


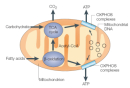
 Figure 1
Figure 1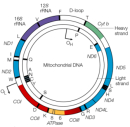

















« Prev Next »
Did you know that your cells contain several thousand copies of an organelle that maintains its own genome? And that instead of being linear, like the chromosomes found in the nucleus, the genome of this organelle is circular? This organelle is the mitochondrion, the powerhouse of eukaryotic cells. In contrast to the human nuclear genome, which consists of 3.3 billion base pairs of DNA, the human mitochondrial genome is built of a mere 16,569 base pairs. Despite its small size, the mitochondrial genome can be used to establish maternal family ties, thanks to its maternal pattern of inheritance. Mutations in the mitochondrial genome have also been associated with diverse forms of human disease and aging.
Mitochondrial Anatomy and Physiology
Mitochondria are spherical, double-membrane-bound organelles that rely on the distinct functions of their two membranes: the outer mitochondrial membrane and the inner mitochondrial membrane (Figure 1). The outer mitochondrial membrane contains porins that allow the passage of molecules smaller than 5 kilodaltons; it also contains a large multiprotein translocase complex that recognizes mitochondrial signal sequences on larger proteins and permits their passage. The inner mitochondrial membrane contains all of the components of the electron transport system and the ATP synthase complex; it also has many invaginations, called cristae, that greatly increase its total surface area. The double membranes form two mitochondrial compartments: the intermembrane space, located between the inner and outer mitochondrial membranes, and the matrix, located inside the inner membrane. Mitochondrial DNA is housed in the mitochondrial matrix.
Mitochondrial vs. Nuclear DNA
Similar to the nuclear genome, the mitochondrial genome is built of double-stranded DNA, and it encodes genes (Figure 2). However, the mitochondrial genome differs from the nuclear genome in several ways (Taylor & Turnbull, 2005). Many interesting features distinguish human mitochondrial DNA from its nuclear counterpart, including the following:
- The mitochondrial genome is circular, whereas the nuclear genome is linear (Figure 3).
- The mitochondrial genome is built of 16,569 DNA base pairs, whereas the nuclear genome is made of 3.3 billion DNA base pairs.
- The mitochondrial genome contains 37 genes that encode 13 proteins, 22 tRNAs, and 2 rRNAs.
- The 13 mitochondrial gene-encoded proteins all instruct cells to produce protein subunits of the enzyme complexes of the oxidative phosphorylation system, which enables mitochondria to act as the powerhouses of our cells.
- The small mitochondrial genome is not able to independently produce all of the proteins needed for functionality; thus, mitochondria rely heavily on imported nuclear gene products.
- One mitochondrion contains dozens of copies of its mitochondrial genome. In addition, each cell contains numerous mitochondria. Therefore, a given cell can contain several thousand copies of its mitochondrial genome, but only one copy of its nuclear genome.
- The mitochondrial genome is not enveloped, and is it not packaged into chromatin.
- The mitochondrial genome contains few, if any, noncoding DNA sequences. (Three percent of the mitochondrial genome is noncoding DNA, whereas 93% of the nuclear genome is noncoding DNA).
- Some mitochondrial coding sequences (triplet codons) do not follow the universal codon usage rules when they are translated into proteins.
- Some mitochondrial nucleotide bases exhibit functional overlap between two genes; in other words, the same nucleotide can sometimes function as both the last base of one gene and the first base of the next gene.
- The mitochondrial mode of inheritance is strictly maternal, whereas nuclear genomes are inherited equally from both parents. Therefore, mitochondria-associated disease mutations are also always inherited maternally.
- Mitochondrial genes on both DNA strands are transcribed in a polycistronic manner: Large mitochondrial mRNAs contain the instructions to build many different proteins, which are encoded one after the next along the mRNA. In contrast, nuclear genes are usually transcribed one at a time from their own mRNA.
Mitochondrial DNA Mutations
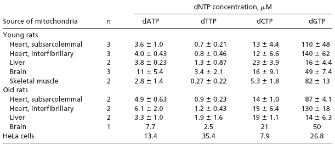
© 2005 National Academy of Sciences, USA Song, S. et al., DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity, PNAS 102, 4990-4995 (2005). All rights reserved. 
Why do mitochondria have such a high mutation rate? A nuclear gene, called DNA polymerase gamma (POLG), encodes the DNA polymerase responsible for replicating the mitochondrial genome. The POLG protein consists of two domains: a catalytic domain that exhibits polymerase activity, and an exonuclease domain that is involved in the recognition and removal of DNA base-pair mismatches that occur during DNA replication. A recent study suggests that mitochondria may have a nucleotide imbalance that leads to decreased POLG fidelity and higher mitochondrial DNA mutation rates (Song et al., 2005).
In the aforementioned study, Song and colleagues (2005) measured the mitochondrial levels of free deoxynucleotide triphosphates (dNTPs), the building blocks of new DNA strands made during DNA replication, in tissues from young and old rats. As shown in Table 1, the researchers did not detect differences in the nucleotide levels in tissues taken from young versus old rats. However, they found that mitochondrial dNTP levels were highly divergent, and that dGTP was by far the most abundant nucleotide in the mitochondria of most tissues. In heart muscle and skeletal muscle, for example, they found that dGTP represented 85% to 91% of the mitochondrial nucleotide pool, whereas dTTP was present at 0.5%. Similarly, in the brain, dGTP represented 62% of the mitochondrial nucleotide pool, whereas dTTP constituted only 4%. In the liver, dGTP, dCTP, dATP, and dTTP made up 37%, 51%, 9%, and 3% of the mitochondrial nucleotide pool, respectively. The abundant dGTP levels in mitochondria were in stark contrast to the dGTP levels in the rest of the cell: In whole rat embryos, dGTP represented only 10% of the free nucleotide pool.
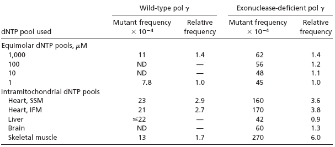
© 2005 National Academy of Sciences, USA Song, S. et al., DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity, PNAS 102, 4990-4995 (2005). All rights reserved.
The researchers next hypothesized that tipping the balance in dNTP levels may cause the mitochondrial DNA polymerase to make more mistakes during DNA replication. To test this hypothesis, they carried out experiments to determine whether POLG fidelity was compromised when dNTP levels were unequal (Song et al., 2005). The research team used purified human POLG with or without the proofreading exonuclease domain (Exo+ or Exo-, respectively) to attempt to fill in a gap of DNA sequence corresponding to the lacZ gene when provided with either equal levels of dNTPs or dNTP levels that mimicked those found in mitochondria. When the filled-in lacZ gene was introduced into bacterial host cells, the resulting color of the bacterial cell colony was used to report whether a mutation had occurred: blue colonies represented the wild-type lacZ gene, whereas light blue or white colonies represented clones that had acquired a mutation in the filled-in lacZ gene.
The DNA sequence of individual lacZ clones was also determined, so that the nature of the mutation could be deduced (Song et al., 2005). As shown in Table 2, when Exo- POLG was provided with dNTP pools mimicking those of mitochondria in subsarcolemmal heart muscle (SSM), intrafibrillary heart muscle (IFM), or skeletal muscle (SM), the lacZ mutation frequencies were between two- and sixfold higher. Notably, DNA sequence analysis of the lacZ templates showed a 30-fold increase in the rate of T to C substitutions. Furthermore, when Exo+ POLG was provided with these same dNTP pools, the researchers noted a four- to eightfold increase in T to C substitutions. These results suggested that increased dGTP levels compromised the fidelity of both Exo- and Exo+ POLG. Song et al. (2005) concluded that this imbalance in the mitochondrial dNTP pool was a likely contributor to the relatively high mitochondrial DNA mutation rate.
Clinical Manifestations of Mitochondrial Mutations
Due to the maternal pattern of mitochondrial inheritance, males with a mitochondrial disease are not considered to be at risk for transmitting the disorder to their offspring. It's important to remember that there are many mitochondria within a cell, each with its own mtDNA and potential mutations. Thus, when discussing mitochondrial mutations, it is necessary to think of mutations present across the entire mitochondrial population rather than in a single mitochondrion. Although mitochondrial populations are considered heteroplasmic, with variations among the many mtDNA genomes, mothers can have mitochondrial populations that are homoplasmic for a given mitochondrial mutation; in this case, the majority of their mitochondrial genome would harbor the mutation. Homoplasmic mitochondrial mutations will be transmitted to all maternal offspring; however, due to the complex interplay between the mitochondrial and nuclear genomes, it is often difficult to predict disease outcomes, even with homoplasmic mitochondrial populations.
Classic Mitochondrial Syndromes
A list of clinical disorders associated with mitochondrial mutations is provided in Table 3. One of these mitochondria-associated disorders is Leber hereditary optic neuropathy (LHON), which leads to a loss of vision in both eyes and is most commonly associated with a homoplasmic mitochondrial DNA mutation, although heteroplasmic transmission also occurs (Man et al., 2003). While all of the children of a homoplasmic mother will inherit the LHON mutation, not all will develop the disease; in fact, only 50% of the male offspring and 10% of female offspring will suffer from optic nerve disease. These findings point to the likely involvement of other genes and environmental factors.
Similarly, a homoplasmic mutation in a mitochondrial genome-encoded ribosomal RNA, called RNR1, causes postlingual deafness (deafness that occurs after three years of age, when a child has already learned to speak). The clinical symptoms of this disease are associated with the administration of a particular type of antibiotic (Prezant et al., 1993). Therefore, environmental factors also contribute to the phenotypes associated with this mitochondrial mutation.
Clinical Syndromes with a High Probability of Mitochondrial DNA Involvement
What are some clues that may suggest a mitochondrial link to disease? Some clinical features include a maternal family history and the involvement of several different tissues. Furthermore, because mitochondria function as the powerhouses of our cells, mitochondrial mutations often lead to more pronounced phenotypes in tissues that have high energy demands, such as brain, retinal, skeletal muscle, and cardiac muscle tissues. A number of clinical syndromes are currently believed to be associated with mitochondrial disease. Possible examples include Pearson syndrome, Leigh syndrome, progressive external ophthalmoplegia, exercise-induced muscle pain, fatigue, and rhabdomyolysis.
Mitochondrial Mutations That Contribute to Common Disease Phenotypes
Mitochondrial mutations are also likely to contribute to a number of common clinical diseases. One example is diabetes, which is the most prevalent metabolic disease affecting humans. It is also likely that mitochondrial mutations may predispose individuals to Alzheimer's disease and Parkinson's disease.
Mitochondrial Inheritance
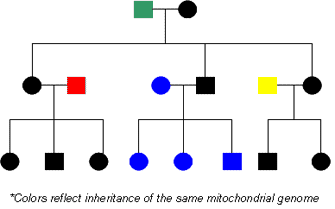
Figure 4: Inheritance of the maternal mitochondrial genome.
The mitochondrial genome is inherited from the mother in each generation.
© 2008 Nature Education All rights reserved.
As previously mentioned, mitochondrial DNA in humans is always inherited from a person's mother (Figure 4). As a result, we share our mitochondrial DNA sequence with our mothers, brothers, sisters, maternal grandmothers, maternal aunts and uncles, and other maternal relatives. Due to the high mutation rates associated with mitochondrial DNA, significant variability exists in mitochondrial DNA sequences among unrelated individuals. However, the mitochondrial DNA sequences of maternally related individuals, such as a grandmother and her grandson or granddaughter, are very similar and can be easily matched.
Mitochondrial DNA sequence data has proved extremely useful in human rights cases, as it is a great a tool for establishing the identity of individuals who have been separated from their families. This approach has been very successful for the following reasons (Owens et al., 2002; Schubert, 2003):
- A person's mitochondrial DNA sequence is shared with all of his or her maternal relatives, allowing a genetic match even with few surviving relatives.
- Mitochondrial DNA varies greatly between unrelated families, but it should be nearly identical among closely related individuals.
- A given cell contains many more copies of its mitochondrial DNA than its nuclear DNA, which allows researchers to more easily obtain and analyze mitochondrial DNA samples from deceased relatives.
One of the most prominent researchers to use mitochondrial
DNA sequence data to tackle human rights issues is Dr. Mary-Claire King, who
has undertaken such efforts in numerous countries, including Argentina,
Croatia, El Salvador, Guatemala, Haiti, Rwanda, Mexico, Chile, Honduras,
Ethiopia, and the Philippines (Owens et al., 2002; Schubert, 2003).
A particularly interesting example of Dr. King's work occurred in Argentina. As a result of a military dictatorship that overthrew the existing Argentinean government in 1976, thousands of citizens disappeared between 1976 and 1983, including infants and children who were abducted along with their parents. In addition, some children were born to women who were pregnant at the time of their kidnappings. After the military dictatorship was defeated, a new government commission predicted that at least 8,800 and possibly as many as 30,000 people had been kidnapped, including 220 documented infants and children. In 1977, the grandmothers of these orphans formed the Associacion de Abuelas de Plaza de Mayo in an effort to identify their missing grandchildren, many of whom were illegally adopted by military families.
In 1984, King used mitochondrial DNA sequence data to reunite some of these Argentinean orphans with their grandmothers. King collected blood samples from orphaned children and from women who had lost their children and grandchildren. Using mitochondrial DNA sequence data, she then matched more than 60 orphans with their biological families. In fact, as recently as 1999, a young Argentinean man named Guillermo was finally reunited with his grandmother and sister. Guillermo's parents were kidnapped by security forces in October 1978; Guillermo's mother, Patricia, was pregnant at the time of her kidnapping, and Guillermo was born one month later. Guillermo provided a blood sample to King's group, and his mitochondrial DNA sequence was a perfect match to that of one woman out of 2,000 in the database: Rosa, the mother of Patricia. As an additional test, the researchers obtained a DNA sample from Mariana, the known daughter of Patricia, who was at a friend's house on the day her parents were kidnapped. Guillermo's mitochondrial DNA sequence was also a perfect match to that of Mariana (Owens et al., 2002; Schubert, 2003). As shown by this example, mitochondrial DNA sequences can be used to establish family ties with maternal relatives, even when both of a person's parents are missing.
Mitochondria in Aging and Cancer
Over the years, a probable role for mitochondria in both aging and cancer has emerged. As a byproduct of their role as powerhouses of our cells, mitochondria generate reactive oxygen species (ROS). ROS production has been proposed to cause somatic mitochondrial mutations. This can lead to a cycle in which ROS generate mutations, which in turn lead to disregulation of respiration and accumulation of more mutations. Indeed, ROS production contributes to tissue aging due to decreased metabolic function and energy production, increased cell death, and a decreased capacity to replicate the genome.
In 1998, a link between colorectal cancer and somatic mitochondrial mutations was established by Polyak and colleagues. These researchers cultured colorectal cancer cells taken from the tumors of 10 colorectal cancer patients. They then compared the mitochondrial DNA sequences of the tumor cell lines to the mitochondrial DNA sequences of cells from neighboring normal colon tissue from the same patient. This side-by-side comparison was used to identify somatic mutations that had occurred in the mitochondrial DNA of the tumor cells. The researchers found that seven cell lines had acquired somatic mutations in their mitochondrial DNA sequences. Three of the cell lines had acquired a single mutation, and four had acquired between two and three mutations, for a total of twelve mutations. Eight of the mutations were in protein-encoding mitochondrial genes, and four of the mutations were in mitochondrial ribosomal RNA (rRNA) genes.
To confirm that the mitochondrial mutations had occurred in the tumors themselves and not during the culturing of the cells, the researchers next isolated DNA from the original tumor tissue and sequenced the mitochondrial DNA directly. Tumor tissue was only available for five out of the seven patients with mitochondrial mutations in their cultured cells. In all five cases, however, the same mutations were present in the primary tumor as in the cultured tumor cells. Furthermore, the mitochondrial mutations were all homoplasmic in both the primary tumor and in the cultured cells.
Based on these findings, Polyak and colleagues (1998) suggested that the somatic mitochondrial mutations might have provided a growth advantage to a single cell that subsequently proliferated more rapidly than the surrounding cells. Furthermore, based on the homoplasmic nature of the mitochondrial DNA mutation, they suggested that the mutation might have provided a replicative advantage to the mutant mitochondrial genome. In the years that followed, a number of other studies also established associations between somatic mitochondrial mutations and various forms of cancer, including leukemias and solid tumors. However, a causative link between mitochondrial mutations and cancer has not yet been firmly established.
Advances and Prospects in Mitochondrial Genetics
Clearly, the role of the mitochondrial genome must be considered with respect to human genetic disease. The heterogeneity of the mitochondrial genome presents many unmet challenges to researchers. However, emerging technologies are likely to aid the discovery of underlying genetic mechanisms linking these powerhouses to neurodegenerative disease, cancer, diabetes, and aging.
Mitochondrial DNA plays important roles in other areas of genetics as well. For example, it has been used to address questions about how the widespread distribution of humans in the world today was established. Because mitochondria are passed exclusively through the maternal lineage and there is little recombination in the mitochondrial genome, variation in the mitochondrial genome (as well as in the Y chromosome in the case of paternal lineages) has been used to delineate how and when humans migrated and occupied the world. Studying the mitochondrial genome in individuals from distinct geographic origins has made it possible to establish that the human populations of today are all derived from a small group of individuals that left Africa approximately 170,000 years ago (Ingman et al., 2000).
Summary
Though small in size, the mitochondrial genome is responsible for ensuring that the powerhouses of our cells function properly. This circular genome is both more plentiful than its nuclear counterpart and more prone to mutation. Currently, it is difficult to predict the way in which mtDNA mutations will pass from mother to child due to the interplay between the mitochondrial and nuclear genomes. It is clear, however, that these mutations are more pronounced in tissues that place high energy demands on mitochondria. In spite of this, mtDNA mutations are not entirely a bad thing. In fact, the variability introduced into mtDNA sequences by these mutations helps link family members to one another and has proven useful in reuniting missing children with their mothers, grandmothers, brothers, and sisters. Much remains to be learned about mtDNA, however, and continued study of this fascinating genome will continue to expand our understanding of human disease and human history for years to come.
References and Recommended Reading
Ingman, M., et al. Mitochondrial genome variation and the origin of modern humans. Nature 408, 708-713 (2000) doi:10.1038/35047064 (link to article)
Man, P. Y., et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. American Journal of Human Genetics 72, 333-339 (2003)
Owens, K. N., et al. Genomic sequencing in the service of human rights. International Journal of Epidemiology 31, 53-58 (2002)
Polyak, K., et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature Genetics 20, 291-293 (1998) doi:10.1038/3108 (link to article)
Prezant, T. R., et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nature Genetics 4, 289-294 (1993) 10.1038/ng0793-289 (link to article)
Schubert, C. Profile: Mary-Claire King. Nature Medicine 9, 633 (2003) doi:10.1038/nm/0603-633 (link to article)
Song, S., et al. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proceedings of the National Academy of Sciences 102, 4990-4995 (2005)
Taylor, R. W., & Turnbull, D. M. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics 6, 389-402 (2005) doi:10.1038/nrg1606 (link to article)










