protein (totERK or totAKT).
Signaling transduction inhibitors
The following chemical inhibitors were used: BIXB1382 (10µM, Cellbiochem); Gefitinib (5µM, American Custom Chemical Corporation); UO126 (10µM, Cell Signaling); PD184552 (2µM, Axonmedchem); LY294002 (10µM, Cell Signaling). Briefly, cells were serum-starved for 18 hours prior to incubation with the inhibitors. Subsequently, cells were stimulated with either SPIONs or EGF for the time points indicated.
Statistical analysis
Significance levels were determined by 2-tailed Student's t test analyses. All tests were 2-sided and results considered significant if P < 0.05. Significance levels and P values for figures 2-5 are included as Supplementary Tables S3-S12.
References
Grecco, H. E., Schmick, M. & Bastiaens, P. I. H. Signaling from the Living Plasma Membrane. Cell 144, 897–909 (2011).
Seshacharyulu, P. et al. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 16, 15–31 (2012).
Eccles, S. A. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol 55, 685–696 (2011).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010).
Citri, A. & Yarden, Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 7, 505–516 (2006).
Mahmoudi, M., Sant, S., Wang, B., Laurent, S. & Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Advanced drug delivery reviews 63, 24–46 (2011).
Nel, A. E. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater 8, 543–557 (2009).
Marano, F., Hussain, S., Rodrigues-Lima, F., Baeza-Squiban, A. & Boland, S. Nanoparticles: molecular targets and cell signalling. Archives of toxicology 85, 733–741 (2011).
Unfried, K., Sydlik, U., Bierhals, K., Weissenberg, A. & Abel, J. Carbon nanoparticle-induced lung epithelial cell proliferation is mediated by receptor-dependent Akt activation. American journal of physiology. Lung cellular and molecular physiology 294, L358–367 (2008).
Sydlik, U. et al. Ultrafine carbon particles induce apoptosis and proliferation in rat lung epithelial cells via specific signaling pathways both using EGF-R. American journal of physiology. Lung cellular and molecular physiology 291, L725–733 (2006).
Mahmoudi, M. et al. Protein−Nanoparticle Interactions: Opportunities and Challenges. Chemical Reviews 111, 5610–5637 (2011).
Verma, A. et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat Mater 7, 588–595 (2008).
Lynch, I., Salvati, A. & Dawson, K. A. Protein-nanoparticle interactions: What does the cell see? Nat Nano 4, 546–547 (2009).
Deng, Z. J., Liang, M., Monteiro, M., Toth, I. & Minchin, R. F. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat Nano 6, 39–44 (2011).
Jiang, W., Kim, B. Y. S., Rutka, J. T. & Chan, W. C. W. Nanoparticle-mediated cellular response is size-dependent. Nature Nanotechnology 3, 145–150 (2008).
Mailander, V. & Landfester, K. Interaction of nanoparticles with cells. Biomacromolecules 10, 2379–2400 (2009).
Laurent, S., Dutz, S., Häfeli, U. O. & Mahmoudi, M. Magnetic fluid hyperthermia: Focus on superparamagnetic iron oxide nanoparticles. Advances in Colloid and Interface Science 166, 8–23 (2011).
Mahmoudi, M., Stroeve, P., Milani, A. S. & Arbab, S. A. Superparamagnetic Iron Oxide Nanoparticles: Synthesis, Surface Engineering, Cytotoxicity and Biomedical Applications;, (Nova Science Publisher, New York, 2011).
Sharifi, S. et al. Toxicity of nanomaterials. Chemical Society Reviews (2012).
Shirasawa, S., Furuse, M., Yokoyama, N. & Sasazuki, T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260, 85–88 (1993).
Santner, S. J. et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat 65, 101–110 (2001).
Karnoub, A. E. & Weinberg, R. A. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9, 517–531 (2008).
Moelling, K., Schad, K., Bosse, M., Zimmermann, S. & Schweneker, M. Regulation of Raf-Akt Cross-talk. J Biol Chem 277, 31099–31106 (2002).
Aksamitiene, E. et al. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal 23, 1794–1805 (2011).
Birtwistle, M. R. et al. Ligand-dependent responses of the ErbB signaling network: experimental and modeling analyses. Mol Syst Biol 3, 144 (2007).
Mahmoudi, M. et al. Effect of Nanoparticles on the Cell Life Cycle. Chemical Reviews, 110314105112023 (2011).
Mahmoudi, M. et al. Magnetic Resonance Imaging Tracking of Stem Cells in Vivo Using Iron Oxide Nanoparticles as a Tool for the Advancement of Clinical Regenerative Medicine. Chemical Reviews 111, 253–280 (2011).
Apopa, P. L. et al. Iron oxide nanoparticles induce human microvascular endothelial cell permeability through reactive oxygen species production and microtubule remodeling. Particle and Fibre Toxicology 6, 1 (2009).
Piao, M. J., Kim, K. C., Choi, J. Y., Choi, J. & Hyun, J. W. Silver nanoparticles down-regulate Nrf2-mediated 8-oxoguanine DNA glycosylase 1 through inactivation of extracellular regulated kinase and protein kinase B in human Chang liver cells. Toxicol Lett 207, 143–148 (2011).
Eom, H. J. & Choi, J. Oxidative stress of CeO2 nanoparticles via p38-Nrf-2 signaling pathway in human bronchial epithelial cell, Beas-2B. Toxicol Lett 187, 77–83 (2009).
Huang, S., Chueh, P. J., Lin, Y. W., Shih, T. S. & Chuang, S. M. Disturbed mitotic progression and genome segregation are involved in cell transformation mediated by nano-TiO2 long-term exposure. Toxicol Appl Pharmacol 241, 182–194 (2009).
Comfort, K. K., Maurer, E. I., Braydich-Stolle, L. K. & Hussain, S. M. Interference of silver, gold and iron oxide nanoparticles on epidermal growth factor signal transduction in epithelial cells. ACS Nano 5, 10000–10008 (2011).
Park, J. et al. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat Mater 3, 891–895 (2004).
Usher, T. C. & Wallis, S. H. Process of making carboxylated dextran. US Patent 6,703,499. (2004).
Martwiset, S., Koh, A. E. & Chen, W. Nonfouling Characteristics of Dextran-Containing Surfaces. Langmuir 22, 8192–8196 (2006).
Saboktakin, M. R., Tabatabaie, R. M., Maharramov, A. & Ramazanov, M. A. A synthetic macromolecule as MRI detectable drug carriers: Aminodextran-coated iron oxide nanoparticles. Carbohydrate Polymers 80, 695–698 (2010).
Xu, Y., Qin, Y., Palchoudhury, S. & Bao, Y. Water-Soluble Iron Oxide Nanoparticles with High Stability and Selective Surface Functionality. Langmuir 27, 8990–8997 (2011).
Aksamitiene, E., Hoek, J. B., Kholodenko, B. & Kiyatkin, A. Multistrip Western blotting to increase quantitative data output. Electrophoresis 28, 3163–3173 (2007).
Rasband, W. & ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/(1997–2011).
Acknowledgements
This work was supported by the Science Foundation Ireland under Grant No. 06/CE/B1129.
Author information
Authors and Affiliations
Contributions
All authors co-designed and co-wrote the paper. MM and JR performed the experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rauch, J., Kolch, W. & Mahmoudi, M. Cell Type-Specific Activation of AKT and ERK Signaling Pathways by Small Negatively-Charged Magnetic Nanoparticles. Sci Rep 2, 868 (2012). https://doi.org/10.1038/srep00868
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00868
This article is cited by
-
Protein Corona Composition of Superparamagnetic Iron Oxide Nanoparticles with Various Physico-Chemical Properties and Coatings
Scientific Reports (2014)
-
Magnetic nanoparticles trigger cell proliferation arrest of neuro-2a cells and ROS-mediated endoplasmic reticulum stress response
Journal of Nanoparticle Research (2014)
-
Effect of surface charge on the colloidal stability and in vitro uptake of carboxymethyl dextran-coated iron oxide nanoparticles
Journal of Nanoparticle Research (2013)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
