


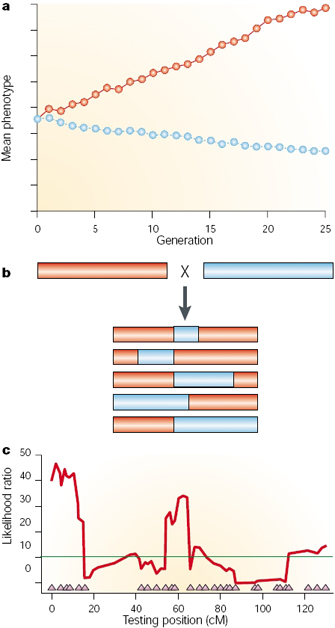
 Figure 1: Quantitative trait locus mapping.
Figure 1: Quantitative trait locus mapping.














« Prev Next »
Quantitative trait locus (QTL) analysis is a statistical method that links two types of information—phenotypic data (trait measurements) and genotypic data (usually molecular markers)—in an attempt to explain the genetic basis of variation in complex traits (Falconer & Mackay, 1996; Kearsey, 1998; Lynch & Walsh, 1998). QTL analysis allows researchers in fields as diverse as agriculture, evolution, and medicine to link certain complex phenotypes to specific regions of chromosomes. The goal of this process is to identify the action, interaction, number, and precise location of these regions.
How Is QTL Analysis Conducted?
© 2001 Nature Publishing Group Mackey, T. F. Quantitative trait loci in Drosophila. Nature Reviews Genetics 2, 13 (2001). All rights reserved. 
In order to begin a QTL analysis, scientists require two things. First, they need two or more strains of organisms that differ genetically with regard to the trait of interest. For example, they might select lines fixed for different alleles influencing egg size (one large and one small). Second, researchers also require genetic markers that distinguish between these parental lines. Molecular markers are preferred for genotyping, because these markers are unlikely to affect the trait of interest. Several types of markers are used, including single nucleotide polymorphisms (SNPs), simple sequence repeats (SSRs, or microsatellites), restriction fragment length polymorphisms (RFLPs), and transposable element positions (Casa et al., 2000; Vignal et al., 2002; Gupta & Rustgi, 2004; Henry, 2006). Then, to carry out the QTL analysis, the parental strains are crossed, resulting in heterozygous (F1) individuals, and these individuals are then crossed using one of a number of different schemes (Darvasi, 1998). Finally, the phenotypes and genotypes of the derived (F2) population are scored. Markers that are genetically linked to a QTL influencing the trait of interest will segregate more frequently with trait values (large or small egg size in our example), whereas unlinked markers will not show significant association with phenotype (Figure 1).
For traits controlled by tens or hundreds of genes, the parental lines need not actually be different for the phenotype in question; rather, they must simply contain different alleles, which are then reassorted by recombination in the derived population to produce a range of phenotypic values. Consider, for example, a trait that is controlled by four genes, wherein the upper-case alleles increase the value of the trait and the lower-case alleles decrease the value of the trait. Here, if the effects of the alleles of the four genes are similar, individuals with the AABBccdd and aabbCCDD genotypes might have roughly the same phenotype. The members of the F1 generation (AaBbCcDd) would be invariant and would have an intermediate phenotype. However, the F2 generation, or the progeny from a backcross of an F1 individual with either parent, would be variable. The F2 offspring would have anywhere from zero to eight upper-case alleles; the backcross progeny would have anywhere from four to eight upper-case alleles.
A principal goal of QTL analysis has been to answer the question of whether phenotypic differences are primarily due to a few loci with fairly large effects, or to many loci, each with minute effects. It appears that a substantial proportion of the phenotypic variation in many quantitative traits can be explained with few loci of large effect, with the remainder due to numerous loci of small effect (Remington & Purugganan, 2003; Mackay, 2004; Roff, 2007). For example, in domesticated rice (Oryza sativa), studies of flowering time have identified six QTL; the sum of the effects of the top five QTL explains 84% of the variation in this trait (Yano et al., 1997; Yamamoto et al., 1998, 2000). Once QTL have been identified, molecular techniques can be employed to narrow the QTL down to candidate genes (a process described later in this article). One important emerging trend in these analyses is the prominent role of regulatory genes, or genes that code for transcription factors and other signaling proteins. For instance, in rice, three flowering time QTL have been identified at the molecular level, and all of these loci encode regulatory proteins known from studies of Arabidopsis thaliana (Remington & Purugganan, 2003).
A meta-analysis of extensive data in pigs and dairy found that QTL effects were skewed towards fewer QTL with large effects (Hayes and Goddard 2001). Orr (2001) addresses the question of defining and distinguishing between "large" and "small" effects. As with all statistical analyses, sample size is a critical factor. Small sample sizes may fail to detect QTL of small effect and result in an overestimation of effect size of those QTL that are identified (Beavis 1994, 1997). This is known at the "Beavis effect". Otto and Jones (2000) suggested a method for comparing detected QTL to a distribution of expected values in order to estimate how many loci might have been missed. Recent studies have taken these biases into account (e.g., Albert et al. 2007).
Another consistent trend in looking at QTL across traits and taxa is that phenotypes are frequently affected by a variety of interactions (e.g., genotype-by-sex, genotype-by-environment, and epistatic interactions between QTL), although not all QTL studies are designed to detect such interactions. Indeed, several complex traits in the fruit fly Drosophila melanogaster have been extensively analyzed, and this research has indicated that the effects of such interactions are common (Mackay, 2001, 2004). For example, detailed examination of life span in D. melanogaster has revealed that many genes influence longevity (Nuzhdin et al., 2005; Wilson et al., 2006). In addition, significant dominance, epistatic, and genotype-by-environment effects have also been reported for life span (Leips & Mackay, 2002; Forbes et al., 2004). Similarly, QTL studies examining plant architecture differences between maize and teosinte have repeatedly shown significant epistatic interactions (Doebley et al., 1995; Lauter & Doebley, 2002). These same types of interactions have additionally been demonstrated in soybeans (Lark et al., 1995).
It is also possible to perform QTL analysis on unmanipulated natural populations using hybrids, sibships (half-sibling or full-sibling families), and/or pedigree information (Lynch & Walsh, 1998; Mott et al., 2000; Slate, 2005). Diverse ecological and evolutionary questions have been addressed using these tools. For example, Shaw and colleagues (2007) identified multiple QTL associated with differences in male calling song between two closely related species of the Hawaiian cricket, a trait involved in rapid speciation. Similarly, Baack and colleagues (2008) addressed the question of possible gene flow between domesticated crops and their wild relatives in contrasting environments using crop-sunflower hybrids. Environmental and conservation questions have also been explored. For instance, Weinig and colleagues (2007) examined various loci that influence invasive success by exotic species, while Pauwels and colleagues (2008) reviewed questions surrounding QTL for tolerance to heavy metal exposure in plants that could contribute to phytoremediation of polluted soils.
Caveats and Qualifications of QTL Analysis
Like most methods, QTL analysis is not without limitations. For instance, QTL studies require very large sample sizes, and they can only map those differences that are captured between the initial parental strains. Because these strains are unlikely to contain segregating alleles of large effect at every locus contributing to variation in natural populations, some loci will remain undetected. Furthermore, the specific alleles that do segregate, particularly in inbred lines, may not be relevant to natural populations. Other alleles at these same loci are likely to be of interest, however. Thus, the goal for many studies is to identify loci rather than particular alleles. (One notable area of exception involves applied studies in medicine and agriculture, which are often interested in specific segregating alleles).
The number of times that individual genes have been identified following a QTL mapping study remains small. Indeed, Roff (2007) lists examples of quantitative traits in which single genes have major effects and their molecular basis has been studied, and he notes that this number is modest relative to the effort invested in QTL studies. One reason for this discrepancy is that many QTL map to regions of the genome of perhaps 20 centimorgans (cM) in length, and these regions often contain multiple loci that influence the same trait (see, however, Price, 2006). Moreover, identifying the actual loci that affect a quantitative trait involves demonstrating causality using techniques like positional cloning (see Clee et al., 2006) followed by targeted gene replacement (see Sullivan et al., 1997). Frequently, the quest for individual genes within a QTL is assisted by the identification of a priori candidate genes using classical reverse genetics or bioinformatics. A functional relationship between the candidate gene and the QTL must then be demonstrated, such as by using functional complementation (the addition of wild-type complementary DNA from the gene in question into the nucleus to rescue a loss-of function mutation or to produce an alternative phenotype; see, for example, Frary et al., 2000). Other techniques, such as deficiency mapping (deletion mapping), are available for specific organisms, including Drosophila (Mackay, 2001).
The Future of QTL Mapping
New permutations of QTL mapping build upon the utility of the original premise: locus discovery by co-segregation of traits with markers. Now, however, the definition of a trait can be broadened beyond whole-organism phenotypes to phenotypes such as the amount of RNA transcript from a particular gene (expression or eQTL; Schadt et al., 2003) or the amount of protein produced from a particular gene (protein QTL or PQL; Damerval et al., 1994). QTL mapping works in these contexts because these phenotypes are polygenic, just like more traditional organismal phenotypes, such as yield in corn. For example, transcript abundance is controlled not just by cis-acting sequences like the promoter, but also by potentially unlinked, trans-acting transcription factors. Similarly, protein abundance is controlled by "local" variation at the coding gene itself, and by "distant" variation mapping to other regions of the genome. Local variation is likely to be composed of cis variants controlling transcript levels (though the correlation between transcript level and protein abundance is often quite low, so this may represent a minority of cases; see Foss et al., 2007). Other local mechanisms might include polymorphisms for the stability or regulation of the protein. In contrast, distant variation could include upstream regulation control regions. Beyond these examples, further extension of QTL analysis includes mapping the contribution of imprinting to size-related traits (Cheverud et al., 2008), and other adaptations of QTL mapping will no doubt follow.
Historically, the availability of adequately dense markers (genotypes) has been the limiting step for QTL analysis. However, high-throughput technologies and genomics have begun to overcome this barrier. Thus, the remaining limitations in QTL analysis are now predominantly at the level of phenotyping, although the use of genomic and proteomic data as phenotypes circumvents this challenge to some extent.
Genome-wide association studies (GWAS) are becoming increasingly popular in genetic research, and they are an excellent complement to QTL mapping. Whereas QTL contain many linked genes, which are then challenging to separate, GWAS produce many unlinked individual genes or even nucleotides, but these studies are riddled with large expected numbers of false positives. Though GWAS remain limited to organisms with genomic resources, combining the two techniques can make the most of both approaches and help provide the ultimate deliverable: individual genes or even nucleotides that contribute to the phenotype of interest.
Indeed, combining different QTL techniques and technologies has great promise. For example, Hubner and colleagues (2005) used data on gene expression in fat and kidney tissue from two previously generated, recombinant rat strains to study hypertension. Alternatively, samples adapted to different environments may be compared, or other populations of interest might be selected for expression analysis. This approach permits measurement of hundreds or even thousands of traits simultaneously. Differences in expression may be co-localized with phenotypic QTL that have been previously determined to create manageable lists of positional candidate genes (Wayne & McIntyre, 2002). Other interesting questions concerning gene regulation can be addressed by combining eQTL and QTL, such as the relative contributions of cis-regulatory elements versus trans-regulatory elements. Regarding hypertension, Hubner et al. (2005) identified 73 candidate genes deemed suitable for testing in human populations, and many of the most highly linked eQTL were regulated in cis. These integrated approaches will become more common, and they promise a deeper understanding of the genetic basis of complex traits, including disease (Hubner et al., 2006). Integrating phenotypic QTL with protein QTL can also give investigators a more direct link between genotype and phenotype via co-localization of candidate protein abundance with a phenotypic QTL (De Vienne et al., 1999). Still more kinds of data can be integrated with QTL mapping for a "total information" genomics approach (e.g., eQTL, proteomics, and SNPs) (Stylianou et al., 2008).
QTL studies have a long and rich history and have played important roles in gene cloning and characterization; however, there is still a great deal of work to be done. Existing data on model organisms need to be expanded to the point at which meta-analysis is feasible in order to document robust trends regarding genetic architecture. Data generated by lab-based QTL studies can also be used to direct and inform other efforts, such as population genomics, wherein a large number of molecular markers are scored in the attempt to identify targets of selection and thus genes underlying ecologically important traits (Stinchcombe & Hoekstra, 2008). Furthermore, QTL studies can inform functional genomics, in which the goal is to characterize allelic variation and how it influences the fitness and function of whole organisms. Thus, although the map between genotype and phenotype remains difficult to read, QTL analysis and a variety of associated innovations will likely continue to provide key landmarks.
References and Recommended Reading
Albert, A. Y. K., et al. The genetics of adaptive shape shift in stickleback: Pleiotropy and effect size. Evolution 62, 76–85 (2007)
Baack, E. J., et al. Selection on domestication traits and quantitative trait loci in crop-wild sunflower hybrids. Molecular Ecology 17, 666–677 (2008)
Beavis, W. D. The power and deceit of QTL experiments: Lessons from comparative QTL studies. In Proceedings of the Forty-ninth Annual Corn and Sorghum Research Conference (Washington, DC, American Seed Trade Association, 1994), 250–266
———. QTL analyses: Power, precision, and accuracy. In Molecular Dissection of Complex Traits, ed. A. H. Paterson (Boca Raton, FL, CRC Press, 1997), 145–162
Casa, A. M., et al. The MITE family Heartbreaker (Hbr): Molecular markers in maize. Proceedings of the National Academy of Sciences 97, 10083–10089 (2000)
Cheverud, J. M., et al. Genomic imprinting effects on adult body composition in mice. Proceedings of the National Academy of Sciences 105, 4253–4258 (2008)
Clee, S. M., et al. Positional cloning Sorcs1, a type 2 diabetes quantitative trait locus. Nature Genetics 38, 688–693 (2006) (link to article)
Damerval, C., et al. Quantitative trait loci underlying gene product variation—A novel perspective for analysing regulation of genome expression. Genetics 137, 289–301 (1994)
Darvasi, A. Experimental strategies for the genetic dissection of complex traits in animal models. Nature Genetics 18, 19–24 (1998) (link to article)
De Vienne, D., et al. Genetics of proteome variation for QTL characterization: Application to drought-stress responses in maize. Journal of Experimental Botany 50, 303–309 (1999)
Doebley, J., et al. Teosinte branched 1 and the origin of maize: Evidence for epistasis and the evolution of dominance. Genetics 141, 333–346 (1995)
Falconer, D. S., & Mackay, T. F. C. Introduction to Quantitative Genetics, 4th ed. (London, Prentice Hall, 1996)
Forbes, S. N., et al. Quantitative trait loci affecting life span in replicated populations of Drosophila melanogaster. I. Composite interval mapping. Genetics 168, 301–311 (2004)
Foss, E. J. et al. Genetic basis of proteome variation in yeast. Nature Genetics 39, 1369–1375 (2007) (link to article)
Frary, A., et al. fw2.2: A quantitative trait locus key to the evolution of tomato fruit size. Science 289, 85–88 (2000)
Gupta, P. K., & Rustgi, S. Molecular markers from the transcribed/expressed region of the genome in higher plants. Functional and Integrative Genomics 4, 139–162 (2004)
Hayes, B., & Goddard, M. E. The distribution of the effects of genes affecting quantitative traits in livestock. Genetics Selection Evolution 33, 209–229 (2001)
Henry, R. J. Plant Conservation Genetics (New York, Haworth Press, 2006)
Hubner, N., et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nature Genetics 37, 243–253 (2005) (link to article)
Hubner, N., et al. Novel integrative approaches to the identification of candidate genes in hypertension. Hypertension 47, 1–5 (2006)
Kearsey, M. J. The principles of QTL analysis (a minimal mathematics approach). Journal of Experimental Botany 49, 1619–1623 (1998)
Lark, K. G., et al. Interactions between quantitative trait loci in soybean in which trait variation at one locus is conditional upon a specific allele at another. Proceedings of the National Academy of Sciences 92, 4656–4660 (1995)
Lauter, N., & Doebley, J. Genetic variation for phenotypically invariant traits detected in teosinte: Implications for the evolution of novel forms. Genetics 160, 333–342 (2002)
Leips, J., & Mackay, T. F. C. The complex genetic architecture of Drosophila life span. Experimental Aging Research 28, 361–390 (2002)
Lynch, M., & Walsh, B. Genetics and Analysis of Quantitative Traits (Sunderland, MA, Sinauer, 1998)
Mackay, T. F. C. Quantitative trait loci in Drosophila. Nature Reviews Genetics 2, 11–20 (2001) (link to article)
———. The genetic architecture of quantitative traits: Lessons from Drosophila. Current Opinion in Genetics and Development 14, 253–257 (2004)
Mott, R., et al. A new method for fine mapping quantitative trait loci in outbred animal stocks. Proceedings of the National Academy of Sciences 97, 12649–12654 (2000)
Nuzhdin, S. V., et al. Survival analysis of life span quantitative trait loci in Drosophila melanogaster. Genetics 170, 719–731 (2005)
Orr, H. A. The genetics of species differences. Trends in Ecology and Evolution 16, 343–350 (2001)
Otto, S. P., & Jones, C. D. Detecting the undetected: Estimating the total number of loci underlying a quantitative trait. Genetics 156, 2093–2107 (2000)
Pauwels, M., et al. Merging methods in molecular and ecological genetics to study the adaptation of plants to anthropogenic metal-polluted sites: Implications for phytoremediation. Molecular Ecology 17, 108–119 (2008)
Price, A. H. Believe it or not, QTLs are accurate! Trends in Plant Science 11, 213–216 (2006)
Remington, D. L., & Purugganan, M. D. Candidate genes, quantitative trait loci, and functional trait evolution in plants. International Journal of Plant Sciences 164, S7–S20 (2003)
Roff, D. A. A centennial celebration for quantitative genetics. Evolution 61, 1017–1032 (2007)
Schadt, E. E., et al. Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302 (2003) (link to article)
Shaw, K. L., et al. QTL analysis of a rapidly evolving speciation phenotype in the Hawaiian cricket Laupala. Molecular Ecology 16, 2879–2892 (2007)
Slate, J. Quantitative trait locus mapping in natural populations: Progress, caveats and future directions. Molecular Ecology 14, 363–379 (2005)
Stinchcombe, J. R., & Hoekstra, H. E. Combining population genomics and quantitative genetics: Finding the genes underlying ecologically important traits. Heredity 100, 158–170 (2008)
Stylianou, I. M., et al. Applying gene expression, proteomics and single-nucleotide polymorphism analysis for complex trait gene identification. Genetics 178, 1795–1805 (2008)
Sullivan, P. M., et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and artherosclerosis. Journal of Biological Chemistry 272, 17972–17980 (1997)
Vignal, A., et al. A review on SNP and other types of molecular markers and their use in animal genetics. Genetics Selection Evolution 34, 275–305 (2002)
Wayne, M. L., & McIntyre, L. M. Combining mapping and arraying: An approach to candidate gene identification. Proceedings of the National Academy of Sciences 99, 14903–14906 (2002)
Weinig, C., et al. Resolving the genetic basis of invasiveness and predicting invasions. Genetica 129, 205–216 (2007)
Wilson, R. H., et al. High-resolution mapping of quantitative trait loci affecting increased life span in Drosophila melanogaster. Genetics 173, 1455–1463 (2006)
Yamamoto, T., et al. Fine mapping of quantitative trait loci Hd-1, Hd-2 and Hd-3, controlling heading date of rice, as single Mendelian factors. Theoretical and Applied Genetics 97, 37–44 (1998)
———. Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 154, 885–891 (2000)
Yano, M., et al. Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theoretical and Applied Genetics 95, 1025–1032 (1997)
Zeng, Z. B. Precision mapping of quantitative trait loci. Genetics 136, 1457–1468 (1994)










