


















« Prev Next »

Sorptives and Sorbents in Soils
Most relevant chemicals can be grouped into three sorptive categories: (a) anionic sorptives that are negatively charged because they have more electrons than protons (e.g., the nutrient orthophosphate, PO43-, is an anion); (b) cationic sorptives that are positively charged because they have fewer electrons than protons (e.g., the divalent cations Ca2+ and Pb2+); and (c) uncharged organic sorptives that exhibit a range of polarities (non-polar to polar) based on the distribution of electrons across the molecule (e.g., the aromatic ring compound, benzene, C6H6, has uniform electron distribution and hence is non-polar, whereas sucrose, C12H22O11, has a non-uniform distribution of electrons and is highly polar). These categories can also be applied to portions of large sorptives that contain a diversity of functional groups. For instance, the antibiotic oxytetracycline contains hydroxyl (R-OH) groups that can lose a proton (deprontonate) to become negatively charged (R-O-), amino (R-NH2) groups that can protonate to form positively-charged functional groups (R-NH3+), and un-charged, non-polar aromatic (-Ar-) ring structures. Thus, depending on the solution pH, oxytetracycline can exhibit behavior consistent with any of these three sorptive categories.In a similar manner, we can broadly conceptualize the reactive surface sites on sorbents as positively charged, negatively charged, or un-charged (polar or non-polar) in nature. The major solid phase materials (sorbents) in soils are layer silicate clays, metal-(oxyhydr)oxides, and soil organic matter (SOM). Layer silicate clays are primarily negatively charged because their stacks of aluminum-oxygen and silicon-oxygen sheets are often chemically substituted by ions of lower valance. In many soils, they represent the largest source of negative charge. Metal-(oxyhydr)oxides are variably charged because their surfaces become hydroxylated when exposed to water (Liu et al. 1998) and assume anionic, neutral, or cationic forms based on the degree of protonation (≡M-O-, ≡MOH or ≡MOH2+, where ≡M represents a metal bound at the edge of a crystal structure), which varies as a function of solution pH. Thus, these variably charged minerals adopt a net positive surface charge at low pH and a net negative surface charge at high pH (see Qafoku et al. 2004 for a recent review of variably charged soil minerals). SOM includes living and partially decayed (non-living) materials as well as assemblages of biomolecules and transformation products of organic residue decay known as humic substances. SOM contains a multitude of reactive sites ranging from potentially anionic hydroxyls (R-OH) and carboxylic groups (R-COOH) to cationic sulfhydryl (R-SH) and amino groups (R-NH2), as well as aromatic (-Ar-) and aliphatic ([-CH2-]n) moieties that are the principal un-charged, non-polar regions of the soil solid phase. Variations in the abundance, surface area and chemical composition of these three sorbents significantly influence the sorption characteristics of a given soil (Johnston & Tombácz 2002).
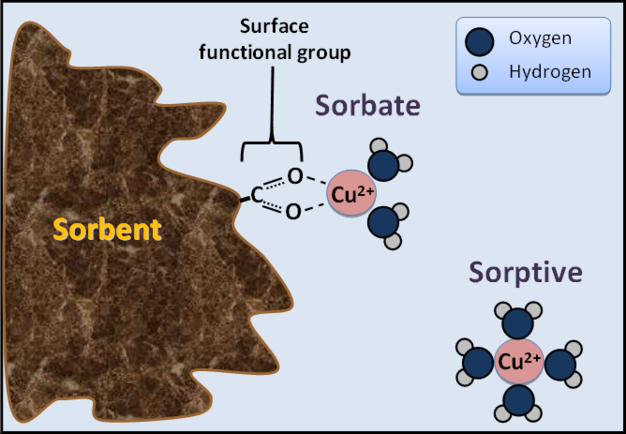
Figure 1: Sorptive, sorbate, and sorbent.
Schematic diagram illustrating the location and differences between a sorptive ion (hydrated copper ion), sorbate ion (copper ion complexed with carboxylate surface functional group on soil organic matter), and sorbent (soil organic matter).
© 2013 Nature Education All rights reserved. 
Principal Factors Governing Sorption in Soils
Sorptive Concentration
Despite heterogeneity in the chemical composition of sorbents (solids), sorptive concentration has perhaps the most significant influence on the accumulation of a sorbate on or within a sorbent. At low sorptive concentration, Le Chatelier's Principle of chemical equilibrium predicts that increasing (or decreasing) sorptive concentration will result in an increase (or decrease) in sorbate concentration. For instance, adding fertilizer to a soil will increase the solution potassium (K+) concentration and subsequently increase the amount of K sorbed by the solid phase. Conversely, as growing plants uptake K+ from the soil solution, this will drive desorption of the sorbate K+ from the soil. In this way, the soil serves as a nutrient reserve for plants and soil organisms. 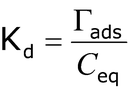
A common method for assessing sorption characteristics of a soil is to measure the relationship (or distribution) between the equilibrium concentration of the sorptive (Ceq in mol L-1) and the sorbate (Γads in mol kg-1) across a range of sorptive concentrations while holding temperature and other parameters constant. The resulting dataset is termed a sorption isotherm (Figure 2) and is often used in a predictive fashion to describe sorption behavior. For many organic compounds and ions in solution at low concentration (Chiou 2002), this relationship is linear and summarized by a distribution coefficient (Kd; L kg-1):
[1]
However, at higher ion concentrations the relationship between Γads and Ceq can exhibit significant nonlinearity (Huang et al. 2002), often approaching a maximum sorption plateau after which subsequent increases in sorptive concentration do not result in additional sorption. Many sandy agricultural soils are near their maximum sorption capacity for phosphorus (P) as a result of long-term fertilization practices and/or a naturally low affinity for P.
Figure 2: Adsorption isotherms.
Adsorption isotherms illustrating phosphorus (P) sorption to B horizon soils with differing clay content.
© 2012 Nature Education Adapted from Goyne et al. 2008. All rights reserved.
Sorptive and Sorbent Charge
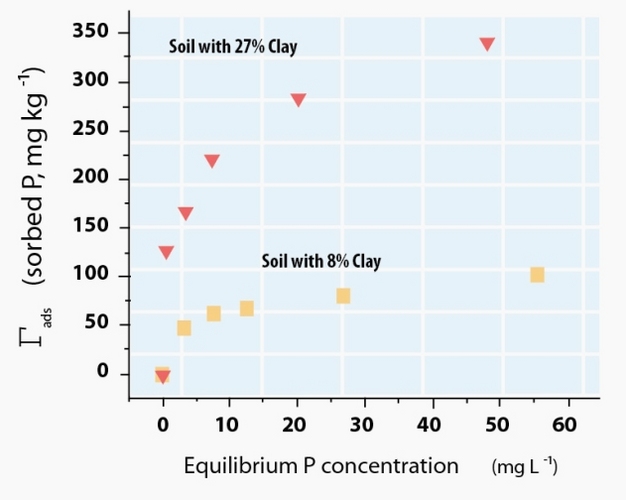
For ionic sorptives, the sign and magnitude of electrical charge on the sorptive and sorbent are important determinates of sorptive fate. Anions will be attracted to positively charged sorbents and cations will be attracted to negatively charged sorbents. This is illustrated well by the differential mobility in soils of the common nuclear fission products radioactive cesium-137 (137Cs), strontium-90 (90Cs), technetium-99 (99Tc) and iodine-129 (129I). In aqueous environments 137Cs and 90Sr ionize to the monovalent (singly-charged) 137Cs+, and divalent (doubly-charged) 90Sr2+ cations, while 129I and 99Tc ionize to the monovalent anions (129I- or 129IO3-) and 99TcO4-. If accidental release of fission products occurs in soils dominated by negatively charged sorbents (i.e., layer silicate clays), such as at the Department of Energy (DOE) Hanford Site in the northwestern U.S., the cations 137Cs+ and 90Sr2+ are retained closer to the point of contamination, whereas the anions 129I- and 99TcO4- pass rapidly through the soil and can threaten groundwater sources (Zachara et al. 2007). In contrast, anionic fission products released at the DOE Savannah River Site in the southeastern U.S. are comparatively less mobile due to the prevalence of metal (oxyhydr)oxide sorbents, which possess significant positively charged sites in acidic (low pH) soils of this region (Kaplan 2003). Similar sorptive-sorbent interactions have been noted for plant nutrients (Sollins et al. 1988).Ionic sorptives are attracted to oppositely charged sorbents by long-range electrostatic forces. For example, on negatively charged sorbents, these forces result in a preferential accumulation of cations relative to anions near the sorbent surface. However, diffusive and dispersive forces act to homogenize this ion distribution such that the ratio of cations to anions decreases exponentially with distance from the sorbent until the bulk solution ion balance is obtained. A precise description of the resulting ion distribution is complex and remains incomplete, but is commonly approximated by concepts involving an inner layer of cations immediately adjacent the sorbent surface — often termed the Stern layer — and a diffuse collection of cations and anions — termed the diffuse layer — which undergo diffusive exchange with the bulk solution (Figure 3). Sorbates within the Stern layer that shed one or more of their surrounding water molecules (i.e., waters of hydration) and form direct ionic and/or covalent (electron sharing) bonds with the sorbent are considered inner-sphere adsorption complexes, while those ions retaining their hydration spheres and maintaining proximity to the surface solely through electrostatic interactions are considered outer-sphere adsorption complexes (Sposito 2004). Characterizing the ion distribution within these layers presents a significant challenge (Brown Jr & Calas 2011) that is perhaps best approached by combining synchrotron-based x-ray adsorption spectroscopy (Brown & Sturchio 2002, Hesterberg et al. 2011, Singh & Gräfe 2010) with molecular modeling (Zhang et al. 2004).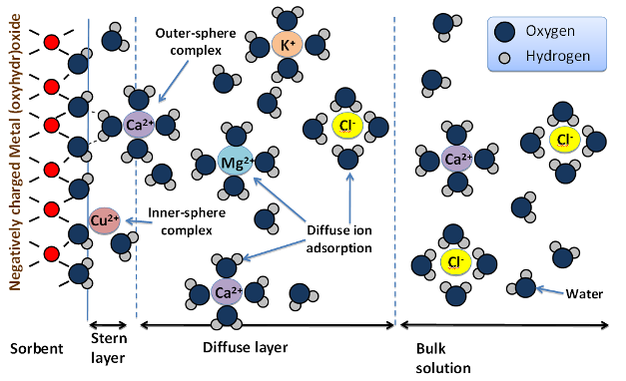
Figure 3: Schematic arrangement of sorbates at a charged sorbent interface.
To accommodate a negatively charged sorbent, cations accumulate closer to the surface than anions. Inner-sphere and outer-sphere complexes occupy the first layer of sorbates, with the remainder of the sorbent charge balanced by preferential cation accumulation in the diffuse layer.
© 2012 Nature Education Adapted from Maurice 2009, Chorover & Brusseau 2009. All rights reserved.
Solution pH
Solution pH can have a pronounced effect on sorption by influencing both the magnitude and sign of sorptive and sorbent charge. As the solution pH increases, sorbent hydroxyl and carboxyl functional groups of metal-(oxyhydr)oxides and SOM deprontonate. In turn, this increases negative charge density on the sorbent thus facilitating cation adsorption (Figure 4), while decreasing anion adsorption. Consequently, the ability of a soil to retain cations (e.g., Ca2+, Mg2+, K+, etc. — the sum of which represents the cation exchange capacity) generally improves when soil pH is increased through liming. Sorptives that can undergo acid dissociation or hydrolysis reactions also exhibit strong pH-dependent sorption trends (Essington 2004). For instance, the previously mentioned antibiotic oxytetracycline (OTC) is predominately positively charged at pH values below its first acid-dissociation constant (pKa1 = 3.3), net neutral between pH 3.3 to 7.3 (pKa2 = 7.3), and negatively charged above 7.3. Consequently, OTC sorption is greater at lower pH than at higher pH in soils containing layer silicate and organic matter sorbents (Sassman & Lee 2005), which can remain negatively charged below pH 3. In contrast, OTC sorption to metal-(oxyhydr)oxides-which are generally positively charged below pH 8 — is often greatest between pH 7.3 and 8 when OTC is negatively charged and the metal-(oxyhydr)oxide is still positively charged (Figueroa & MacKay 2005).
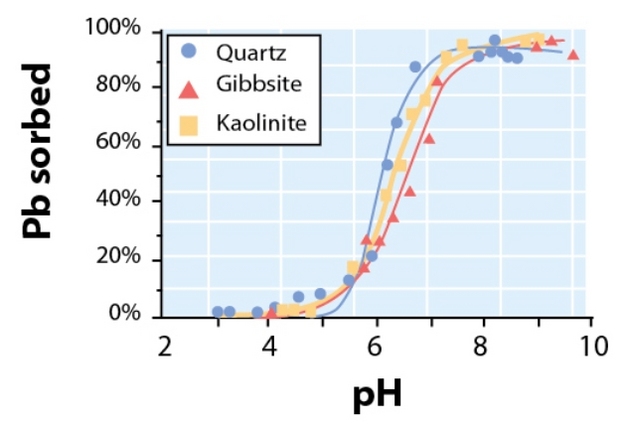
Figure 4
Adsorption of Pb2+ by quartz, gibbsite, and kaolinite as a function of pH.
© 2012 Nature Education Adapted from Essington 2004. All rights reserved.
Sorptive Size
For inorganic sorptives of equivalent charge and concentration, the ion size becomes an important determinate of their comparative sorption behavior. This is conveniently encapsulated within the concept of ionic potential (IP), defined as the ratio of ion charge to ion radius (IP = Z/r). It is generally observed that affinity for a sorbent increases with decreasing ionic potential for a given valence (e.g., IPCa2+ = 20 and IPMg2+ =48). This derives from two general principles: (1) It requires less energy to remove a water molecule from a hydration sphere of a large ion than it does for an equivalently charged ion of smaller size. This provides larger ions with an energetic advantage in forming inner-sphere bonds with the sorbent. (2) As ionic radius increases the electron cloud extends further away from the nucleus and can more easily distort and engage in covalent (electron sharing) bonds with the sorbent. As a consequence, sorptive ion affinity for many sorbents follows common trends based on ionic radius within a single group of the periodic table, and such ionic radius based selectivity is most readily observed for Group IA, IIA, and IIB elements. For example, Group IA element affinity decreases as a function of ionic radii: Cs+ (0.167 nm)> Rb+ (0.148 nm) > K+ (0.138 nm)> Na+ (0.102 nm) > Li+ (0.059 nm) (Sparks 2003). Such relationships can be useful for predicting cation behavior in soil, as constituents bound in inner-sphere surface complexes are inherently more difficult to desorb, hence they are less mobile than ions sorbed via outer-sphere complexes. 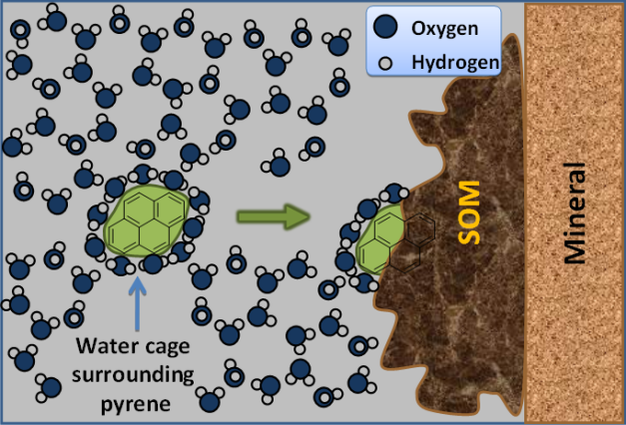
Figure 5: Sorption driven by hydrophobicity.
Development of a water cage (clathrate structure) around hydrophobic molecule disrupts bulk liquid water structure. Sorption of pyrene to hydrophobic moieties in soil organic matter (SOM) minimizes this disruption.
© 2012 Nature Education All rights reserved.
Sorption of Organic Molecules: Additional Considerations
For ionizable organic constituents, sorption is largely governed by sorptive/sorbent charge characteristics similar to those discussed for inorganic cations and anions. However, these same principles do not hold for sorption of un-charged organics and additional factors must be considered for large organic molecules (e.g., humic substances). Un-charged compounds that are non-polar cannot bond effectively with the polar water molecule and thus minimizing the interfacial area between the water and the non-polar sorptive is energetically favorable. This leads to the common segregation of non-polar compounds from water commonly referred to as the hydrophobic effect. When portions of a non-polar compound come in contact with a non-polar sorbent they are no longer exposed to water and thus the overall non-polar/polar interfacial area is reduced. Reduction in this interfacial area is further enhanced if the sorbate binds to a non-polar (or hydrophobic) region of SOM. Thus, sorption of non-polar compounds is well correlated with SOM content (Figure 6). However, the molecular-scale structure of SOM is exceedingly complex (Sutton & Sposito 2005; Lehmann et al. 2008) and variations in the chemical structure of SOM strongly influence sorption (Young & Weber 2005). Once non-polar organics are ‘driven' from solution and into contact with hydrophobic regions within SOM, short-range van der Waals forces can enhance sorbate-sorbent interactions (Pignatello & Xing 1995). Van der Waals forces arise from resonating polarity fluctuations within non-polar moieties of the sorbent and sorbate. 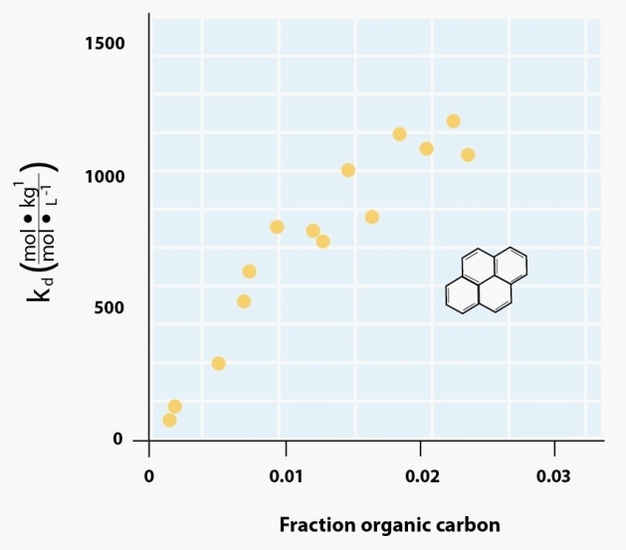
Figure 6: Pyrene sorption to a variety of soils and sediments.
Increase in the distribution coefficient (Kd) for pyrene, an non-ionic, non-polar sorptive, as a function of the fraction of organic carbon content (fOC) in the solid phase.
© 2012 Nature Education Adapted from Schwarzenbach 2003; data from Means et al. 1980. All rights reserved.
Sorption of un-charged compounds with polar functional groups — particularly those containing oxygen or nitrogen atoms — are strongly influenced by hydrogen bonding between the sorbate and sorbent. Hydrogen bonding develops due to interaction of a hydrogen atom in a polar bond and an unshared electron pair in an electronegative atom in close proximity. For example, a sorbate ketone group (C=O) can form a hydrogen bond with a hydroxylated surface functional group on a mineral surface. Both van der Waals forces and hydrogen bonding are additive across the molecule. Thus, as size of the sorbate increases, these attractive forces become progressively more important for sorbate retention to the soil (Schwarzenbach 2003). For example, the soil distribution coefficient (Kd) for the single aromatic ring compound benzene is smaller than the Kd value for pyrene, which has four fused aromatic rings (Chiou et al. 1998). The preferential sorption of humic substances with high molecular weight over those with lower molecular weight can be explained in similar terms (Zhou et al. 2001).
Conclusion
Sorption is arguably the most important chemical processes governing the retention of nutrients, pollutants and other chemicals in the soil. We have touched on the fundamental aspects of sorption in soils, but this review is by no means a comprehensive treatise on the subject. In particular, the importance of time (kinetics) and precipitation (the formation of 3-D solid phases) has not been addressed. We refer interested readers to excellent recent reviews by Borda & Sparks (2008) and Chorover & Brusseau (2009), both of which discuss the kinetics of sorption with a treatment of surface precipitation. Finally, we emphasize that in soils, sorption occurs concurrently with biotic and abiotic transformation reactions in an open hydrodynamic system. Therefore, a comprehensive understanding of sorptive fate requires integration across multiple Earth system sciences.
Aaron Thompson1* and Keith W. Goyne2
[1]Department of Crop and Soil Sciences, University of Georgia, Athens, Georgia 30602, USA.
[2]Department of Soil, Environmental and Atmospheric Sciences, University of Missouri, Columbia, MO 65211, USA
* Corresponding author: AaronT@uga.edu; 706-410-1293.
Glossary
absorption: The process through which ions or molecules are removed from solution and accumulate within existing solid constituents.
adsorption: The process through which ions or molecules are removed from solution and accumulate on solid constituents at the solid-liquid interface.
Cation Exchange Capacity (CEC): Sum of cations that can be displaced (exchanged) into solution from the soil solid phase when the soil is equilibrated with a dilute salt solution; typically expressed in units of cmolc kg-1.
desorption: The release of ions or molecules from solids into solution.
diffuse layer: The region of ion adsorption near a sorbent surface that is subject to diffusion with the bulk solution. Diffuse layer ions are not immediately adjacent the surface, but rather are distributed between the inner, Stern layer ions and the bulk solution by balance of electrostatic attraction to the sorbent and diffusion away form the sorbent.
functional group: Atom or group of atoms in a chemical structure that impart a particular chemical characteristic to the compound. A specific functional group (e.g., R-COOH) typically behaves in a similar manner on all chemical structures, although its behavior is modified to a degree by the bonding environment.
humic substances: Dark, complex, heterogeneous mixtures of organic materials that form in geomedia from microbial transformations and chemical reactions that occur during the decay of organic biomolecules, polymers, and resides.
hydration sphere: Shell of water molecules surrounding an ion in solution.
hydrogen bond: Intermolecular attraction between a hydrogen atom in a polar bond with an unshared electron pair of an electronegative atom in close proximity.
hydrophobic effect: The attraction of nonionic, non-polar compounds to surfaces that occurs due to the thermodynamic drive of these molecules to minimize interactions with water molecules.
inner-sphere adsorption complex: Sorption of an ion or molecule to a solid surface where waters of hydration are distorted during the sorption process and no water molecules remain interposed between the sorbate and sorbent.
layer silicate clay: Clay minerals composed of planes of aluminum (AlIII) or magnesium (MgII) in octahedral coordination with oxygen and planes of silica (SiIV) in tetrahedral coordination to oxygen. Substitution of AlIII for SiIV in the tetrahedral plane or substitution of MgII or FeII for AlIII in the octahedral plane (isomorphic substitution) results in a permanent charge imbalance (i.e., structural charge) that must be satisfied through cation adsorption.
metal (oxyhydr)oxide: Minerals composed of various structural arrangements of metal cations-principally AlIII, FeIII and MnIV-in octahedral coordination with oxygen or hydroxide anions. These minerals are dissolution byproducts of mineral weathering and they are often found as coatings on layer silicates and other soil particles.
outer-sphere adsorption complex: Sorption of an ion or molecule to a solid surface where waters of hydration are interposed between the sorbate and sorbent.
precipitation: Formation of an insoluble product that occurs via reactions between ions or molecules in solution.
soil organic matter: Living and partially decayed (non-living) materials as well as assemblages of biomolecules and transformation products of organic residue decay known as humic substances.
sorbate: Ions or molecules that have accumulated on or within a solid due to a sorption reaction.
sorbent: The solid phase constituent participating in a sorption reaction. The solid phase may be more specifically referred to as an absorbent or adsorbent if the mechanism of removal is known to be absorption or adsorption, respectively.
sorption: Removal of a compound from solution by solid phase constituents. This term is often used when the mechanism of removal (adsorption, absorption, or precipitation) is unknown.
sorption isotherm: Graphical representation of surface excess (i.e., the amount of substance sorbed to a solid) relative to sorptive concentration in solution after reaction at fixed temperature, pressure, ionic strength, pH, and solid-to-solution ratio.
sorptive: Ions or molecules in solution that could potentially participate in a sorption reaction.
Stern layer: The layer of ions adsorbed immediately adjacent to a charged sorbent surface. Ions in the Stern layer can be directly bonded to the sorbent through covalent and ionic bonds (inner-sphere complexes) or held adjacent to a sorbent through strictly electrostatic forces in outer-sphere complexes.
van der Waals forces: Intermolecular attractive forces that arise between nonionic, non-polar molecules due to dipole-dipole interactions and instantaneous dipole interactions (London dispersion forces).
References and Recommended Reading
Borda, M. J. & Sparks, D. L. "Kinetics and mechanisms of sorption-desorption in soils: A multiscale assessment," in Biophysical-Chemical Processes of Heavy Metals and Metalloids in Soil Environments, 97-124, eds. A. Violette, P. M. Huang, & G. M. Gadd, New York: Wiley, 2008.
Brown Jr, G. E. & Calas, G. Environmental mineralogy - Understanding element behavior in ecosystems. Comptes Rendus Geoscience 343, 90-112 (2011).
Brown, G. E. & Sturchio, N. C. "An overview of synchrotron radiation applications to low temperature geochemistry and environmental science," in Applications of Synchrotron Radiation in Low-Temperature Geochemistry and Environmental Sciences, Volume 49, 1-115, Reviews in Mineralogy & Geochemistry, eds. P. A. Fenter et al., Chantilly, VA: Mineralogical Society of America, 2002.
Chiou, C. T. et al. Partition characteristics of polycyclic aromatic hydrocarbons on soils and sediments. Environmental Science & Technology 32, 264-269 (1998).
Chiou, C. T. Partition and Adsorption of Organic Contaminants in Environmental Systems. New York, NY: Wiley, 2002.
Chorover, J. & Brusseau, M. L. "Kinetics of sorption-desorption," in Kinetics of Water-Rock Interaction, eds. S. L. Brantley, J. D. Kubicki, & A. F. White, 109-149, New York: Springer, 2009..
Essington, M. E. Soil and Water Chemistry. Boca Raton, FL: CRC Press, 2004.
Figueroa, R. A. & MacKay, A. A. Sorption of oxytetracycline to iron oxides and iron oxide-rich soils. Environmental Science & Technology 39, 6664-6671 (2005).
Goyne, K. W. et al. Phosphorus and nitrogen sorption to soils in the presence of poultry litter-extracted dissolved organic matter. Journal of Environmental Quality 37, 154-163 (2008).
Huang, W. et al. Effects of organic matter heterogeneity on sorption and desorption of organic contaminants by soils and sediments. Applied Geochemistry 18, 955-972 (2003).
Hesterberg, D. et al. X-ray microspectroscopy and chemical reactions in soil microsites. Journal of Environmental Quality 40, 667-678 (2011).
Johnston, C. T. & Tombácz, E. "Surface chemistry of soil minerals," in Soil Mineralogy With Environmental Applications, eds. J. B. Dixon & D. G. Schulze, 37-67, Madison, WI: Soil Science Society of America, 2002.
Kaplan, D. I. Influence of surface charge of an Fe-oxide and an organic matter dominated soil on iodide and pertechnetate sorption. Radiochimica Acta 91, 173-178 (2003).
Lehmann, J. et al. Spatial complexity of soil organic matter forms at nanometre scales. Nature Geoscience 1, 238-242 (2008).
Liu, P. et al. Reaction of water vapor with α-Al2O3(0001) and α -Fe2O3(0001) surfaces: Synchrotron X-ray photoemission studies and thermodynamic calculations. Surface Science 417, 53-65 (1998).
Maurice, P. A. Environmental Surfaces and Interfaces From The Nanoscale To The Global Scale. New York, NY: Wiley, 2009.
Means, J. C. et al. Sorption of polynuclear aromatic hydrocarbons by sediments and soils. Environmental Science & Technology 14, 1524-1528 (1980).
Pignatello, J. J. & Xing, B. Mechanisms of slow sorption of organic chemicals to natural particles. Environmental Science and Technology 30, 1-11 (1995).
Qafoku, N. P. et al. "Variable charge soils: Their mineralogy, chemistry and management," in Advances in Agronomy, Volume 84, ed. D. L. Sparks, 159-215, New York: Academic Press, 2004.
Sassman S. A. & Lee, L. S. Sorption of three tetracyclines by several soils: Assessing the role of pH and cation exchange. Environmental Science & Technology 39, 7452-7459 (2005).
Schwarzenbach, R. P., Gschwend, P. M. & Imboden, D. M. Environmental Organic Chemistry, 2nd ed. New York, NY: Wiley, 2003.
Singh, B. & Gräfe, M. Synchrotron-Based Techniques in Soils and Sediments. New York, NY: Elsevier, 2010.
Sollins, P. et al. Nutrient mobility in variable and permanent-charge soils. Biogeochemistry 6, 181-199 (1988).
Sparks, D. L. Environmental Soil Chemistry, 2nd ed. San Deigo, CA: Academic Press, 2003.
Sposito, G. The Surface Chemistry of Natural Particles. New York, NY: Oxford University Press, 2004.
Sposito, G. The Chemistry of Soils, 2nd ed. New York, NY: Oxford University Press, 2008.
Sutton, R. & Sposito, G. Molecular structure in soil humic substances: The new view. Environmental Science & Technology 39, 9009-9015 (2005).
Thompson, H. S. On the absorbent power of soils. Journal of the Royal Agricultural Society England 11, 68-74 (1850).
Way, J. T. On the power of soils to absorb manure. Journal of the Royal Agricultural Society England 11, 313-379 (1850).
Young, T. M. & Weber Jr, W. J. "Sorption and desorption rates for neutral organic compounds in soils," in Chemical Processes in Soils, 519-562, eds. M. A. Tabatabai & D. L. Sparks, Madison, WI: Soil Science Society of America, 2005.
Zachara, J. M. et al. Geochemical processes controlling migration of tank wastes in Hanford's vadose zone. Vadose Zone Journal 6, 985-1003 (2007).
Zhang, Z. et al. Ion adsorption at the rutile-water interface: Linking molecular and macroscopic properties. Langmuir 20, 4954-4969 (2004).
Zhou, Q. et al. Size fractionation upon adsorption of fulvic acid on goethite: Equilibrium and kinetic studies. Geochimica et Cosmochimica Acta 65, 803-812 (2001).










