
Abstract
The accessibility of skin makes it an ideal target organ for nucleic acid-based therapeutics; however, effective patient-friendly delivery remains a major obstacle to clinical utility. A variety of limited and inefficient methods of delivering nucleic acids to keratinocytes have been demonstrated; further advances will require well-characterized reagents, rapid noninvasive assays of delivery and well-developed skin model systems. Using intravital fluorescence and bioluminescence imaging and a standard set of reporter plasmids we demonstrate transfection of cells in mouse and human xenograft skin using intradermal injection and two microneedle array delivery systems. Reporter gene expression could be detected in individual keratinocytes, in real-time, in both mouse skin as well as human skin xenografts. These studies revealed that non-invasive intravital imaging can be used as a guide for developing gene delivery tools, establishing a benchmark for comparative testing of nucleic acid skin delivery technologies.
Similar content being viewed by others

Introduction
The impressive progress in identifying the underlying causes of many epidermal disorders, including genodermatoses, have led to potential therapeutic targets and promising intervention strategies; however, translation of such novel therapies into the clinic has not been achieved.1 Nucleic acid-based therapies are strong candidates for many skin disorders that lack effective treatment options.2,3,4 However, realization of this goal will require new methods for efficient delivery through the stratum corneum and efficient uptake and utilization of the nucleic acid therapeutic by targeted cells. At present, no nucleic acid skin delivery methodologies exist that result in safe and efficient delivery across these biological barriers without causing pain or tissue damage. To address this need, a consortium of researchers with diverse yet complementary expertise in skin delivery was established5 to address the following specific aims: 1. develop and test patient-friendly skin delivery technologies that effectively and efficiently deliver nucleic acids to skin; 2. combine appropriate and promising technologies to overcome the delivery barriers and; 3. enable direct comparison of existing delivery technologies through developing and making available to the research community, standardized and validated tools, including intravital imaging systems, mouse and human xenograft skin models and common reagents. Validated reagents and verified skin models that allow meaningful comparisons are required for effective development of nucleic acid delivery systems.
Successful development of nucleic acid therapies would clearly benefit from non-invasive imaging modalities that enable visualization of expression patterns in skin strata in real time. The use of a dual reporter system (e.g., expression of luciferase and fluorescent protein from a single plasmid construct) allows for increased data acquisition since both bioluminescence and fluorescence imaging modalities can be utilized. In vivo bioluminescence imaging (BLI) has been shown to be a versatile and sensitive technique for tracking and quantifying gene expression and assessing delivery efficiency using reporter genes such as firefly luciferase.6,7,8 Despite its strengths, BLI has relatively low resolution and therefore detection and identification of individual reporter-positive cells requires fluorescence imaging. Imaging of fluorescent reporters offers a complementary approach to BLI for spatiotemporal analysis of reporter gene expression with the possibility of cellular and sub-cellular resolution.9,10 Real-time confocal scanning microscopy has been adapted to image cells in living tissue and is being applied to basic research problems and is finding utility in clinical studies. For example, confocal reflectance imaging is used to diagnose skin cancers, as well as other skin disorders, using intrinsic optical properties of tissue.11,12,13,14,15 High-resolution fluorescence microscopes capable of intravital skin imaging have been described.6,9,16 For these studies, we chose to use a single axis confocal fluorescence imaging system to take advantage of its high resolution and real time data acquisition capabilities.
To test the utility of combining bioluminescence imaging and fluorescence microscopy to study plasmid delivery and expression in vivo, common stocks of standardized dual-reporter (luciferase and GFP) plasmids were evaluated. The plasmids were administered via intradermal (ID) injection and delivery and expression was compared to that of two different microneedle technologies: soluble protrusion array devices (PADs)17 and coated metal microneedles.18,19 Overall plasmid expression was quantified at the tissue level in vivo using bioluminescence (luciferase), which served as a guide for fluorescence microscopy that was used to localize individual GFP-positive cells (keratinocytes) within the luminescent regions of mouse footpad. This combination of technologies and standard reagents establishes the foundation of an approach for rapidly testing and validating other nucleic acid delivery technologies.
Results
Dual-mode reporter plasmids enable localization of regions of gene transfer and allows single keratinocyte detection in mouse footpad skin
In an effort to standardize gene delivery tools, a series of expression plasmids utilizing different constitutive human promoters was prepared and evaluated. These vectors co-express a modified version of the bioluminescent firefly luciferase enzyme (luc2, for quantitative analysis) fused to enhanced green fluorescent protein (eGFP), which allows localization and characterization of the transfected cells within the epithelium. The pUbc-luc2/eGFP plasmid (Fig. 1A), which utilizes the human ubiquitin C (Ubc) promoter, was chosen for further study based on the strength and uniformity of reporter expression throughout the various layers of the epidermis (Fig. 1B,C). This construct was evaluated against pCMV-hMGFP/CBL (Fig. S1A), a construct previously developed to monitor gene transfer to skin that expresses click beetle luciferase (CBL) fused to humanized Monster green fluorescence protein (hMGFP) under the control of the cytomegalovirus (CMV) promoter.6,17

Detection of reporter plasmid expression in mouse footpad skin.
(A) Schematic map of pUbc-luc2/eGFP plasmid. (B) Bioluminescence imaging of pUbc-luc2/eGFP and pCMV-hMGFP/CBL (left and right paws, respectively) expression 24 h after intradermal paw injection. (C) Fluorescence microscopy of a skin section obtained from a paw previously imaged for bioluminescence (see Panel B, mouse 3, left paw) injected with pUbc-luc2/eGFP (scale bar is 50 µm). DAPI stain (blue) was used to identify cellular nuclei. Left panel shows brightfield overlay. Abbreviations: Ampr, ampicillin resistance; eGFP, enhanced green fluorescence protein; luc2, luciferase 2; Ubc, ubiquitin c; p, photons; sr, steradian.
The pUbc-luc2/eGFP and pCMV-hMGFP/CBL plasmids were introduced into mouse footpad skin (left and right, respectively) via ID injection and expression levels were imaged 24 h later. Bioluminescent signals were initially imaged and quantified to localize regions of interest and guide the fluorescence microscopy. Although some variability was observed, both constructs expressed similar levels (n = 6 mice, p-value = 0.38) of the reporter (Fig. 1B). The distribution of pUbc-luc2/eGFP expression through all layers of the epidermis was confirmed by traditional fluorescence microscopy of frozen skin sections prepared from treated mice (Fig. 1C). In contrast, pCMV-hMGFP/CBL expression was confined to the upper layers (granular and stratum corneum) of the epidermis (Fig. S1), as previously reported.6,20,21,22,23,24,25
Following BLI, the mice with the paws expressing the highest levels of luciferase were subsequently imaged with a confocal intravital fluorescence/reflectance Lucid VivaScope® imaging system. This desktop confocal scanning laser microscope was initially designed for reflectance imaging of skin for clinical diagnostic applications and was recently modified, at our request, to allow fluorescence imaging via a 488 nm laser (see Materials and Methods). Using this imaging system, mouse paw skin was initially imaged in the reflectance mode to identify the various skin layers by cell morphology and tissue structure (see Materials and Methods). Once the desired layer for analysis was localized, the tissue was scanned using the 488 nm laser (see Fig. 2A for a schematic describing the system's capabilities).

In vivo visualization of eGFP-positive keratinocytes in mouse footpad skin.
(A) Schematic representation of the scanning capabilities of the Lucid VivaScope 2500 intravital imaging system. The scanning dimensions (left panel, top) are 1.2×1.2 cm. A 4×3 VivaBlock (right panel, top) is depicted with a depth capability of 250 μm. A single VivaBlock (bottom panel) is shown with 10 slices (each 5 μm thick) containing a 750×750 μm field of view. (B,C) Visualization of individual eGFP-positive cells. In vivo imaging of eGFP-expressing keratinocytes 24 h following ID injection of pUbc-luc2/eGFP into mouse footpad skin. Mouse paws were intravitally imaged in fluorescence (488 nm laser) mode (left panels) at 20.8 (B) and 32 (C) µm depths and their corresponding reflectance mode images are shown (right panels). In reflectance mode (right panels), the microscope allows visualization of cell morphology and skin layers. Co-localization of cells expressing eGFP (left panels) with skin structure facilitates identification of the keratinocyte differentiation state. Scale bar is 100 µm. (D) 3D reconstructed (40 slices) image showing top (from stratum corneum) and bottom (from dermis) views. Cells are pseudocolored green. SC, stratum corneum. (B–D) Field of view is 750×750 µm.
After fluorescing cells were detected, a stack (Z-map) of 40 slices (each 5 µm thick) was obtained, first in fluorescence mode and then in reflectance mode, with a separation of 1.6 µm between slices. Individual cells expressing eGFP in mouse footpad skin were clearly detected (Fig. 2B,C, left panels). Using the reflectance mode of the instrument allows discrimination of epidermal layers based on depth, size and granularity. Granular cells and corneocytes are larger, irregular and granular (Fig. 2B, right panel) while basal cells and lower spinous cells are small and regular (Fig. 2C, right panel). Squame-like flattened cells are visualized when the tissue is visualized from above the epidermis (Fig. 2D, top image) while small and rounded cells (with basal or spinosum layer characteristics) are also readily distinguished in a 3D reconstruction rendered from the Z-stack series (Fig. 2D, bottom image, viewed from below the epidermis).
Clear differences in pCMV-CBL/hMGFP and pUbc-luc2/eGFP expression profiles were observed. For example, while eGFP expression (from pUbc-luc2/eGFP) tended to distribute homogenously through the entire cell (Fig. 1C , 2B and C and Supplemental Video 1), including the nucleus, hMGFP expression (from the pCMV-CBL/hMGFP plasmid), which has been previously shown to form aggregates when fused with CBL in mouse keratinocytes,6,9 preferentially accumulated in the cytoplasm, with little or no nuclear localization (Fig. S1B and Supplemental Video 2). Furthermore, the confocal fluorescence intravital imaging of Ubc promoter-expressed eGFP resulted in a wide distribution through all layers of the epidermis, including the basal layer keratinocytes (Fig. 2C and D [bottom] and Supplemental Video 1), while CMV-mediated expression led to localized hMGFP expression in the upper (mainly granulosum) layers of the epidermis (Fig. S1D) and was rarely detectable in deeper layers (Fig. S1 and Supplemental Video 2). A similar CMV promoter-mediated hMGFP distribution pattern was observed by traditional fluorescence microscopy of skin sections (Fig. 1C and Fig. S1D, respectively), corroborating the intravital imaging results.
Visualization of eGFP-positive cells in human skin xenografts
For a predictable and uniform human skin target, we utilized a commercially available organotypic 3D human skin culture (StrataTest®, see Materials and Methods). Four weeks after transplantation of the human skin equivalent onto immunocompromised mice, the grafted skin showed little or no evidence of inflammation and the human epidermis could be readily distinguished from mouse epidermis by a human keratin 1-specific antibody (Fig. S2).
To determine if reporter gene expression could be imaged in human skin xenografts with cellular resolution, the pUbc-luc2/eGFP plasmid was injected intradermally into transplanted human skin. After 24 h, the human skin, on live anaesthetized mice, was imaged by confocal fluorescence microscopy. Individual keratinocytes expressing the fluorescence reporter were found at or near the injection site and Z-stack images were acquired from the top of the skin to a depth of approximately 65 μm (see Supplemental Video 3). EGFP-positive epidermal keratinocytes were clearly visualized (Fig. 3A) and remained detectable to a depth of approximately 32 μm. The mice were sacrificed 48 h after treatment and the human skin was removed and cryosectioned for analysis by direct fluorescence microscopy and immunostaining. The eGFP-expressing cells were identified as human keratinocytes by colocalization following immunostaining with a human keratin 1-specific antibody (Fig. 3C–F).

Fluorescence microscopic imaging of eGFP reporter plasmid expression in human skin xenografts.
(A) Following ID injection (24 h) of reporter plasmid (pUbc-luc2/eGFP) into human xenografts (grafted on immunocompromised mice), individual keratinocytes expressing eGFP (left panel) are readily observed by intravital microscopy (Lucid system) using the fluorescence mode at 13 µm depth (the corresponding reflectance mode image is shown in the right panel). The scale bar is 50 µm. (B) Fluorescence microscopy of frozen sections from skin taken from previously imaged mice (see panel A), 48 h after injection (scale bar is 20 µm). Brightfield-fluorescence overlay (top left panel). Fluorescence overlay (top right and bottom panels). eGFP and human keratin 1 are shown as green and red, respectively. DAPI staining (blue) shows nuclei.
Plasmid expression following delivery by PAD or coated metal microneedles to epidermal keratinocytes can be repeatedly monitored in vivo over time
Once the utility of the imaging systems and reporter plasmids were validated as outcome measures, two distinct microneedle designs (coated metal microneedles and PADs) were utilized. Coated metal microneedles (Fig. 4A, bottom) or soluble PADs (Fig. 4A, top) of similar needle lengths (see Fig. 4B and C for side-by-side comparison) were loaded with plasmid DNA cargo and applied to mouse footpad skin. Bioluminescent signals from microneedle-delivered plasmid were monitored over time in live animals. Initially, signals from tissues treated with both types of microneedle arrays were similar (Fig. 5A–D). However, while mouse paws treated with PAD arrays maintained constant reporter gene expression for at least 24 days (Fig. 5A,B), the paws treated with coated metal microneedles showed increased bioluminescence during the first week ( Fig. 5C,D ) and then reached a higher plateau. In both cases, metal (Fig. S3, top) and PAD (Fig. S3, bottom), bioluminescent signals were detected after 72 days following application. Similar expression, albeit lower (2–6 fold lower expression for metal coated needles from day 2 until plateau; 2-fold from days 1 to 14 for PADs), was observed following delivery of pCMV-CBL/hMGFP using both types of microneedle arrays (Fig. S4).

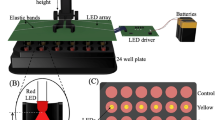
Protrusion array device (PAD) and coated metal microneedle arrays.
(A) Photographic comparison of PADs and metal microneedle arrays. PADs containing 3×5 needles per array (top photograph) are manufactured from polyvinyl alcohol and are designed to dissolve and release cargo when applied to skin. Metal microneedles (1×5 needles per array) are coated with cargo to allow rapid delivery to skin following penetration (bottom photograph). Scale bars = 1,000 µm. (B) Front (left) and side (right) magnified view of PAD microneedle. Scale bar = 500 μm. (C) Magnified view of metal microneedle. Scale bar = 500 µm.

Intravital imaging of reporter expression in mouse skin following delivery of plasmid by soluble and steel microneedles.
One microneedle array loaded with reporter plasmid was applied per paw and subjected to in vivo imaging. Reporter expression (bioluminescence signal) was monitored in vivo over time following administration of pUbc-luc2/eGFP either with PAD (A,B) or steel (C,D) microneedles. Bioluminescent images (A,C) were generated and the amount of luciferase signal quantified in photons/second/cm2/steradian and plotted (B,D) Mouse left (LP) and right (RP) paws were treated (n = 4) with pUbc-luc2/eGFP and control DNA, respectively. Mouse paws treated with PADs were further in vivo imaged with the VivaScope confocal microscope, acquiring image stacks from the skin surface to a depth of ∼65 µm. (E) En face image of a Z-stack (merge of 40 slices). (F) Frozen sections were prepared from paw skin and imaged by traditional fluorescence microscopy as in Fig. 1 . Scale bar is 20 µm. (G) 3D reconstruction of 40 slices showing top view from stratum corneum (top 3D image) and bottom view from dermis (bottom image). Cells are pseudocolored green.
EGFP gene expression in individual epidermal cells following microneedle application could also be visualized with the confocal microscope. Paws exhibiting high bioluminescent signals (indicating functional plasmid delivery) were selected and subjected to confocal fluorescence imaging. As an example, eGFP-positive cells in mouse epidermis are shown in Fig. 5E (merged stack of 40 slices) and Fig. 5G (3D reconstruction) extending from the granulosum [top] to the basal [bottom] layer, 13 to 64 μm depth, 24 h after applying PAD microneedles loaded with plasmid. Fluorescence microscopy of frozen skin sections confirmed that eGFP-expressing keratinocytes were localized in the epidermis of PAD-treated paws (Fig. 5F). EGFP-expressing cells were also observed following delivery of plasmid coated metal microneedles (data not shown). Cells expressing eGFP were also visualized using the fluorescence imaging system in human xenografts 24 h following PAD application (Fig. 6A). These cells are likely keratinocytes based on their morphology and location as determined using the reflectance mode of the VivaScope imaging system (Fig. 6B).

Fluorescence imaging of eGFP reporter plasmid expression in human skin xenografts after PAD application.
Following PAD application (24 h) of reporter plasmid (pUbc-luc2/eGFP) into human xenografts (grafted on immunocompromised mice), individual keratinocytes expressing eGFP (left) are observed by intravital microscopy (VivaScope) using the fluorescence mode at 5–10 µm depth (left). The corresponding reflectance mode image is shown at the right. Images show the merge of slices 3 to 6 (of a 40-slice Z-stack). The scale bar is 100 µm.
Discussion
A plethora of nucleic acid skin delivery technologies have been described and evaluated by numerous research groups with varying success. Physical methods include direct hypodermic needle injection,6,26,27 electroporation,28,29,30 gene gun,31,32 magnetofection,33 iontophoresis,34 radio frequency35 and microneedle36,37 applications.38 Topical formulations that deliver nucleic acids through the stratum corneum barrier have also been reported.39,40,41 In addition, some of these methods are being combined with chemically-modified nucleic acids to improve tissue distribution and cellular uptake.6,17,42,43 These technologies have been tested in a variety of animal models, organotypic skin cultures and skin explants, including those from human. The 2009 Annual Meeting of the International Pachyonychia Congenita Consortium (IPCC) focused on issues surrounding effective delivery of nucleic acids to skin. Although wide-ranging innovative physical and chemical methods were discussed with the goal of enhancing delivery, it was concluded that there was no existing systematic way to study and compare the various methods designed to achieve or enhance delivery.5 The absence of such validated systems complicates comparative evaluation of methods designed to improve delivery, which in turn, impedes progress.
The first step towards comparative evaluation of different delivery technologies was to identify plasmid vectors in which reporter gene expression in keratinocytes is both strong and uniform throughout the epidermal layers. Among the promoters traditionally utilized to express reporter genes in the skin, CMV is the most commonly used, likely due to its promoter strength and ability to express in a wide variety of tissue and cell types. However, we confirmed what we and others have previously reported: use of the CMV promoter in epidermis is problematic because the majority of expression is found in the granular and stratum corneum layers6,20,22,24,25,44,45 (see also Fig. S1). The reason for this restricted expression pattern is not known, although several mechanisms have been postulated.6 Nevertheless, the asymmetrical expression resulting from the CMV promoter or CMV enhancer elements complicates side-by-side comparison of nucleic acid delivery technologies.
As a first step to identify appropriate delivery tools, reporter expression plasmid constructs were designed and tested to evaluate their ability to express reporter uniformly throughout the epidermis with the intent to use the best performing plasmids to evaluate delivery technologies.6 A luc2/eGFP dual reporter fusion construct, driven by the Ubc promoter was chosen (see Figs. 1 and 2 ) as it was previously shown to drive strong detectable reporter gene expression through all layers of the epidermis, avoiding the non-uniform expression associated with the CMV promoter as described above. The reporter luc2 allows for macroscopic in vivo bioluminescence imaging and in vivo quantification of reporter expression, making this approach useful for initial screening of functional plasmid delivery and localizing regions of interest for further study. Detection of GFP by in vivo confocal fluorescence microscopy allows for imaging of tissues at cellular (and even sub-cellular) resolution and localization of GFP-positive keratinocytes in mouse or human skin.
Sub-cellular resolution was observed with the fluorescence imaging system; nuclei were clearly identified by the absence of GFP signal (dark region). Interestingly, differential patterns of GFP cellular distribution were observed for the two fusion proteins utilized. While hMGFP, which was previously shown to form aggregates in the cytoplasm when fused with CBL,9 is mostly absent from the nuclei of murine keratinocytes, eGFP tended to uniformly distribute between cytoplasm and nuclei. This is consistent with a previous study describing that eGFP, due to its low molecular weight (27 kDa), diffuses into the nuclei of various cell types.46,47 This mechanism may involve the ability of proteins less than ∼50 kDa (e.g., eGFP) to passively transit through the nuclear membrane pores in the absence of a nuclear localization signal(s).47,48 It has also been described that the 50 kDa limit can be exceeded,47,49 which may explain why luc2 (∼61 kDa) does not impede the diffusion of this fusion protein into the nuclei of mouse keratinocytes. In contrast, many human keratinocytes did not exhibit nuclear eGFP, although it is unclear whether the size or another parameter is responsible for this differential nuclear localization pattern.
Expression of reporter plasmid was readily observed in human skin equivalents grafted on immunocompromised mice, suggesting this system may be useful for screening in a human background. The xenografts utilized in this study confirmed that delivery of functional reporter plasmid is achievable and readily visualized by in vivo imaging in human skin (see Figs. 3 and 6). Xenografts were used as opposed to human skin explants (e.g., from abdominoplasty or foreskin surgeries) in these studies to take advantage of their uniformity, having been made from a cloned, spontaneously immortalized keratinocyte and that they are commercially available. The usefulness of human skin xenografts, such as the full thickness skin equivalents used in this study, may represent an intermediate step, facilitating the translation of skin delivery methodologies to patients.
Despite the efforts of many groups to achieve nucleic acid skin delivery, only partial progress has been made to effectively translate these technologies to the clinic.50,51 For example, intratumoral injection of plasmid expressing interleukin 2 followed by electroporation was demonstrated to be safe, effective (resulting in tumor regression for stage III/IV melanoma patients), reproducible and titratable with transient pain during and after electroporation being the major adverse event reported.50 Additionally, an siRNA was successfully evaluated in a small Phase Ib trial in which the inhibitor was administered by ID injection into foot sole lesions of a pachyonychia congenita (PC) patient.52 This first-in-skin clinical trial, using a single nucleotide, mutation-specific siRNA (TD101, targets a mutant form of keratin 6a [K6a]), resulted in a positive clinical response in which the treated skin reverted to “normal” (non-tender) at the siRNA-treated site, but not the contralateral site injected with vehicle alone.52 The mechanism of delivery was likely facilitated by “pressure-fection”, based on recently published results showing that increasing volumes of a fixed amount of intradermally-injected plasmid led to enhanced delivery of functional nucleic acid.6 Unfortunately, the intense pain and highly localized delivery associated with these intralesional injections preclude future widespread use in the clinic. Both of these examples resulted in substantial pain for the patients, underscoring the need for further development of patient-friendly technologies.
Among the most promising technologies to deliver nucleic acids (or any cargo that contains large and/or charged molecules) through the stratum corneum are microneedle patches.53 The microneedle protrusions efficiently penetrate the stratum corneum barrier and are less painful compared to traditional hypodermic needle injections as they are designed to only penetrate and deliver cargo to the non-innervated epidermis and upper dermis.54,55 Recent reports have demonstrated that microneedles can deliver both functional plasmid DNA17,36 and siRNAs.17
Using BLI initially, we investigated the ability to detect plasmid delivery using two distinct microneedle designs (see Figs. 4 and 5). The typical signal intensity (i.e., amount of functional plasmid delivered) observed was one (for metal coated microneedles) or two (for PADs) orders of magnitude lower than achieved by ID injection of 20 µg of plasmid as measured by BLI at 24 h (compare Figs. 1 and 6, see also6). Increased levels of reporter expression (e.g., 1–2 orders in magnitude higher than observed in Fig. 1) can be achieved by ID injection of larger amounts (30–60 µg) of pUbc-luc2/eGFP plasmid (data not shown). It should be noted that due to limited loading capacity and the restricted area of the target site (mouse footpad), microneedle array application results in delivery of much smaller amounts of plasmid cargo (0.1 µg for PADs and 3 µg for metal coated microneedles). In an effort to increase DNA plasmid delivery, multiple sequential microneedle array applications were attempted, but no significant improvement was observed (data not shown).
The mechanism of keratinocyte DNA uptake following microneedle application is not known but may involve cellular membrane disruption and/or damage caused during microneedle insertion. It should be noted that microneedle delivery of unmodified siRNA did not result in detectable target knockdown when applied to mouse skin in preliminary experiments (data not shown). The use of siRNAs with so-called self-delivery enhancements (i.e., contain chemical modifications that enhance cellular uptake) was shown, however, to result in target reduction.17 Taken together, these results suggest that cellular membrane damage or disruption may be required for plasmid uptake in the absence of pressure or other delivery agents.7,17,56
The coated metal microneedles exhibited higher reporter plasmid expression than PADs, consistent with their higher DNA payload (i.e., 3 µg for an array of five coated metal microneedles vs. 0.1 µg for five PAD microneedles, see Materials and Methods). It should also be noted that metal microneedles deposit over 80% of their coated payload within 1 minute when inserted in fresh human skin57 and over 90% within 5 minutes when inserted into pig18 or mouse58 skin. Given the extended insertion period used in this study, 20 minutes and the high solubility of the DNA cargo in an aqueous milieu, near complete payload deposition is anticipated. In contrast, PAD microneedles have been described to deliver only ∼10% of cargo to mouse skin.17 Furthermore, the thin structure and increased strength of the metal microneedles allow for much deeper penetration (up to 700 µm59). The length, conical shape and skin deformation characteristics of PADs appear to limit penetration to the epidermal and top dermal layers (about 50–100 µm17 and data not shown), despite a similar needle length as the metal microneedles of up to 700 µm. Although microneedle payload capacity limits its usefulness to potent molecules (i.e., nanogram to microgram doses), the decreased pain associated with microneedle delivery systems, coupled with the ability to directly traverse the stratum corneum barrier, warrants further development of these technologies.
The standardization and use of molecular tools, nucleic acid reagents including plasmids and skin model systems, coupled with bioluminescence and fluorescence imaging systems as described herein should allow for direct comparisons of existing and future nucleic acid delivery technologies. Indeed, this project was funded by the American Recovery and Reinvestment Act of 2009 (Grand Opportunity award) with the explicit aim to make these resources available to the research community to allow comparative testing of nucleic acid skin delivery methodologies. The identification of the most promising delivery technologies will allow researchers to focus efforts on optimization of the most promising approaches with an eye towards rapid translation to the clinic.
Methods
Animals
6–8 week old female CD1 (Charles River, Wilmington, MA) and B6.Cg-Foxn1nu/J mice (The Jackson Laboratory, Bar Harbor, ME) were used according to procedures approved in animal protocol numbers 10526 and 21627 by the Administrative Panel for Laboratory Animal Care at Stanford University and animal protocol number A3230-01 at Yale University, using the guidelines set by the Institutional Animal Care and Use Committees of the National Institutes of Health, Stanford University and Yale University.
Plasmids constructions
The pCMV-hMGFP/CBL (clone 138) plasmid was previously described9 and contains a fusion of humanized Montastrea cavernosa (“monster”) green fluorescence protein (hMGFP) and click beetle luciferase (CBL), expressed under the control of the cytomegalovirus (CMV) promoter. pUbc-luc2/eGFP was generated by ligation of the Ubc promoter and the luc2/eGFP coding region of pFULG60 into the backbone of pCMV-hMGFP/CBL. The 3.7 Kb fragment, generated from Pac I digestion of pFULG followed by Klenow fill-in to generate a “blunt end” and further digestion with Not I, was ligated into the 3.7 Kb fragment generated by digestion of pCMV-hMGFP/CBL with Bgl II followed by Klenow fill in to generate a “blunt end” and further digestion with Not I. All enzymes were purchased from New England Biolabs (Ipswich, MA).
Microneedle fabrication
Protrusion array devices. PADs were produced as previously described.17 Briefly, a template pattern (square grid, 2 mm spacing) of projecting metal pins was brought into momentary (1 s) contact with a 1 mm thick film of 20% aqueous polyvinyl alcohol polymer solution supported on a glass substrate. The pins were then withdrawn a distance of 1 cm over 13 s under a uniform airflow of 3.0 m/s at 45°C, creating strands of viscous polymer solution, which were dried in place forming an array of fiber-like protrusions extending from the resulting dry polymer film. Each fiber structure created had a roughly rectangular base of about 200 by 500 μm, tapering to a fiber waist width of approximately 40 μm at 1 mm above the film surface.
The protrusions were next mechanically separated from the template array with a razor blade. Each fiber structure was mechanically trimmed with fine scissors at a nominal 45-degree bevel at approximately 750 μm above the film surface (across the 40 μm waist) to produce an array of sharp, needle-like projections ( Fig. 4A,B ). Each of these needles was loaded manually from a micropipet tip charged with plasmid solution (2 mg/ml). The amount of DNA loaded per needle was 0.02 µg based on the average amount of DNA solution taken up by each needle (10 nl). The dry polymer needles rapidly imbibe and swell with the charging solution, but retain shape and sharpness after drying. Subsequent to loading, PADs were further processed in a 50°C vacuum oven evacuated to −18 inches mercury pressure for 4 h to dry and “harden” the needle structures to facilitate skin penetration. The integrity of the plasmid DNA following PAD loading and processing was confirmed by eluting the plasmid payload from microneedle tips (in 50 µl PBS) and analysis by agarose gel 1% (w/v) electrophoresis. No differences between PAD-loaded plasmid and the original stock DNA was observed (data not shown).
Coated metal microneedles. Stainless steel microneedles (∼700 µm long, with a base cross section of 75 by 150 µm) were fabricated using laser cutting and electropolishing, as previously described.18,59 To apply the plasmid DNA coating solution, the microneedles were dipped 12 times at 25°C into DNA coating solution (7 mg/ml) in deionized water using a dip-coating device described previously,18,59,61 with air drying between immersions. Plasmid integrity was confirmed by agarose gel (1% w/v) electrophoresis after coating and drying procedures by dissolving the DNA on the microneedles in water (data not shown). Negative control steel microneedles were coated with salmon sperm DNA (7 mg/ml, Boehringer Mannheim, Penzberg, Germany). The amount of DNA coated on the microneedle array was determined by soaking the DNA-coated microneedles in deionized water for 12 h at 4°C and the amount of DNA eluted was measured using a NanoDrop instrument (Thermo Scientific, Wilmington, DE). The amount of DNA coated was calculated to be 3 µg per microneedle array. Each microneedle array consists of 5 single microneedles. ( Fig. 4A,C )
Human skin equivalent xenografts on immunodeficient mice
Human skin equivalents (StrataTest®) were obtained from StrataTech (Madison, WI). These are produced in vitro by seeding immortal keratinocytes (NIKS®) on a matrix, comprised of human fibroblasts embedded in type I collagen.62 Grafting of StrataTest skin equivalents was performed under sterile conditions onto the back of B6.Cg-Foxn1nu/J mice anesthetized by intraperitoneal injection of 100 mg/kg ketamine (Hospira, Lake Forrest, IL) mixed with 10 mg/kg xylazine (Sigma-Aldrich St Louis, Mo). Eyes were protected with antibiotic ointment (Vetropolycin, Pharmaderm, Melville, NY). Prior to surgery, mouse back skin was cleaned with betadine followed by 70% ethanol.
A square region of mouse back skin (dorsal midline, approximately 1.8×1.8 cm) was removed and a 2×2 cm piece of StrataTest skin equivalent was placed over the defect, carefully aligning mouse and human skin edges. After waiting five minutes (to aid adherence of the graft to the mouse), the grafted area was covered with two layers of Vaseline gauze (The Kendall Company, Mansfield, MA), pre-coated with Bacitracin cream (CVS pharmacy, Woonsocket, RI), a layer of Tegaderm (3 M Health Care, St. Paul, MN), two bandages (Derma Sciences, Princeton, NJ) covering the size of the wound and finally wrapped with Coban 3 M tape (Andover, Salisbury, MA). Ibuprofen (Motrin, Upjohn, USA, 0.128 mg/ml) was administered for 48 h in the water.
Following surgery, the mice were monitored daily for the first week and the grafts were unbandaged at 14 days. Mice were treated no sooner than two months following surgery at which time the grafts had shrunken to about 0.5 cm2. The presence of human skin was confirmed by immunofluorescence using human or mouse specific antibodies as described below and examined by histology stained with Harris Hematoxylin with glacial acetic acid, (Ploy Scientific, Bay Shore, NY) and Eosin Y 1% Alcoholic (Poly Scientific) (see Figs. S2 and S3 ).
Application of microneedle arrays to mouse and human skin
Mouse skin. Four cohorts of mice (5 mice per group) were treated with metal or PAD microneedles loaded with plasmid DNA. PAD arrays (3×5 needles per array), coated with pUbc-luc2/eGFP (first cohort) or pCMV-hMGFP/CBL (second cohort), were applied by flicking the array with a finger to seat the needle tips into the left paw followed by holding the PAD for 1 minute in contact with the skin. Control PADs, loaded with β-gal expression plasmid DNA (pCMVsport-β-gal, Invitrogen), were applied to the right paws.
Coated metal microneedle arrays (1×5 needles per array) coated with pUbc-luc2/eGFP (third cohort) or pCMV-hMGFP/CBL (fourth cohort) were tightly held within forceps and inserted in the skin of the left paw by pushing until full penetration was achieved. As a negative control, the right paw of each mouse was treated with metal microneedle arrays coated with salmon sperm DNA.
The applied arrays were left seated in the skin for 20 minutes to give the DNA payloads the opportunity to dissolve inside the skin. After removal, PAD arrays were observed under an inspection Amscope SM1-BZ microscope (Iscope Corp, Chino, CA) to assess skin penetration and needle dissolution. Although the footpad is only large enough to receive about 5 needles simultaneously, the 3×5 rectangular PAD needle array served to stabilize and orient the microneedles normal to the footpad surface. Post-insertion inspection of the array confirmed penetration of ∼5 needles per array per application, as determined by the absence of tip of the needle due to dissolution upon insertion and removal. Treated paws were analyzed by in vivo bioluminescence imaging, fluorescence imaging using the VivaScope system or by traditional microscopy of frozen skin sections as described below.
Human skin equivalents. PAD arrays (5×5 microneedles) were applied to human skin equivalents using a vacuum channel plate made from a modified 0.2 μm sterile syringe filter (Whatman, Florham Park, NJ), with one face and filter removed, leaving a channeled plate with an exhaust port. Attachment of this exhaust port to a vacuum pressure station (Bernant Company, Barrington, IL) provided a stable flat surface with sufficient airflow to hold a PAD over the skin surface. Upon contact with the plate, the mouse skin was abruptly seated against the channeled plate and the needles inserted in the skin. The vacuum was continued for 1 minute and the channeled plate removed. PADs were left 20 minutes within the skin to allow needle dissolution and then removed. Coated metal microneedles were applied to human skin equivalents in a similar fashion as described above for mouse paws.
Analysis of reporter expression
In vivo bioluminescence imaging (BLI)
Mice were imaged at the indicated time points over a 3-month period following microneedle application. D-luciferin (Biosynth International, Inc., Naperville, IL) was injected (100 µl of a 30 mg/ml solution; ∼150 mg/kg body weight) into the peritoneal cavity of the mice under isoflurane anesthesia. After 10 min, the live anaesthetized mice were imaged in the IVIS Spectrum Imaging System (Xenogen product from Caliper LifeSciences, Alameda, CA) as previously described.63 The resulting light emission was quantified using LivingImage software 3.1 (Caliper LifeSciences, Alameda, CA), written as an overlay on Igor image analysis software (WaveMetrics, Inc; Lake Oswego, Oregon). The data from six mice were statistically analyzed using a paired T-test.
Fluorescence imaging
Plasmids (20 µg of pUbc-luc2/eGFP or pCMV-hMGFP in 100 µl PBS) were intradermally injected or applied via microneedle arrays as described above to footpads of anesthetized (2–3% isoflurane) CD1 mice.64 Mice were anesthetized 24 hours later with isoflurane and GFP reporter expression in paws was imaged using the Lucid VivaScope 2500 System (Lucid Inc., Rochester, NY).9,10 Aquasonic® 100 (Parker laboratories, INC. Fairfield, NJ) ultrasound transmission gel was used as immersion medium between the objective lens and the tissue cassette. The mouse paw to be imaged was placed over the glass window of the tissue cassette after adding CrodamolTM STS (Croda Inc., Edison, NJ) as index matching fluid (index of refraction = 1.47). The Z-depth position was adjusted by identifying the surface of the tissue using the 630 nm reflectance laser.
Using this same laser, the position in the skin (e.g. stratum corneum, granulosum, spinal layer, basal layer, etc.) was identified.65 After switching to the 488 nm fluorescence excitation laser, Vivablocks (XY position map) were used to screen the tissue to locate GFP-positive cells (the signal displays as white colored over a black background) at this selected depth. Once cells expressing eGFP or hMGFP were found, Vivastack (Z-axis map) images were obtained, first in fluorescence mode and then in reflectance mode (40 slices with a Z separation of 1.6 µm between slices [5 µm thick], see Fig. S4 for details on VivaScope settings when scanning skin tissue). The field of view was 750×750 µm and the images were collected at 9 frames per second. Image files were processed and 3D volumes and video files were reconstructed using ImageJ (National Institutes of Health, Bethesda, MD).
Skin sectioning and immunofluorescence
Following fluorescence imaging, intradermally-injected and microneedle-treated mice were sacrificed. Skin tissues were removed from the footpad, embedded in O.C.T. compound (Tissue-Tek®, Torrance, CA) and frozen in dry ice. Vertical cross sections (10 µm) were obtained using a microtome and mounted with HydromountTM (National Diagnostic, Highland Park, NJ) containing 4′,6-diamidino-2-phenylindole (DAPI, 1 µg/ml, Sigma) for nuclear staining. Tissue sections were imaged by fluorescence microscopy as previously described9.
Human skin xenografts were harvested 48 h following injection of pUbc-luc2/eGFP. Cryosections were fixed with acetone/methanol at −20 degrees for 10 min, washed in PBS and incubated at room temperature with 10% goat serum/1% BSA/0.025% Triton X-100 in PBS for 1 h, followed by incubation with primary antibody (e.g. mouse or human keratin 1 [Covance, Emeryville, CA]) in 10% goat serum/1% BSA Triton X-100 (in PBS) for 1 h at room temperature. After washing with PBS, the skin sections were incubated with secondary antibody (goat anti-rabbit Cy3, Covance) in 10% goat serum/1% BSA/0.025% Triton X-100 for 1 h at room temperature. Nuclei were stained with DAPI for 3 minutes and washed again in PBS, mounted and stored in the dark. Images were acquired with a ZEISS Axiovert 200 microscope (Carl Zeiss, Inc., Thornwood, NY).
References
Lane, E. B. & McLean, W. H. I. Broken bricks and cracked mortar – epidermal diseases resulting from genetic abnormalities. Drug Discov Today: Disease Mechanisms 5, 93–101 (2008).
Lewin, A. S., Glazer, P. M. & Milstone, L. M. Gene therapy for autosomal dominant disorders of keratin. J Investig Dermatol Symp Proc 10, 47–61 (2005).
Geusens, B., Sanders, N., Prow, T., Van Gele, M. & Lambert, J. Cutaneous short-interfering RNA therapy. Expert Opin Drug Deliv 6, 1333–1349 (2009).
Leachman, S. A., et al. Therapeutic siRNAs for dominant genetic skin disorders including pachyonychia congenita. J Dermatol Sci 51, 151–157 (2008).
Kaspar, R., McLean, W. & Schwartz, M. Achieving Successful Delivery of Nucleic Acids to Skin: 6th Annual Meeting of the International Pachyonychia Congenita Consortium. J. Invest. Derm. 129, 2085–2087 (2009).
Gonzalez-Gonzalez, E., et al. Increased interstitial pressure improves nucleic acid delivery to skin enabling a comparative analysis of constitutive promoters. Gene Ther 17, 1270–1278 (2010).
Siprashvili, Z., et al. Gene transfer via reversible plasmid condensation with cysteine-flanked, internally spaced arginine-rich peptides. Hum Gene Ther 14, 1225–1233 (2003).
Hickerson, R. P., et al. Single-nucleotide-specific siRNA targeting in a dominant-negative skin model. J Invest Dermatol 128, 594–605 (2008).
Gonzalez-Gonzalez, E., et al. siRNA silencing of keratinocyte-specific GFP expression in a transgenic mouse skin model. Gene Ther 16, 963–972 (2009).
Ra, H., et al. Assessing delivery and quantifying efficacy of small interfering ribonucleic acid therapeutics in the skin using a dual-axis confocal microscope. J Biomed Opt 15, 036027 (2010).
Gerger, A., et al. Diagnostic applicability of in vivo confocal laser scanning microscopy in melanocytic skin tumors. J Invest Dermatol 124, 493–498 (2005).
Pellacani, G., et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions. J Invest Dermatol 127, 2759–2765 (2007).
Astner, S., Gonzalez, E., Cheung, A., Rius-Diaz, F. & Gonzalez, S. Pilot study on the sensitivity and specificity of in vivo reflectance confocal microscopy in the diagnosis of allergic contact dermatitis. J Am Acad Dermatol 53, 986–992 (2005).
Gerger, A., et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer 107, 193–200 (2006).
Ulrich, M., et al. Actinic keratoses: non-invasive diagnosis for field cancerisation. Br J Dermatol 156 Suppl 3, 13–17 (2007).
Li, Y., et al. Dual mode reflectance and fluorescence confocal laser scanning microscopy for in vivo imaging melanoma progression in murine skin. J Invest Dermatol 125, 798–804 (2005).
Gonzalez-Gonzalez, E., et al. Silencing of reporter gene expression in skin using siRNAs and expression of plasmid DNA delivered by a soluble protrusion array device (PAD). Mol Ther 18, 1667–1674 (2010).
Gill, H. S. & Prausnitz, M. R. Coated microneedles for transdermal delivery. J Control Release 117, 227–237 (2007).
Prausnitz, M. R. Microneedles for transdermal drug delivery. Adv Drug Deliv Rev 56, 581–587 (2004).
Cao, Y. A., et al. Molecular Imaging Using Labeled Donor Tissues Reveals Patterns of Engraftment, Rejection and Survival in Transplantation. Transplantation 80, 134–139 (2005).
Ghazizadeh, S., Katz, A. B., Harrington, R. & Taichman, L. B. Lentivirus-mediated gene transfer to human epidermis. J Investig Dermatol Symp Proc 9, 269–275 (2004).
Hengge, U. R., Chan, E. F., Foster, R. A., Walker, P. S. & Vogel, J. C. Cytokine gene expression in epidermis with biological effects following injection of naked DNA. Nat Genet 10, 161–166 (1995).
Sawamura, D., et al. In vivo transfer of a foreign gene to keratinocytes using the hemagglutinating virus of Japan-liposome method. J Invest Dermatol 108, 195–199 (1997).
Sawamura, D., et al. The majority of keratinocytes incorporate intradermally injected plasmid DNA regardless of size but only a small proportion of cells can express the gene product. J Invest Dermatol 118, 967–971 (2002).
Lin, M. T., Wang, F., Uitto, J. & Yoon, K. Differential expression of tissue-specific promoters by gene gun. Br J Dermatol 144, 34–39 (2001).
Hengge, U. R., Walker, P. S. & Vogel, J. C. Expression of naked DNA in human, pig and mouse skin. J Clin Invest 97, 2911–2916 (1996).
Sawamura, D., Akiyama, M. & Shimizu, H. Direct injection of naked DNA and cytokine transgene expression: implications for keratinocyte gene therapy. Clin Exp Dermatol 27, 480–484 (2002).
Heller, L. C., Jaroszeski, M. J., Coppola, D. & Heller, R. Comparison of electrically mediated and liposome-complexed plasmid DNA delivery to the skin. Genet Vaccines Ther 6, 16 (2008).
Favard, C., Dean, D. S. & Rols, M. P. Electrotransfer as a non viral method of gene delivery. Curr Gene Ther 7, 67–77 (2007).
Zhang, L., Nolan, E., Kreitschitz, S. & Rabussay, D. P. Enhanced delivery of naked DNA to the skin by non-invasive in vivo electroporation. Biochim Biophys Acta 1572, 1–9 (2002).
Yang, C. H., et al. Seeing the gene therapy: application of gene gun technique to transfect and decolour pigmented rat skin with human agouti signalling protein cDNA. Gene Ther 11, 1033–1039 (2004).
Yang, N. S., Burkholder, J., McCabe, D., Neumann, V. & Fuller, D. Particle-mediated gene delivery in vivo and in vitro. Curr Protoc Hum Genet Chapter 12, Unit 12 16 (2001).
Holzbach, T., et al. Non-viral VEGF gene therapy - Magnetofection of acoustically active magnetic lipospheres (“Magnetobubbles”) increases tissue-survival in an oversized skin flap model. J Cell Mol Med (2008).
Kigasawa, K., et al. Noninvasive delivery of siRNA into the epidermis by iontophoresis using an atopic dermatitis-like model rat. Int J Pharm 383, 157–160 (2010).
Birchall, J., et al. Cutaneous gene expression of plasmid DNA in excised human skin following delivery via microchannels created by radio frequency ablation. Int J Pharm 312, 15–23 (2006).
Gill, H. S., Soderholm, J., Prausnitz, M. R. & Sallberg, M. Cutaneous vaccination using microneedles coated with hepatitis C DNA vaccine. Gene Ther 17, 811–814 (2010).
Birchall, J., et al. Cutaneous DNA delivery and gene expression in ex vivo human skin explants via wet-etch micro-fabricated micro-needles. J Drug Target 13, 415–421 (2005).
Arora, A., Prausnitz, M. R. & Mitragotri, S. Micro-scale devices for transdermal drug delivery. Int J Pharm 364, 227–236 (2008).
Badea, I., Wettig, S., Verrall, R. & Foldvari, M. Topical non-invasive gene delivery using gemini nanoparticles in interferon-gamma-deficient mice. Eur J Pharm Biopharm 65, 414–422 (2007).
Takanashi, M., et al. Therapeutic silencing of an endogenous gene by siRNA cream in an arthritis model mouse. Gene Ther 16, 982–989 (2009).
Yi, X., et al. MITF-siRNA formulation is a safe and effective therapy for human melasma. Mol Ther 19, 362–371 (2011).
Hickerson, R. P., et al. Use of Self-Delivery siRNAs to Inhibit Gene Expression in an Organotypic Pachyonychia Congenita Model. J Invest Dermatol, In press (2011).
Kaspar, R. L., Leachman, S. A., McLean, W. H. I. & Schwartz, M. E. Towards a treatment for PC – Report on the 7th Annual International Pachyonychia Congenita Consortium Meeting. J Invest Dermatol, In press (2011).
Sawamura, D., et al. In vivo gene introduction into keratinocytes using jet injection. Gene Ther 6, 1785–1787 (1999).
Brocard, J., et al. Spatio-temporally controlled site-specific somatic mutagenesis in the mouse. Proc Natl Acad Sci U S A 94, 14559–14563 (1997).
Wei, X., Henke, V. G., Strubing, C., Brown, E. B. & Clapham, D. E. Real-time imaging of nuclear permeation by EGFP in single intact cells. Biophys J 84, 1317–1327 (2003).
Seibel, N. M., Eljouni, J., Nalaskowski, M. M. & Hampe, W. Nuclear localization of enhanced green fluorescent protein homomultimers. Anal Biochem 368, 95–99 (2007).
von Arnim, A. G., Deng, X. W. & Stacey, M. G. Cloning vectors for the expression of green fluorescent protein fusion proteins in transgenic plants. Gene 221, 35–43 (1998).
Macara, I. G. Transport into and out of the nucleus. Microbiol Mol Biol Rev 65, 570–594, table of contents (2001).
Daud, A. I., et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol 26, 5896–5903 (2008).
Heller, L. C. & Heller, R. Electroporation gene therapy preclinical and clinical trials for melanoma. Curr Gene Ther 10, 312–317 (2010).
Leachman, S. A., et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 18, 442–446 (2010).
Prausnitz, M. R., Mikszta, J. A., Cormier, M. & Andrianov, A. K. Microneedle-based vaccines. Curr Top Microbiol Immunol 333, 369–393 (2009).
Haq, M. I., et al. Clinical administration of microneedles: skin puncture, pain and sensation. Biomed Microdevices 11, 35–47 (2009).
Gill, H. S., Denson, D. D., Burris, B. A. & Prausnitz, M. R. Effect of microneedle design on pain in human volunteers. Clin J Pain 24, 585–594 (2008).
Gupta, A. K., Eshraghi, Y., Gliniak, C. & Gosain, A. K. Non-Viral Transfection of Mouse Calvarial Organ In Vitro using Accell-Modified si RNA. Plast Reconstr Surg (2009).
Pearton, M., et al. Influenza virus-like particles coated onto microneedles can elicit stimulatory effects on Langerhans cells in human skin. Vaccine 28, 6104–6113 (2010).
Zhu, Q., et al. Immunization by vaccine-coated microneedle arrays protects against lethal influenza virus challenge. Proc Natl Acad Sci U S A 106, 7968–7973 (2009).
Kim, Y. C., Quan, F. S., Compans, R. W., Kang, S. M. & Prausnitz, M. R. Formulation and coating of microneedles with inactivated influenza virus to improve vaccine stability and immunogenicity. J Control Release 142, 187–195 (2010).
Liu, H., et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A 107, 18115–18120 (2010).
Kim, Y. C., et al. Improved influenza vaccination in the skin using vaccine coated microneedles. Vaccine 27, 6932–6938 (2009).
Rasmussen, C., et al. The StrataTest(R) human skin model, a consistent in vitro alternative for toxicological testing. Toxicol In Vitro (2010).
Contag, C. H. & Bachmann, M. H. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng 4, 235–260 (2002).
Wang, Q., et al. Delivery and inhibition of reporter genes by small interfering RNAs in a mouse skin model. J Invest Dermatol 127, 2577–2584 (2007).
Rajadhyaksha, M., Grossman, M., Esterowitz, D., Webb, R. H. & Anderson, R. R. In vivo confocal scanning laser microscopy of human skin: melanin provides strong contrast. J Invest Dermatol 104, 946–952 (1995).
Acknowledgements
The authors would like to thank all of the participants of the GO Delivery! Project consortia for their input and support. We thank Manny Flores, Andrea Burgon and Heini Ilves for technical support, Jennifer Prescher for the plasmid pFULG and Robert Kaspar for help in data processing. This work was supported by NIH grants RC2ARO58955 (RLK, CHC and LMM) and U54 CA105296 (CHC). E.G.G. is the recipient of a Pachyonychia Congenita Project fellowship.
Author information
Authors and Affiliations
Contributions
All authors reviewed the manuscript and participated in experimental design. E. G. G. performed experiments, wrote the main text of the article and prepared and formatted figures performed Y. K. provided loaded steel microneedles. T. J. S. provided loaded PAD arrays. R. P. H. engineered and prepared DNA plasmid vectors. R. S. assisted with mouse imaging. J. C. B. concept, design and oversight of experiments. M. F. L. helped with tissue section preparation and analysis. R. H. prepared and analyzed skin grafts. Y. L. performed experiments in human skin grafts. N. K. S. prepared human skin grafts. M. R. P., L. M. M., C. H. C. and R. L. K. helped write the manuscript and participated in study concept, experimental design and interpretation of the results.
Ethics declarations
Competing interests
R. L. K., R. P. H., T. J. S. and M. F. L. are employees of TransDerm Inc., which has a patent application for the PAD technology. C. H. C. is a founder of Xenogen Corp. now part of Caliper LifeSciences. M. R. P. serves as a consultant and is an inventor on patents licensed to companies developing microneedle-based products. This potential conflict of interest has been disclosed and is being managed by Georgia Tech and Emory University.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY FIGURES
Supplementary Information
SUPPLEMENTAL VIDEO FILE 1: Video of eGFP expression 24 h after intradermal (ID) injection of pUbc-luc2/eGFP plasmid in mouse footpad using the VivaScope 2500 (Lucid) confocal fluorescence microscope.
Supplementary Information
SUPPLEMENTAL VIDEO FILE 2: Video of hMGFP expression 24 h after ID injection of pCMV-hMGFP/CBL plasmid in mouse footpad using the VivaScope 2500 (Lucid) confocal fluorescence microscope.
Supplementary Information
SUPPLEMENTAL VIDEO FILE 3: Video of eGFP expression 24 h after injection of pUbc-luc2/eGFP plasmid in human skin xenografts using the VivaScope 2500 (Lucid) confocal fluorescence microscope.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
González-González, E., Kim, YC., Speaker, T. et al. Visualization of plasmid delivery to keratinocytes in mouse and human epidermis. Sci Rep 1, 158 (2011). https://doi.org/10.1038/srep00158
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00158
This article is cited by
-
Non-Invasive Intravital Imaging of siRNA-Mediated Mutant Keratin Gene Repression in Skin
Molecular Imaging and Biology (2016)
-
Microneedles: an innovative platform for gene delivery
Drug Delivery and Translational Research (2015)
-
Local Delivery of Gene-Modifying Triplex-Forming Molecules to the Epidermis
Journal of Investigative Dermatology (2013)
-
Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence
Nature Medicine (2013)
-
Gene Silencing in Skin After Deposition of Self-Delivery siRNA With a Motorized Microneedle Array Device
Molecular Therapy - Nucleic Acids (2013)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
