
Abstract
During development, axons of neurons in the mammalian central nervous system lose their ability to regenerate. To study the regeneration process, axons of mouse hippocampal neurons were partially damaged by an UVA laser dissector system. The possibility to deliver very low average power to the sample reduced the collateral thermal damage and allowed studying axonal regeneration of mouse neurons during early days in vitro. Force spectroscopy measurements were performed during and after axon ablation with a bead attached to the axonal membrane and held in an optical trap. With this approach, we quantified the adhesion of the axon to the substrate and the viscoelastic properties of the membrane during regeneration. The reorganization and regeneration of the axon was documented by long-term live imaging. Here we demonstrate that BDNF regulates neuronal adhesion and favors the formation of actin waves during regeneration after axonal lesion.
Similar content being viewed by others


Introduction
Understanding the molecular mechanisms involved in neuronal regeneration is crucial for the development of well-targeted treatments for human neurological diseases1. Several studies on nerve regeneration in complex organisms employed different techniques to perform axon severing (axotomy)2,3. Studies in PC12 cells4, chicken neurons5 and Aplysia neurons6, in which the neurites were completely transected, analyzed the involvement of the cytoskeleton in the re-growth of the proximal stump. A recent study investigated the regeneration behavior in goldfish axons upon induction of a partial damage with no rupture of the cell membrane7.
However, the regeneration of transected or damaged neurites can be impaired due to the collateral damage induced by the laser in the surrounding tissue8,9 and the results from pharmacological approaches applied to whole tissue can be misleading because of the reaction by the neighboring cells10. Also in a simplified in vitro system it is mandatory to quantify, characterize8,11 and control12 the extent of the damage induced by laser axotomy, in order to obtain a condition in which the axonal recovery is not impaired13. In addition, high-throughput methods are necessary to assess the reliability of any tested parameter, such as compounds affecting neurite regeneration in models with wild-type and mutant genetic background2,14.
Cytoskeletal elements and molecular motors responsible for cell growth and motility during exploratory motion and differentiation15,16,17 have a key role in the regeneration process. The distinct mechanical properties and dynamics of cytoskeletal filaments provide clues for understanding the cytoskeletal functions18,19,20. Intermediate filaments are the most rigid components, which stabilize the overall cell shape21. Microtubules form a polarized filament network allowing intracellular organelle positioning and vesicle transport through interactions with motor proteins22. Actin microfilaments provide the protrusive forces for the formation of filopodia and lamellipodia15,23. Moreover, in complex with myosin motors, they generate traction forces between focal adhesion contacts and the extracellular matrix (ECM)24. Such strain is regulated in space and time to maintain a constant membrane tension and a balanced deformation of the ECM25. Moreover, it contributes to the accumulation of vesicles at presynaptic terminals26 and stabilizes the neuromuscular junctions27. Axotomy by laser dissection leads to depolymerization of cytoskeletal filaments28, release of equilibrium tension and disassembly of adhesion contacts29. During axonal regeneration, the cell has to restore the disrupted structures, to elongate the dissected neurite, to initiate growth cone navigation30 and to re-establish the homeostatic compression-tension equilibrium31 in order to recover the functionality of the connection with its targets.
In the present work, we report an in vitro model of axonal regeneration based on a sub-nanosecond pulsed UVA laser. With an average power of a few microwatts delivered to the sample, our system allows induction of a partial lesion in the axons of cultured mouse hippocampal neurons in a highly controlled and reproducible manner without affecting the regeneration process.
A versatile, custom-built cell incubator stage adaptable for upright or inverted microscopes allowed the continuous long-term monitoring of the regeneration process of an injured neuron following the dynamics of axonal re-growth. Here we analyzed the formation of actin waves before and after the partial axonal damage and investigated the effect of brain-derived neurotrophic factor (BDNF) on their number and rate of movement along the axon.
Integrated holographic optical tweezers (OTs) were used for interferometric force spectroscopy during neurite ablation to quantify the release of tension in the dissected process with a sub-piconewton resolution. This system was used, during the regeneration of the neurite, to observe the plasma membrane dynamics, the strength of the interaction between the axon and the extracellular matrix and their regulation by BDNF, with a sub-millisecond time resolution.
Results
Ablation efficiency and regeneration
The laser ablation system allows to selectively perturb subcellular compartments in a highly reproducible way. Therefore, it is recognized as an invaluable tool for studying axonal regeneration after injury32. Figure 1a displays a neural network cultured on a glass support. To demonstrate the confined ablation volume, the position of the focus was moved 2 μm below the cells; with an average power of 6 μW, an ablation track was generated in the glass support (middle frame). Although the laser light went through the cell before focusing on the glass, the neurons and their connections were unaffected (right frame). Upon delivering a laser power of 4 μW focused on the sample, the neurite was completely dissected (figure 1b). In contrast, the damage to the neurite appeared restricted to the formation of a neck when the pulse energy was reduced to 2.3 μW (figure 1c).

Laser ablation efficiency.
(a) Mouse hippocampal neurons were plated on a glass support and images were taken in bright field. Neural network at 10 DIV in focus (left frame). The focus was then moved 2 μm below the cells and with an average power of 6 μW, the glass support was ablated (middle frame) without affecting the network structure (right frame). The white arrow indicates the position of the UVA laser spot. Bar is 10 μm. (b) With an average power of 4 μW at the sample, a neural connection is completely transected (middle frame, arrow) and subsequently retracts without affecting the neighboring neurites (right frame). The white arrow indicates the transected neurite. Bar is 7 μm. (c) With an average power at the sample of 2.3 μW, the axon of a 3 DIV neuron is partially damaged without disruption of the membrane. The white arrows indicate the ablation site (middle frame) and the formation of a neck (right frame). Bar is 5 μm.
The partial damage inflicted to the neurite using such a low laser power, triggers the activation of biological pathways responsible for wound healing and re-growth without inducing cell death. This is particularly important when using mouse neurons and when the ablation is performed very close to the cell body on short neurites present at an early stage of development. With the optical parameters used in figure 1c, the axons of hippocampal neurons between 1 and 10 days in vitro (DIV) were dissected and their regeneration was followed by long-term bright field time-lapse imaging. In neurons injured in the first 3 DIV, all the dissected neurites regenerated, 46% of those from neurons at 4 DIV were able to regenerate, while no regeneration was observed in neurons older than 6 DIV onwards within the timeframe of the experiment (not shown). A total of 20 neurons were analyzed for each experimental group. When the distal part of the neurite degenerated, re-growth always followed the previous path.
Long-term imaging of dissected neurites in immature neurons: actin waves
To investigate the process of axonal regeneration, axons of neurons at 2 and 3 DIV were partially damaged. At these DIVs, the neuron is at stage 2 of polarization and presents a longer neurite, which invariably will become the axon33. This was confirmed by retrospective tau immunostaining of the neurons that was consistently identifying the dissected process as the axon (not shown). After axotomy, regeneration of the transected axon was monitored by time-lapse bright field imaging at 1 Hz for 1 hr. Three distinct behaviors were observed. A very fast regeneration, with disappearance of the neck in the axon, could occur within few minutes (figure 2a), or a longer regeneration process involved the formation of lamellipodia and was completed in about 1 hr. The lamellipodium could form on the lesion site engulfing the lesion and leaving an intact neurite after its re-absorption, (figure 2b), or could form close to the cell body travelling along the neurite toward the lesion site (figure 2c). We next tested the effects of BDNF on the regeneration process. Neurons incubated with (n = 20) or without BDNF (n = 20) were monitored for one hour after laser axotomy on an upright microscope. BDNF-treated neurons regenerated slightly faster (the average time of healing of neurons incubated with and without BDNF were respectively 31±6 and 38±7 minutes, data are the mean ± standard errors) and the regeneration process was occurring more frequently by the formation of a lamellipodium (1.4 fold over control), indicating a direct effect of BDNF on the growth capacity of the neurons.

Healing behaviours.
The axons of mouse hippocampal neurons at 2 DIV were ablated with an average power of the UVA laser of 2.5 μW at a pulse rate of 100 Hz. The white arrows in the frames at 10 seconds and 14 minutes indicate the ablation sites. (a) Fast healing with no formation of lamellipodia. Bar is 8 μm. (b) Formation of a lamellipodium at the site of lesion (black arrow), that reabsorbs leaving a healed axon. Bar is 5 μm. (c) Formation of a lamellipodium close to the cell body (black arrow) that reaches the site of lesion. Bars are 10 μm.
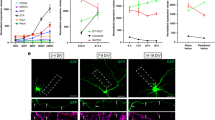
Growth cone-like structures (so-called actin waves) originating at the soma have been described as a transport mechanism toward the tip of the neurite necessary to assist its pulsatile growth34. Therefore, the axons of neurons in the first 3 DIV were lesioned to monitor the formation of the actin waves35 and the effect of BDNF on cytoskeletal dynamics. Neurons were plated on a gridded Petri dish, the nascent axons were lesioned and the dishes were then moved to an inverted microscope for long-term imaging. The number of actin waves was counted and compared between lesioned and control neurons. Figure 3 shows an example of a control and BDNF-treated injured neurons, analyzed over a time of 68 minutes. Along the axon of the control neuron (figure 3a), one single actin wave was observed, while, upon BDNF incubation (figure 3b), three distinct actin waves moving toward the tip of the axon were visible. Quantification of the number of actin waves per hour (figure 3c) and of their rate of propagation along the axon (figure 3d), indicate a significant difference between control and injured neurons and between injured neurons incubated with or without BDNF. The rate of actin waves movement was further analyzed on both sides of the lesion revealing that in injured neurons the average velocity was reduced 2.5 fold downstream of the lesion site, while in BDNF-treated samples the velocity did not decrease as the wave passed the lesion (figure 3e).

Actin waves formation in injured axons increase in the presence of BDNF.
(a,b) Bright field images of the axon of an untreated (a) or BDNF-treated (b) hippocampal neuron after partial axotomy. The white arrows and numbers indicate the actin waves moving along the axon. Numbers in the upper left corner indicate minutes. Bars are 8 μm. (c) Quantification of the number of actin waves per hour in control and injured axons of neurons incubated with or without BDNF (ctrl n = 21; lesion n = 19; BDNF n = 16; lesion with BDNF n = 22; p-values: * = 0.039, ** = 0.011) (d) Quantification of the propagation rate of actin waves in control and injured axons of neurons incubated without or with BDNF (ctrl n = 108; lesion n = 86; BDNF n = 34; lesion with BDNF n = 91; p-values: * = 0.029, ** = 0.001). (e) Quantification of the velocity of the actin waves upstream (before) and downstream (after) the lesion site in the injured axons as in d, control and BDNF-treated (p-values: * = 0.02, ** = 0.0017). The significance with an alpha value of 0.05 was calculated by an unpaired two-tailed t-test for (c) (d) and (e). Data are the mean ± standard errors.
Effect of laser induced damage on the cytoskeleton
In order to analyze the damage induced by the low intensity laser, the nascent axons of neurons plated on gridded Petri dishes were lesioned and the neurons were then fixed for immunohistochemistry analysis. Microfilaments, microtubules and neurofilaments were labeled by specific antibodies (figure 4a). In the staining of all three cytoskeletal elements, a gap is appreciable where the filaments appeared interrupted, suggesting that re-polymerization of the cytoskeleton and transport of monomers is necessary for the lesion to be repaired. In our experiments BDNF had no effect on the number or the propagation of actin waves in neurons that had not been injured. However, it is known that actin turnover is increased upon BDNF treatment. In fact, BDNF treatment induced an increase in the number and length of filopodia in steady state condition. Neurons at 3DIV were treated for 30 minutes with BDNF and labeled with phalloidin recognizing F-actin and with antibodies against actin and Abi-1, a protein localized at actin barbed ends when bound to the capping protein Eps836. Control neurons (figure 4b, left panel) show less filopodia when compared to BDNF-treated neurons (figure 4c, left panel). The right panels in figure 4b and c show magnifications of the growth cones with phalloidin staining being more evident in the BDNF-treated neurons. The barbed ends of the phalloidin-stained filopodia are labeled by Abi-1; actin monomers are also concentrated along the same regions and more evident in the control neurons, but are not labeled by Abi-1.

The laser damage induces a partial dissection of the three cytoskeletal elements.
BDNF increases actin filopodia at steady state. (a) Immunostaining of neurofilament subunit L (green), βIII-tubulin (blue) and F-actin (red) in the axon of a hippocampal neuron at 3DIV immediately after laser injury. Bar is 2 µm. (b) Immunostaining of actin (green), F-actin (blue) and Abi-1 (red) in a hippocampal neuron at 3DIV and (c) in a hippocampal neuron at 3DIV treated with 100 ng/ml BDNF for 30 minutes. The right panels, of b and c, show magnifications of the growth cones (white square box in the left panels). Bars are 10 µm in the left panels and 2 µm in the right panels.
Force spectroscopy of tension release
To quantify the neuron's adhesion force to the substrate, the tension released in the damaged neurite was analyzed by force spectroscopy measurements during laser dissection using the previously described method12,16. Figure 5a shows an axon from a 2 DIV neuron with a fibronectin-coated bead kept in an optical trap and attached to its membrane. The kymograph in figure 5b shows that, once the axon had been cut, cellular material retracted faster below the lesion site (increase of grey levels correlate with decrease in light absorption). On the site of cut (figure 5b, right panel), a reduction of material over time is appreciable, that correspond to the thinning of the lesioned axon. Figure 5c shows the recorded z trace of the bead held away from the axon where the oscillation range is between ±40 nm and the total variance of the x, y and z traces is 232 nm2. When the bead adhered to the axon, the oscillation range of the z trace is between ±20 nm and the total variance decreased to 120 nm2 (figure 5d) indicating that the bead is attached to the cell membrane and presumably to the underlying actin cortex.

Measurements of the release of tension during laser ablation of hippocampal axons.
(a) Bright field images acquired during ablation of an axon. A fibronectin-coated bead is attached to the membrane (silica bead, Ø 4 μm) and held in an optical trap. Average power of the IR laser at the sample is 35 mW. White arrows indicate the lesion site. Bar is 5 μm. (b) Kymograph analysis along lines parallel and perpendicular to the axon (white lines, upper panel), upstream and downstream of the lesion site (bottom left panel) and on the lesion site (bottom right panel), before and after the cut (pink lines on kymograph: xcut, position of the lesion; tcut, time of the cut). (c) Filtered z trace of the bead held away from the axon. (d) Filtered z trace of the bead attached to the axon. The high pass filter with cutoff at 10 Hz was previously described37. (e) Recorded traces by back focal plane interferometry of the bead position in the optical trap (upper panel). Blue, green and red traces represent the bead position along x, y and z axis, respectively (Black numbers on the right vertical graph axis indicate the force traces in the x, y directions; red numbers the force along the z direction). Sampling rate is 4 kHz. Black and brown traces are the x and y positions obtained by video tracking. (f) Total amplitude of the force measured in in the x, y and z axis shown in panel e (blue trace: Ftot = √(Fx2 + Fy2 + Fz2)). Total amplitude of the displacement of a scratch performed on the culture support measured by video tracking (in pN), to monitor the stage drift (black trace). (g) Z trace (shown in panel e) filtered with a high pass Butterworth filter with cutoff at 10 Hz. (h) Total variance of the Brownian motion of the bead attached to the axon (t0 and t1 indicate respectively the beginning and the end of tension release in the axon after dissection).
Figure 5e shows the traces of the force measurements (x, y, z components) performed by back focal plane interferometry during laser ablation. With such tracking method, we can measure the tension release in the three orthogonal direction of space with high sensitivity (sub-pN) and high temporal resolution (sub-millisecond). Figure 5f shows the total amplitude of the forces measured in figure 5e (calculated as Ftot = √(Fx2 + Fy2 + Fz2)) and the control trace in black represents the video tracking of a scratch on the culture support, used to correct the force measurements for the stage drift. A relevant advantage of interferometric versus video tracking is its kHz temporal bandwidth, which allows us to detect changes in the Brownian motion of the bead due to local variation of environment around the probe.
In figure 5g, the filtered z component trace (high pass filter with cutoff at 10 Hz as previously described37) is illustrated, showing the changes in the oscillation range of the trapped probe attached to the axon. Figure 5h indicates the total variance of the Brownian motion of the trapped bead determined in 50 ms steps for overlapping time windows of 500 ms (2000 data points)38 after high pass filtering of the x, y, z displacement traces. By filtering the traces and monitoring only the changes for noise, the component due to the membrane flow or cortical actin motion are excluded. Thus, the measurements represent the Brownian motion of the bead which is influenced by its coupling to the cell membrane. The decrease of Brownian motion detected immediately after ablation is unlikely to be due to a tighter coupling of the bead to the surface but rather, it suggests an increase in the viscosity of the membrane due to its strain39. After the delivery of UV laser energy, the variance of the Brownian motion of the bead indicates the beginning (decrease of the variance, t0 in figure 5h) and the end (start of the plateau, t1 in figure 5h) of the membrane strain. Therefore, the duration of tension release can be accurately detected by a decrease of the Brownian motion fluctuations and it is composed of a fast phase of few tens of seconds and a slow phase that could last few minutes. The force measured before t0 is due to the displacement of the probe from the trap centre, when the bead contacts the neurite and it is subtracted from the measure of the release of tension after ablation. Therefore, the release of tension was quantified as the difference in the total force measured at t0 and t1 (figure 5f).
Such measurements were performed on a total of nine neurons from three preparations incubated in the absence or presence of BDNF at 3 DIV (figure 6). As a control of the experimental protocol, we show the lack of correlation between the measured tension release and the UVA energy delivered at the sample, for an energy range below the one needed to completely sever the axon (figure 6a). The measured tension was normalized with respect to the neurite area. The magnitude of the released tension was 2.5 fold higher in neurons incubated with BDNF with respect to that of control neurons (figure 6b), suggesting a decrease in the number of contact points with the ECM.

The release of tension during laser ablation of hippocampal neurons is higher in the presence of BDNF.
(a) Plot of the tension released versus UVA energy delivered to the sample. (b) Quantification of the released tension, normalized for the neurite-substrate contact area between the UVA spot and the bead position, in control and BDNF-incubated neurons (1-3DIV n = 9; 1-3DIV BDNF n = 9; p-value: ** = 0.0037). The significance with an alpha value of 0.05 was calculated by an unpaired two-tailed t-test. Data are the mean ± standard errors.
Force spectroscopy during regeneration
During regeneration of an injured axon, the cytoskeletal components of the neuron re-organize to rebuild the intracellular architecture and re-establish the lost contacts with the ECM. To study the dynamics of such process, force spectroscopy measurements were performed on the bead attached to the membrane of the axon during and after laser dissection on 23 neurons. The fibronectin-coated bead behaves as a local adhesion site with known stiffness40. Because re-growth of the neurite is always occurring along the same path of the degenerated axon and the bead is very close to the lesion site, it is possible to monitor the forces that the cell exerts on the contact with the ECM during axonal re-growth41. Figure 7a shows the damage site (white arrow) and the morphology of the regenerating axon. An actin wave has formed 8 minutes after the lesion (black arrows). Figure 7b shows the force traces recorded in two subsequent phases. The force spectroscopy measurements obtained after neurite injury correlated with the magnitude of the forces exerted by the cell on newly formed adhesion contacts (figure 7b, left panels). During regeneration, the neuron reconstructs de novo its focal adhesions and establishes a contact between fibronectin on the bead and the cytoskeletal network. The relaxation of the membrane was monitored as an increase in the total variance (after t1 in figure 7b left panels), which correlates with the wave approaching the bead (figure 7a, upper right panels at 8 and 9.4 minutes). In the measurements performed between 11 and 20 minutes after the lesion (figure 7b, right panels), an increase followed by a decrease in the variance was detected.

Force spectroscopy measurements during axonal regeneration.
(a) Bright field images acquired during ablation and regeneration of the injured axon. A fibronectin coated bead (silica bead, Ø 4 μm) attached to an axon of a hippocampal neuron (2 DIV) and held in an optical trap. The average power of the IR laser at the sample is 35 mW. The white arrow indicates the site of damage and black arrows indicate the formation of a lamellipodium. Bar is 5 μm. (b) Traces obtained by back focal plane interferometry of the bead position in the optical trap (upper panels). Blue, green and red traces are from the x, y and z position of the bead, respectively. Right panel only: black numbers on right vertical graph axis indicate the force traces in the x and y direction; red numbers the force in the z axis). Total variance of the Brownian motion of the bead attached to the axon during and after UVA laser dissection. t0 and t1 indicate the beginning and the end of tension release, respectively (lower panels). Sampling rate is 4 kHz. (c) Bright field image of a hippocampal neuron with a fibronectin coated bead (silica bead, Ø 4 μm) attached to the axon. The time-lapse imaging started 5 minutes after the axon lesion (white arrow). Bar is 5 μm. (d) Magnification of the bead position (square box in panel c) taken at 20, 80 and 185 seconds. The upper right panel shows the intensity profile along the green line (top left to bottom right, 1 μm) in the first frame to detect the bead displacement. The dashed black, red and blue lines correspond to intensity profile calculated on the three frames (from left to right, respectively). The lower panel shows the total variance of the bead Brownian motion recorded between 0 and 200 seconds, 5 minutes after axon lesion. The red and blue arrows indicate the level of the variance corresponding to the events shown in the upper panel.
Figure 7c shows a bead attached to an injured axon and the bead position at different times is evident in the magnifications in figure 7d. At 80 seconds, the bead is displaced toward the cell body, while, at 185 seconds, it is displaced toward the tip of the axon (figure 7d, upper panels). The bead movements correspond to changes in the total variance (figure 7d, lower panel, red and blue arrows) recorded during the regeneration phase (5 minutes after the axon ablation). The variance at 20 seconds is 1113 nm2, at 80 seconds it decreases to 837 nm2 and at 185 seconds increases to 2307 nm2. The variance reduction corresponds to the displacement of the bead toward the cell body, whereas its increase to a displacement of the bead toward the tip.
Discussion
The response to axotomy varies and depends on factors such as age42, species43, nature of the injury and distance of the injury from the soma44. Many studies focused on the pharmacological or genetic blockade of the inhibitory activity of myelin45 to enable axon regeneration. However, not all neurons are able to regenerate even in a minimized inhibitory environment. Thus, the understanding of the molecular mechanisms involved in the intrinsic ability of the axon to re-grow plays a key role in the identification of new strategies promoting nerve regeneration.
A high ablation efficiency can be achieved with short wavelength illumination sources and with a lower average power reaching the sample, thereby reducing the collateral damage to the neighboring structures caused by heat transfer28. With the optical parameters used to obtain a partial damage of the axon and of its cytoskeleton without inducing a complete separation of the membrane, we observed that neurons between 1 and 3 DIV display a higher regeneration capacity. These results confirm that the protocol of laser dissection is not affecting neuronal regeneration even in neurons at early DIVs, in which ablation is performed closer to the cell body because of their shorter neurites. Outgrowth of axons during development, as well as regeneration after injury in the nervous system, is controlled by specific extracellular cues that are either diffusible or bound to cell membranes or to the extracellular matrix. These extracellular signals are integrated by the axon through distinct intracellular pathways that converge onto the cytoskeleton. During regeneration of the injured axons, we observed the formation of travelling growth cone-like structures along the dissected neurite. Different works point out the role of actin waves in neurite outgrowth in vivo and in vitro35,46 and hypothesize on their role in axonal regeneration. Moreover, it has been shown that neurons in the dentate gyrus of the hippocampus, the main site of adult neurogenesis, present robust actin waves35. Consistently, in our result, only neurons at 1–3 DIV show high probability of axonal regeneration. However, after axonal injury, the number and rate of propagation of the actin waves dramatically decreased, possibly due to the ablation of actin microfilaments. BDNF does not appear to influence the formation of actin waves during the physiological process of axon elongation under steady state conditions. However, upon injury, it induces a 4 fold increase in the number of actin waves and a 1.6 fold increase in their rate of propagation in comparison to neurons lesioned in the absence of BDNF. It is thus conceivable that, in response to the lesion, the cell would elicit a signaling pathway activated by BDNF to induce actin waves formation, which are necessary for the transport of new material to initiate the process of healing and re-growth. Cofilin, an actin severing protein, induces actin depolymerization and increases the dynamics of microfilaments in the growth cone. BDNF has been shown to induce an increase in growth cone filopodial length that could be mimicked by inhibition of ROCK, a cofilin inactivator47. Immunostaining for inactive phosphorylated cofilin was reduced after BDNF treatment, suggesting that cofilin activation may transduce the BDNF effect48. ROCK also regulates the activity of myosin light chain (MLC). MLC induces inhibition of neurite outgrowth49 through actomyosin contraction, a key step in cytoskeletal rearrangement50. Cdc42 and RhoA pathways converge on the regulation of myosin activity51 and BDNF activates cdc42 and rac1. RhoA activation and MLC phosphorylation were inhibited by pretreatment with BDNF52 indicating MLC as another possible downstream target of BDNF. In order to regenerate after axotomy, neurons should activate a cell-autonomous program that will end in plastic changes in their network. While a complete actin depolymerization, such as that induced by latrunculin treatment, induces collapse of the growth cone and prevents the formation of filopodia or lamellipodia16, a high rate of actin turnover is nevertheless necessary for lamellipodia formation and neurite elongation, transforming the dissected axon into a highly motile tip. Indeed, it has been shown in Aplysia neurons that cytoskeleton reorganization promotes growth cone formation and elongation of the new axon6. Thus it is plausible that, in the damaged axons, the increase in the number of actin waves induced by pretreatment of neurons with BDNF, would be due to higher actin dynamics mediated through the cofilin or the MLC pathway. The finding that BDNF rescues the rate of propagation of actin waves specifically downstream from the lesion suggests that it may act on the molecular machinery involved in actin turnover and axonal re-growth at a site where transport and possibly accumulation of regulatory molecules is disrupted.
We also showed that BDNF-treated neurons released a larger amount of tension with respect to control neurons. Cytoskeletal filaments arrange in a tensegrity structure where tension and compression forces are in equilibrium and the cell exerts tension on the substrate through the focal adhesion complexes connected to the cytoskeleton. Laser axotomy, inducing structural changes in the cytoskeleton, generates a release of tension in the injured neurite. Experimental and theoretical models indicate that, when a traction force is applied at the tip of a neurite, the force dissipated along the neurite is proportional to the number of adhesion contacts with the ECM53,54,55. A high turnover of focal adhesion complexes, which could also be explained in terms of an enhanced activity of actin filament-severing proteins, would lead to a reduction in the average number of adhesion contacts between the cell and the substrate over time56. Less adhesion contacts would decrease the dissipation of the released tension thereby increasing the force measured during laser axotomy, as we observed upon incubation with BDNF.
Force spectroscopy on the adherent bead was continued during axonal regeneration after the dissection. Before axotomy, the bead was in contact with the apical surface of the axon where no adhesion sites are present. Under these conditions, the ECM-mimicking bead is in a suitable position to sense the forces involved in the formation of new adhesion contacts during regeneration. With our experimental model, these forces can be measured with high sensitivity57. Optical tweezers have already been employed for quantifying forces between the cell and the ECM58,59 that depend on the mechanical properties of the cell membrane and the cytoskeleton60,61 and for modulating the stiffness of the mimetic ECM. From the total variance of the trapped bead, we deduced the strain of the membrane and its subsequent relaxation due to membrane enlargement following vesicle exocytosis62. We detected an increase of the variance when an actin wave was approaching the bead and alternating phases of increase and decrease of the variance, which correlated with the direction of the forces on the bead (toward the neurite tip or the cell body, respectively). The increase in the variance can be correlated to the actin dynamics, which sustain lamellipodia protrusion37. The decrease of the variance, when the force is directed toward the cell body, indicate a distinct cytoskeleton dynamic, such as actin retrograde flow due to myosin II contraction.
The variance after dissection increased non monotonically, albeit without reaching the value observed before axotomy. This could be explained by the mechanical constraint of a known stiffness, provided by the trapped bead that induces the formation of new adhesion contacts.
Therefore, considering that optical tweezers is a well-established technique to study the conversion of chemical energy into mechanical energy in polymer63 and molecular motors, its application in neuronal regeneration studies allows quantifying various parameters such as the released tension after axonal damage and the strength of neurite adhesion to the substrate. Furthermore, following injury, it allows the measuring of forces involved in the reorganization of the cytoskeleton and the formation of new adhesion contacts. By the use of the laser axotomy technique described here, we were able to analyze the axonal regeneration of mouse neurons at early DIV64. Actin waves formed at the cell bodies of damaged neurons are necessary for transport of material, healing of the damage and re-growth of the lesioned axons. A BDNF signaling pathway, activated in response to axonal damage, appears to favor actin waves formation possibly by opposing the dispersion of molecules involved in these processes and by facilitating a rapid turnover of actin. Thus, in the near future it will be of interest to investigate the role of single molecules that affect neuronal regeneration and development, such as cytoskeleton-interacting proteins, in neurons from knock-out and transgenic mice models.
Methods
Optical setup
The entire optical system was described earlier12. Briefly, the trapping source was an ytterbium continuous wave (CW) fiber laser operating at 1064 nm (IPG Laser GmbH). The phase of the IR laser beam was modulated through a spatial light modulator (SLM) (LCOS-SLM, model X10468-07 – Hamamatsu). The interferometer for force spectroscopy measurements was based on a four-quadrant photodiode (QPD, S5980 with C5460SPL 6041 board – Hamamatsu) and a photodiode PD2 (PDA100A-EC - Thorlabs).
The laser dissection source was a pulsed sub-nanosecond UV Nd:YAG laser at 355 nm (PNV-001525-040, PowerChip nano-Pulse UV laser – Teem Photonics).
The holographic tweezers and laser micro-dissector were integrated on a modified upright microscope (BX51 – Olympus) equipped with a 60x, 0.9 NA water dipping objective.
Long-term bright field time-lapse experiments (longer than one day) were performed on a commercial inverted microscope (Eclipse Ti E; Nikon Instruments Inc.) with laser-based autofocus and motorized stage for multipoint sequential image acquisition. The microscope was equipped with a charge-coupled device (CCD) camera (Andor DU-897D-C00), Plan Fluor 40x, 0.75 NA DIC objective and imaging software (Nis elements AR; Nikon Instruments Inc.).
Incubator system
For long-term time-lapse imaging, the neurons must be kept at stable temperature, pH and osmolarity. The temperature was kept at 35°C by a feedback system coupled to a thermocouple in contact with the sample holder (TC-324B, Warner Instruments Corporation). To maintain the physiological pH, a gas of 5% CO2 balanced with air was mixed by a flow meter (CO2BX, Okolab) and injected in the sample holder. To minimize medium evaporation and stabilize its osmolarity, the gas was bubbled through a water reservoir warmed through a load resistance (10 W, 12 V) before being fluxed into the stage incubator. Moreover, a film of oil (polydimethylsiloxane 200 Fluid, 0.913 g/ml, Sigma Aldrich), permeable to CO2 but not to water, was deposited on the culture media. The same system was adapted to the upright microscope, where pH and humidity were controlled by aerating a custom-designed polydimethylsiloxane sleeve, which enclosed the objective and the culture support.
Cell cultures
All the experimental protocols were approved by the Italian Ministry of Health.
Primary cultures were obtained from hippocampi of mice (C57BL6J, Charles River) at embryonic day 18 (E18). Embryos were removed and dissected under sterile conditions. Hippocampi were dissociated by enzymatic digestion in trypsin (0.125% for 20 min. at 37°C). Trypsin activity was blocked by adding complete media (Neurobasal™ (Gibco) supplemented by B27 (2%, Gibco), alanyl-glutamine (2 mM, Gibco), penicillin/streptomycin (both 1 mM, Sigma) containing 10% fetal bovine serum (FBS, Gibco). After trypsinization, tissues were rinsed in complete media without FBS and dissociated with a plastic pipette. Neurons were plated at a concentration of 0.25–1·105 cells/ml on glass-bottom Petri dishes (P35G-0-14-C – MaTek Corporation). After 2 hrs in the incubator to allow cell attachment, the dishes were filled up to 2/3 of their volume with serum-free medium.
To study the effect of BDNF on axonal regeneration, hippocampal neurons were incubated with 100 ng/ml BDNF for 30 minutes.
Silica beads (Ø 4 µm, COOH-terminated– Bangs Laboratories) were coated with fibronectin following the procedure described in the PolyLink Protein Coupling Kit (Polysciences).
Immunohistochemistry
Neurons were fixed in 4 % paraformaldehyde/4 % sucrose in PBS and were incubated with primary antibodies followed by fluorescently conjugated secondary antibodies in 5% goat serum/0.1% Triton in PBS. Antibodies used were: polyclonal anti-NFL (Chemicon), monoclonal anti-βIII-Tubulin (Covance), polyclonal anti-actin (Sigma), phalloidin-Texas Red X (Molecular Probes) and monoclonal anti-Abi-1 (kind gift from Dr. G. Scita, The FIRC Institute of Molecular Oncology, Milan, Italy). The neurons were mounted in ProLong Gold Antifade Reagent with DAPI (Invitrogen). Confocal images were acquired on a Leica TCS SP5 microscope using a 63 x objective (Leica Microsystems).
Measurements
Fibronectin-coated beads were optically trapped and moved into close proximity to the neurite to be dissected (IR power at the sample 35 mW). The position of the trapped probe with respect to the neurite geometry and position of the UV laser focal spot was controlled by displaying a dynamically-generated hologram on the SLM. The trapped probe was positioned 2 μm above the coverslip and about 10 μm away from the UV laser spot. The interference fringes on the back focal plane of the condenser were imaged onto a QPD and PD and the light was converted to voltage outputs filtered and digitized at 5 and 10 kHz, respectively, to calibrate the trap stiffness kx,y,z = (kx, ky, kz) and the detector sensitivity using the power spectrum method16. The calibration of the trap stiffness was repeated for every new position of the trap determined by the SLM, before the attachment of the probe to the neurite.
After calibration, the bead was brought into contact with the neurite to be ablated. After 15 seconds to ensure fibronectin-integrin attachment65, the recording of QPD and PD signals (digitized at 4 kHz and low-pass filtered at 2 kHz) was started simultaneously with the image acquisition by two CCD cameras at different magnification factors and frame rates (1x at 2.5 fps and 0.35x at 1 fps, respectively). About 200–300 UVA laser pulses with energy of 25 nJ per pulse were delivered to inflict the damage to the neurite and in parallel force spectroscopy was performed. A scratch on the culture support was video tracked (in the x and y direction) to quantify the stage drift during the force measurements. The measured displacement (in nm) was multiplied by the stiffness of the optical trap to result in the equivalent of a force (in pN). The release of tension was quantified as the difference in the total force measured at t1 and t0 by interferometric tracking and corrected by the drift of the stage. A measurement session lasted for approximately 20 minutes.
For long-term time lapse imaging, axons of neurons cultured on a gridded Petri dish (MatTek) were dissected and the dish was then moved to the inverted microscope. The injured neurons were localized on the grid and bright field time lapse imaging was continued for about 2 days (with one image every 2 minutes for each of the ten dissected axons).
References
Horner, P. J. and Gage, F. H. Regenerating the damaged central nervous system. Nature 407, 963–970 (2000).
Ghosh-Roy, A. and Chisholm, A. D. Caenorhabditis elegans: a new model organism for studies of axon regeneration. Dev. Dyn. 239, 1460–1464 (2010).
Fleisch, V. C., Fraser, B. and Allison, W. T. Investigating regeneration and functional integration of CNS neurons: Lessons from zebrafish genetics and other fish species. Biochim. Biophys. Acta 1812, 364–380 (2011).
Joshi, H. C., Chu, D., Buxbaum, R. E. and Heidemann, S. R. Tension and compression in the cytoskeleton of PC 12 neurites. J. Cell Biol. 101, 697–705 (1985).
Baas, P. W. and Heidemann, S. R. Microtubule reassembly from nucleating fragments during the regrowth of amputated neurites. J. Cell Biol. 103, 917–927 (1986).
Sahly, I., Khoutorsky, A., Erez, H., Prager-Khoutorsky, M. and Spira, M. E. On-line confocal imaging of the events leading to structural dedifferentiation of an axonal segment into a growth cone after axotomy. J. Comp Neurol. 494, 705–720 (2006).
Wu, T., et al. Neuronal growth cones respond to laser-induced axonal damage. J. R. Soc. Interface (2011).
Bourgeois, F. and Ben-Yakar, A. Femtosecond laser nanoaxotomy properties and their effect on axonal recovery in C. elegans. Opt. Express 16, 5963 (2008).
Kuetemeyer, K., Rezgui, R., Lubatschowski, H. and Heisterkamp, A. Influence of laser parameters and staining on femtosecond laser-based intracellular nanosurgery. Biomed. Opt. Express 1, 587–597 (2010).
Usher, L. C., et al. A chemical screen identifies novel compounds that overcome glial-mediated inhibition of neuronal regeneration. J. Neuroscience 30, 4693–4706 (2010).
Colombelli, J., et al. Mechanosensing in actin stress fibers revealed by a close correlation between force and protein localization. J. Cell Science 122, 1665–1679 (2009).
Difato, F., et al. Combined optical tweezers and laser dissector for controlled ablation of functional connections in neural network. J. of Biomed. Opt. 16 (2011).
Guo, S. X., et al. Femtosecond laser nanoaxotomy lab-on-a-chip for in vivo nerve regeneration studies. Nature Methods 5, 531–533 (2008).
Samara, C., et al. Large-scale in vivo femtosecond laser neurosurgery screen reveals small-molecule enhancer of regeneration. Proc. Natl. Acad. Sci. U. S. A. 107, 18342–18347 (2010).
Mogilner, A. On the edge: modeling protrusion. Current Opinion in Cell Biology 18, 32–39 (2006).
Cojoc, D., et al. Properties of the force exerted by filopodia and lamellipodia and the involvement of cytoskeletal components. PLoS. ONE 2, e1072 (2007).
Kress, H., et al. Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc. Natl. Acad. Sci. U. S. A. 104, 11633–11638 (2007).
Carlsson, A. E. Growth of branched actin networks against obstacles. Biophysical J. 81, 1907–1923 (2001).
Janson, M. E. and Dogterom, M. Scaling of microtubule force-velocity curves obtained at different tubulin concentrations. Physical Review Letters 92 (2004).
Footer, M. J., Kerssemakers, J. W. J., Theriot, J. A. and Dogterom, M. Direct measurement of force generation by actin filament polymerization using an optical trap. Proc. Natl. Acad. Sci. U. S. A. 104, 2181–2186 (2007).
Yuan, A., et al. Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J. Neuroscience 29, 11316–11329 (2009).
Svoboda, K., Schmidt, C. F., Schnapp, B. J. and Block, S. M. Direct observation of kinesin stepping by optical trapping interferometry. Nature 365, 721–727 (1993).
Pantaloni, D., Le Clainche, C. and Carlier, M. F. Mechanism of actin-based motility. Science 292, 2012 (2001).
Giannone, G., et al. Periodic lamellipodial contractions correlate with rearward actin waves. Cell 116, 431–443 (2004).
Allioux-Guerin, M., et al. Spatio-temporal analysis of cell response to a rigidity gradient: a quantitative study by multiple optical tweezers. Biophysical J. (2008).
Siechen, S., Yang, S., Chiba, A. and Saif, T. Mechanical tension contributes to clustering of neurotransmitter vesicles at presynaptic terminals. Proc. Natl. Acad. Sci. U. S. A. 106, 12611–12616 (2009).
Jagannathan Rajagopalan,. Alireza Tofangchi,. and Saif, T. Drosophila Neurons Actively Regulate Axonal Tension In Vivo. Biophysical J. 99 (2010).
Colombelli, J., Reynaud, E. G. and Stelzer, E. H. Investigating relaxation processes in cells and developing organisms: from cell ablation to cytoskeleton nanosurgery. Methods Cell Biol. 82, 267–291 (2007).
Tanner, K., Boudreau, A., Bissell, M. J. and Kumar, S. Dissecting regional variations in stress fiber mechanics in living cells with laser nanosurgery. Biophysical J. 99, 2775–2783 (2010).
Wen, Z. and Zheng, J. Q. Directional guidance of nerve growth cones. Current Opion in Neurobiol. 16, 52–58 (2006).
Ingber, D. E. Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Science 116, 1157–1173 (2008).
Tsai, P. S., et al. Plasma-mediated ablation: an optical tool for submicrometer surgery on neuronal and vascular systems. Current Opinion in Biotechnology 20, 90–99 (2009).
Goslin, K. and Banker, G. Experimental observations on the development of polarity by hippocampal neurons in culture. J. Cell Biol. 108, 1507–1516 (1989).
Ruthel, G. and Banker, G. Role of moving growth cone-like wave structures in the outgrowth of cultured hippocampal axons and dendrites. J. Neurobiol. 39, 97–106 (1999).
Flynn, K. C., Pak, C. W., Shaw, A. E., Bradke, F. and Bamburg, J. R. Growth cone-like waves transport actin and promote axonogenesis and neurite branching. Dev. Neurobiol. 69, 761–779 (2009).
Disanza, A., et al. Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nature Cell Biol. 6, 1180–1188 (2004).
Shahapure, R., et al. Force generation in lamellipodia is a probabilistic process with fast growth and retraction events. Biophysical J. 98, 979–988 (2010).
Kress, H., Stelzer, E. H., Griffiths, G. and Rohrbach, A. Control of relative radiation pressure in optical traps: application to phagocytic membrane binding studies. Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys. 71, 061927 (2005).
Togo, T., Krasieva, T. B. and Steinhardt, R. A. A decrease in membrane tension precedes successful cell-membrane repair. Mol. Biol. Cell 11, 4339–4346 (2000).
Emiliani, V., Sanvitto, D., Zahid, M., Gerbal, F. and Coppey-Moisan, M. Multi force optical tweezers to generate gradients of forces. Opt. Express 12, 3906–3910 (2004).
Vogel, V. and Sheetz, M. P. Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways. Current Opinion in Cell Biol. 21, 38–46 (2009).
Lagunowich, L. A., Schneider, J. C., Chasen, S. and Grunwald, G. B. Immunohistochemical and biochemical analysis of N-cadherin expression during CNS development. J. Neuroscience Res. 32, 202–208 (1992).
Rossi, F., Buffo, A. and Strata, P. Regulation of intrinsic regenerative properties and axonal plasticity in cerebellar Purkinje cells. Restor. Neurol. Neurosci. 19, 85–94 (2001).
Fernandes, K. J., Fan, D. P., Tsui, B. J., Cassar, S. L. and Tetzlaff, W. Influence of the axotomy to cell body distance in rat rubrospinal and spinal motoneurons: differential regulation of GAP-43, tubulins and neurofilament-M. J. Comp Neurol. 414, 495–510 (1999).
Giger, R. J., et al. Mechanisms of CNS myelin inhibition: evidence for distinct and neuronal cell type specific receptor systems. Restor. Neurol. Neurosci. 26, 97–115 (2008).
Bretschneider, T., et al. The three-dimensional dynamics of actin waves, a model of cytoskeletal self-organization. Biophysical J. 96, 2888–2900 (2009).
Hsieh, S. H., Ferraro, G. B. and Fournier, A. E. Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and Slingshot phosphatase. J. Neuroscience 26, 1006–1015 (2006).
Gehler, S., Shaw, A. E., Sarmiere, P. D., Bamburg, J. R. and Letourneau, P. C. Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J. Neuroscience 24, 10741–10749 (2004).
Wylie, S. R. and Chantler, P. D. Myosin IIA drives neurite retraction. Mol. Biol. Cell 14, 4654–4666 (2003).
Brown, M. E. and Bridgman, P. C. Myosin function in nervous and sensory systems. J. Neurobiol. 58, 118–130 (2004).
Yuan, X. B., et al. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nature Cell Biol. 5, 38–45 (2003).
Zhang, Y., Zhao, J., Wang, J. and Jiao, X. Brain-derived neurotrophic factor inhibits phenylalanine-induced neuronal apoptosis by preventing RhoA pathway activation. Neurochem. Res. 35, 480–486 (2010).
O'Toole, M., Latham, R., Baqri, R. M. and Miller, K. E. Modeling mitochondrial dynamics during in vivo axonal elongation. J. Theor. Biol. 255, 369–377 (2008).
O'Toole, M., Lamoureux, P. and Miller, K. E. A physical model of axonal elongation: force, viscosity and adhesions govern the mode of outgrowth. Biophysical J. 94, 2610–2620 (2008).
O'Toole, M. and Miller, K. E. The role of stretching in slow axonal transport. Biophysical J. 100, 351–360 (2011).
Thoumine, O. Interplay between adhesion turnover and cytoskeleton dynamics in the control of growth cone migration. Cell Adh. Migr. 2, 263–267 (2008).
F. Difato, et al. Optical Tweezers Microscopy: PicoNewton Forces in Cell and Molecular Biology. in Nanoscopy and Multidimensional Optical Fluorescence Microscopy, 2010 ed. edited by A. Diaspro (Taylor & Francis, 2010).
Giannone, G. and Sheetz, M. P. Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends in Cell Biology 16, 213–223 (2006).
Sheetz, M. P., Sable, J. E. and Dobereiner, H. G. Continuous membrane-cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu. Rev. Biophys. Biomol. Struct. 35, 417–434 (2006).
Raucher, D. and Sheetz, M. P. Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol. 148, 127–136 (2000).
Rossier, O. M., et al. Force generated by actomyosin contraction builds bridges between adhesive contacts. EMBO J. 29, 1055–1068 (2010).
Togo, T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J. Cell Science 119, 2780–2786 (2006).
Mogilner, A. and Oster, G. Force generation by actin polymerization II: The elastic ratchet and tethered filaments. Biophysical J. 84, 1591–1605 (2003).
Kim, Y. T., Karthikeyan, K., Chirvi, S. and Dave, D. P. Neuro-optical microfluidic platform to study injury and regeneration of single axons. Lab Chip 9, 2576–2581 (2009).
Thoumine, O., Kocian, P., Kottelat, A. and Meister, J. J. Short-term binding of fibroblasts to fibronectin: optical tweezers experiments and probabilistic analysis. Eur. Biophysical J. 29, 398–408 (2000).
Acknowledgements
We thank A. Diaspro for insightful discussion and F. Succol and M. Nanni for their expert advice and assistance in cell culture preparation. This work was supported by grants from Telethon-Italy (GGP10109 to E. C., GGP09134 to F. B.).
Author information
Authors and Affiliations
Contributions
FD performed the experiments, prepared the figures and wrote the manuscript, HT performed immunohistochemistry experiments and the statistical analysis, MP performed long term imaging, AB developed the long term incubator system and EC conceived the experiments and wrote the main manuscript. All the authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Difato, F., Tsushima, H., Pesce, M. et al. The formation of actin waves during regeneration after axonal lesion is enhanced by BDNF. Sci Rep 1, 183 (2011). https://doi.org/10.1038/srep00183
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00183
This article is cited by
-
The Axonal Actin Filament Cytoskeleton: Structure, Function, and Relevance to Injury and Degeneration
Molecular Neurobiology (2024)
-
Functional tissue-engineered microtissue formed by self-aggregation of cells for peripheral nerve regeneration
Stem Cell Research & Therapy (2022)
-
Caffeic Acid Phenethyl Ester (CAPE) Protects PC12 Cells Against Cisplatin-Induced Neurotoxicity by Activating the AMPK/SIRT1, MAPK/Erk, and PI3k/Akt Signaling Pathways
Neurotoxicity Research (2019)
-
Differentiation of adipose-derived stem cells into Schwann cell-like cells through intermittent induction: potential advantage of cellular transient memory function
Stem Cell Research & Therapy (2018)
-
Mesenchymal stem cells secretome-induced axonal outgrowth is mediated by BDNF
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
