
Abstract
Study design:
Post hoc analysis from a randomized controlled cellular therapy trial in acute, complete spinal cord injury (SCI).
Objectives:
Description and quantitative review of study logistics, referral patterns, current practice patterns and subject demographics.
Setting:
Subjects were recruited to one of six international study centers.
Methods:
Data are presented from 1816 patients pre-screened, 75 participants screened and 50 randomized.
Results:
Of the 1816 patients pre-screened, 53.7% did not meet initial study criteria, primarily due to an injury outside the time window (14 days) or failure to meet neurological criteria (complete SCI between C5 motor/C4 sensory and T11). MRIs were obtained on 339 patients; 51.0% were ineligible based on imaging criteria. Of the 75 participants enrolled, 25 failed screening (SF), leaving 50 randomized. The primary reason for SF was based on the neurological exam (51.9%), followed by failure to meet MRI criteria (22.2%). Of the 50 randomized subjects, there were no significant differences in demographics in the active versus control arms. In those participants for whom data was available, 93.8% (45 of 48) of randomized participants received steroids before study entry, whereas 94.0% (47 of 50) had spine surgery before study enrollment.
Conclusion:
The ‘funnel effect’ (large numbers of potentially eligible participants with a small number enrolled) impacts all trials, but was particularly challenging in this trial due to eligibility criteria and logistics. Data collected may provide information on current practice patterns and the issues encountered and addressed may facilitate design of future trials.
Similar content being viewed by others
Introduction
The history of pharmaceutical research in acute spinal cord injury (SCI) is recent. The Sygen (GM-1 ganglioside)1 and NASCIS (National Acute Spinal Cord Injury Study—methylprednisolone) clinical trials helped pave the way for more recent trials in terms of study design, methodology and outcome assessment.2, 3, 4 Although the efficacy of interventions studied in these trials is still the subject of debate, they were instructive in establishing that multi-center, randomized, controlled trials in this population are feasible, albeit challenging. Owing to the paucity of effective FDA-approved treatments for acute or chronic spinal cord injury, there is a large demand for treatments that will alter the natural history of these patients.
SCI research is now entering the era of cellular therapy. The design and execution of cellular therapy-based trials can build on what has been learned in previous pharmaceutical trials, but may also present unique challenges. The use of cell-based agents in acute human SCI trials consists of a single Phase 1 trial using autologous macrophages.5 The Phase 1 trial (sponsored by Proneuron Biotechnologies) was completed in February, 2002 under a US Food and Drug Administration (FDA) Investigational New Drug (IND) application. Autologous macrophages were co-incubated with autologous skin (a proprietary process called ProCord) and delivered surgically via injection into the spinal cord. The scientific rationale is briefly described by Knoller et al.5 Eight participants were enrolled three of whom improved from American Spinal Injury Association [ASIA] Impairment Scale [AIS]) classification A to C, as measured by the International Standards for Neurological Classification of Spinal Cord Injury (ISNCSCI).6 The Phase 1 trial showed a reasonable safety profile and an encouraging recovery rate within the context of expected natural recovery, considering the small sample size. A Phase 2 trial was initiated in October 2003, with the approval of local Institutional Review Boards (IRB), National Regulatory Boards (Israel) and the FDA under an amended IND.
This article describes the unique challenges in the Phase 2 ProCord trial related to study pragmatics and design, particularly issues unique to an autologous cell-based therapy, proposes potential means to overcome these challenges, and reviews participant recruitment and demographics. Safety, outcomes and efficacy data will be reported separately after data analysis is complete.
Materials and methods
Study design
The study was a Phase 2, multi-center, randomized, controlled trial involving six SCI centers, five in the US, and one in Israel (Table 1). Recruitment began in October 2003 with a target enrollment of 61 participants in a 2:1 active to control allocation. The primary efficacy measure was improvement in AIS by at least one grade at 12 months post-intervention. Secondary efficacy endpoints included improvement in ISNCSCI sensory and motor scores, improvement of at least two motor levels in participants with cervical spine injuries, and improvement in bowel and bladder function.
Before trial initiation, an advisory committee was convened to consult on trial design and sample size, which resulted in the following recommendations:
-
A no-intervention/standard of care control group was included in the trial design recognizing the possibility of natural recovery and the lack of conclusive outcome data in existing databases that matched eligibility criteria. Given the satisfactory safety profile (albeit from a small sample size) from the Phase 1 trial, a 2:1 active to control arm allocation was chosen as a more acceptable randomization scheme than 1:1, for potentially eligible participants;
-
The use of a sham or placebo surgical control group was not considered feasible. The extent of sham required in this trial to mimic the experimental procedure would be ethically questionable as it would necessitate an intra-dural injection of a non-active substance. In considering placebo as an alternative to a sham procedure, a laminectomy would be required to approximate the post-operative recovery of the experimental procedure. In either case, it would be impossible to blind the surgeon; therefore only blinding of the participant would be feasible. The investigators concluded that an effective sham or placebo control would require a degree of surgical exposure that could not be ethically justified in this subject population. While this decision limited the use of double-blinded research design, assessment bias was addressed by the use of single blinded outcome examinations at key time points.
Sample size
A sample size of 61 was chosen based on several factors. As there is no established minimal clinically important difference (MCID-the smallest difference in score that is clinically meaningful to an individual) established for the ISNCSCI sensory/motor scores or AIS, rehabilitation and surgical leaders in the field of SCI were asked what they considered a meaningful improvement in both AIS and success rate. They determined that a 20% or greater improvement in the occurrence of conversions from AIS A to B or greater (using a weighted scale), over a 1-year period would be clinically significant. The rate of success in the Phase I trial and the rate of spontaneous recovery were examined by looking at two databases (Israel and the US National Spinal Cord Injury Database [NSCID]), with as many matched criteria as possible. The trial was powered to show that treatment is superior to control with P<0.20. This level of significance was used rather than the 0.05, frequently used in pivotal trials, owing to the exploratory nature of this Phase 2 trial and the difficulty of recruiting suitable participants. Planning for a 2:1 treatment ratio and assuming a true rate of spontaneous recovery from AIS A at 7 days to AIS C at 1 year in 3% of control participants (based on the databases mentioned above) and a true treatment effect of 23%, a sample of 34 active and 17 controls provide 80% power to achieve this goal. (A total of eight participants were originally enrolled and two additional participants were added to the Phase 1a FDA study, and an additional four participants were enrolled under a 1b non-FDA trial bringing the total number to 14. One participant with spinal cord transection was excluded from this calculation, such that 3 of 13 (23%) recovered from AIS A to C.) Ten participants were added to account for an expected 20% dropout rate for a total of 61.
The study sponsor thought that a trend towards improvement, in conjunction with satisfactory safety data, would be enough to justify a Phase 3 trial in the future.
Inclusion/Exclusion criteria
Inclusion/exclusion criteria are listed in Table 2. Additional information explaining the rationale for specific inclusion/exclusion, related to cellular-based therapy and safety issues in acute SCI trials is available in the Supplement Information.
Training
Training in the ISNCSCI motor/sensory examinations and determination of AIS is recommended in the ICCP (International Campaign for Cures of spinal cord injury Paralysis) guidelines7 and by previous researchers.8, 9, 10, 11 These two aspects of the ISNCSCI assessment require a different set of skills,9 thus both motor/sensory examiners (detailed in a separate publication12) and AIS assessors were trained and inter-rater reliability assessed separately, before the start of the trial. To ensure accuracy of the AIS, examinations were reviewed in a blinded manner by the Chair of the ASIA Standards Committee. Following training ISNCSCI examiners evaluated participants at study sites, remote hospitals or at the subject's home for follow-up as needed.
No formal group training occurred for the spine surgeons or radiologists. However, the spine surgeons met before and during the course of the trial to discuss different aspects of the injection technique including intra-operative imaging, targeting of injection sites, duraplasty and post-operative care. Study radiologists conferred to establish standards for MRI (magnetic resonance imaging) interpretation, such as delineating contusion versus edema, the images and techniques required to measure contusion length, and frequently consulted with one another when reviewing images before the study. All MRI studies screened for trial inclusion and follow-up were read by one of two ‘central’ neuroradiologists (blinded to subject allocation), to minimize variability in image interpretation.
Study logistics
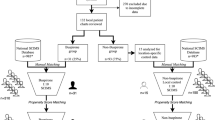
Figure 1 illustrates the study schema and flow.

Study schema.
Referrals
Individuals with SCI became aware of the trial primarily via the internet (42.2%), physician referrals (16.1%) or family members/friends (7.0%) (Table 3). The sponsor informed clinical personnel and the general public about the trial using several methods, including the sponsor and study site's web sites, presentations and/or booths at individual hospitals, rehabilitation, trauma and surgery conferences and registration on www.clinicaltrials.gov. All public information was approved a priori, by the Institutional Review Boards of all participating facilities.
A centralized Call Center (with a toll free number) provided international coverage 24 h a day, 7 days a week. Interested individuals, acute hospital staff and/or family members contacted the Call Center, which used a standardized script for basic screening to minimize the number of calls passed to individual sites for processing by screening out individuals who were ineligible due to age, penetrating trauma or time since injury.
Subject allocation
The Call Center used a geographically based allocation between four of the US sites, which distributed calls to the nearest study site based on subject location, including calls from Canada and Latin America. The fifth US site processed only direct referrals for participants between 16 and 21 years of age. The study site in Israel was allocated participants from the remainder of the world.
Preliminary eligibility assessment and pre-screening
The pre-screening process to determine preliminary eligibility was extensive and thorough for multiple reasons including the long distances frequently traveled by potential participants to study sites. The pre-screening process was initiated after a release of personal health information was obtained by call center or study site personnel. Numerous steps occurred in parallel during the pre-screening process.
One of the first steps in the pre-screening process was to request a copy of all spine MRIs as there were several inclusion/exclusion criteria related to imaging. (The preferred sequences to optimize interpretation of contusion [hemorrhage] length were sagittal T1, T2 FSE, Gradient echo T2, and STIR; axial T1 and gradient echo T2. Frequently, an acute MRI had already been completed by the referring facility; this MRI was accepted, regardless of sequences.) The radiological evaluation included: measuring the length of contusion (excluded if longitudinal dimension of injury [hemorrhage] >3 cm), evidence of anatomical transection/laceration (excluded if present), edema was measured for information only. If the contusion length could not be determined due to metal artifact, or the subject was unable to have an MRI, they were considered ineligible for the trial.
In parallel to the radiological screening criteria, potential participants were screened for eligibility for rehabilitation (for example, pragmatic likelihood that individuals would receive standard of care rehabilitation) and all other inclusion/exclusion criteria. If all criteria appeared to be met, the study protocol was reviewed with the patients and families by phone. For patients who chose to participate, and were located remote from the study site, the patients were transported to a study facility, by air ambulance if necessary (46.7% of participants). Upon arrival at a study site, informed consent was obtained from all participants by the primary investigator or his/her designee. On site screening then began to confirm trial eligibility. Baseline testing occurred just before randomization to re-confirm eligibility.
A repeat (baseline) MRI was obtained (within 48 h before randomization) and reviewed locally or via the centralized MRI center, depending on time constraints. If the contusion had extended beyond three cm, the subject was considered ineligible. By this time, most participants had undergone surgical stabilization of the injury site, and therefore, artifact frequently obscured the contusion. In this case, the screening MRI was used for inclusion/exclusion. An issue encountered during the trial was variability in the study centers application of contraindications for MRI following inferior vena cava filter placement. Thus in some study centers obtaining a baseline MRI in participants with an inferior vena cava filter was problematic.
After screening criteria were met and baseline testing was completed, participants who continued to be eligible for the trial were randomized to either the active or control arm. Randomization envelopes had been prepared centrally in a computerized, blinded fashion by the study statistician using randomized blocks for each study site.
Surgical procedure(s)
As this was an autologous procedure, an initial surgery was necessary to obtain the raw materials (blood and skin) Approximately 230 ml of blood was obtained via central or peripheral venipuncture and a full thickness skin graft (12 × 3 cm) was obtained from the medial aspect of the upper arm. Blood and skin were then delivered to a dedicated cell center located near the clinical site because of the 2 h expiration of the cells (expiration was extended to 8 h toward the end of the study following additional cell stability studies). Blood and skin were then co-incubated for 37 h. The prepared cells were delivered to the operating room where the dura was opened and cells were injected in six locations (20 μl with 0.25 million cells each, total 120 μl and 1.5 million cells) across the caudal border of the lesion, as determined by MRI and intraoperative ultrasound when feasible.
Control participants did not undergo the skin-harvesting procedure nor the injection surgery, but were asked to donate 230 ml of blood, which was activated with allogeneic skin donated by anonymous donors for research purposes. Control participants were followed for 1 year with the same outcome assessments as the active (participants receiving cells) group.
Committees
The study was supervised by the Steering Committee, which met approximately quarterly to advise the sponsor on suggested changes to the protocol or study conduct. An Independent Data Monitoring Committee met twice yearly to monitor safety.
We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during the course of this research.
Results
Pre-screening
Of the 1816 patients pre-screened by the Call Center, 53.7% did not meet initial criteria. The primary reason for ineligibility was time since injury >14 days (46.0%) (Table 4). For patients who met the call center criteria, but failed to meet additional criteria, the primary reason was the neurological level or incompleteness of injury (26.8%), followed by failure to meet MRI criteria (23.8%).
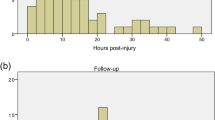
MRIs (n=339) were obtained for pre-screening; 64.3% were completed pre-operatively (before stabilization), 30.1% post-operatively, 5.6% were not identified as pre or post operative by study radiologists. The higher than expected rate of post-operative MRIs (versus pre-operative MRIs) resulted in 11.3% (of ineligible MRIs) diagnostically inadequate MRIs due to artifact. Of the 51.2% of participants deemed ineligible by MRI criteria, the main reason was length of contusion greater than 3 cm (57.9%) (Table 5). Contusion length for all MRIs averaged 2.9 cm±1.7 cm (2.5 cm±1.1 cm in cervical spine injuries [n=142] and 3.4 cm±1.8 cm in thoracic spine injuries [n=162]). The average length of edema for all injuries was 6.8 cm±2.6 cm (6.6 cm±2.7 cm in cervical spine injuries [n=99], and 7.1 cm±2.4 cm in thoracic spine injuries [n=96]). Of all cervical spine films deemed ineligible (n=48), 83% (n=40) were because of a contusion >3 cm. Of all the thoracic spine films deemed ineligible (104), 70% (n=73) were due to a contusion >3 cm.
Screening
Although target enrollment was 61 participants, the unexpected withdrawal of a source of financial support (unrelated to study safety or efficacy issues), compounded by the unforeseen costs due to trial design, forced the sponsor to stop recruitment prematurely. Some of the costly aspects of the trial were related to building or contracting multiple cell centers, medical transportation of participants to clinical trial sites close to the cell centers, and the high rate of screen failure. Therefore, enrollment was suspended in March 2006 after 50 participants were randomized.
Seventy-five of participants who passed initial screening for the trial were enrolled (see Figure 2 for a flow chart of participant enrollment). One-third of these subsequently screen failed (SF) (Screen failure [SF] -failed to meet criteria after signing of informed consent but before randomization. Premature termination [PT]—participants who signed informed consent and were randomized but did not complete the trial. Batch failure—an autologous macrophage preparation that failed to meet microbial and/or release testing criteria, such that the participant was unable to receive the cells) leaving 50 participants in the trial. Enrolled participants screen failed for the following reasons (multiple reasons per participant in some cases): neurologically incomplete (8), neurological level (6), medical reasons (6), MRI (5), concomitant medications (1) (see Table 2 for exclusion criteria related to concomitant medications) and unstable neurological exam (1). Of the 50 eligible participants, eight were considered premature terminations (PT), which occurred due to: batch failure (2), febrile (2), MRI (1), death (1), lost to follow-up (1) and consent withdrawn (1). Of the eight PTs, five occurred before or on the date of injection or the equivalent in the control group (day 0), three occurred after day 0 (day 7, month 3 and month 9). Of the 42 participants remaining, 33 completed the 12-month follow-up in accordance with the study protocol (one completed 9 months, eight completed 6 months) by the end of November 2006, when the financial issues discussed above necessitated canceling follow-up visits.

Flow chart of participant enrollment.
The 50 randomized participants arrived at a study site on average 8.8±3.0 days following injury. Participants were screened on day 10.0±1.7, baseline testing and randomization was performed on day 11.0±1.4 and participants received the macrophages (or the control group equivalent) on day 13.00±1.5 (One participant had a planned injection [protocol deviation] on day 15. The participant had a previous batch failure before day 14 and a second attempt was made on day 15, resulting in a second batch failure and premature termination). Baseline testing and screening were combined because of time constraints in 20 of the 50 (40.0%) participants.
Enrollment Demographics
Of the 50 participants randomized there were 17 controls and 33 active participants. No significant differences were noted between groups in terms of gender, age, body mass index, vertebral level of injury (range C5-T11), cause of injury, spine surgery or use of steroids before study entry (Table 6). Ninety-six percent of participants were Caucasian, 2.0% were Hispanic and 2.0% were African American.
Approximately 94% of randomized participants received steroids following injury. Among the participants for whom type of steroid was known, all but one received methylprednisolone, and the remaining participants received dexamethasone. Three participants did not receive steroids, and for two additional participants, steroid use was not known. Of those five participants, four were from outside of the United States. The one US participant who did not receive steroids was admitted into the trauma system beyond the eight hour window, and therefore, was not eligible for steroids based on the NASCIS trials protocol.3, 4
Ninety-four percent of participants had spine surgery (decompression and/or stabilization) before study entry. Two of the three who did not have previous surgical intervention had decompression (1) or stabilization (1) during the surgical procedure to inject the macrophages. The third participant was allocated to control and did not require surgery. One participant who was eligible based on the pre-stabilization MRI, had stabilization before arrival at the study site with stainless steel instrumentation, impacting the radiographic interpretation of the injury site. The participant's hardware was removed and replaced with titanium screws and rods (reducing ferro-magnetic artifact) during the procedure to inject the autologous macrophages, to minimize the artifact in future MRIs. In comparing vertebral to neurological level of injury (NLI) based on the ISNCSCI at the baseline exam in the 49 participants (24 cervical, 25 thoracic) for whom data was available; in 45.8% of cervical and 8.0% of thoracic the vertebral and NLI were equivalent (26.5% overall), in 8.3% of cervical and16.0% of thoracic the vertebral level of injury was one level higher (12.2% overall) than NLI, and for 45.8% of cervical and 76.0% of thoracic the vertebral level of injury was lower than the NLI (range 1–4 levels) (61.2% overall).
Cerebrospinal fluid was collected for research purposes with limited success. CSF was collected in active participants at day 0, after the dura was opened during the surgical procedure to inject the macrophages and via lumbar puncture at day 7 and month 12 in all participants. Based on available data, CSF was successfully collected at day 0 in 69.0% of active participants (20 of 29), at day 7 and month 12 in 39.1% (18 of 46) and 3.4% (1 of 29), respectively, of all participants.
Discussion
The conundrum in clinical research is the funnel effect, which is particularly challenging with acute SCI. Although there are approximately 11–12 000 new traumatic spinal cord injuries per year in the United States, once strict inclusion criteria for a specific study are applied, the number of potentially eligible participants diminishes substantially. In the United States, patients with SCI are widely scattered between acute care facilities with varying degrees of neurotrauma experience, such that few centers see large numbers of patients with acute SCI. In this trial, 4.1% of individuals who contacted the call center were transported to a study site, 2.8% of whom were randomized. Even eliminating those individuals who did not meet the basic criteria to pass through the Call Center, only 6.0% were randomized. The inclusion/exclusion criteria for this trial were carefully chosen to decrease the impact of confounding independent variables while focusing on patient safety, therefore the criteria were somewhat restrictive until efficacy and safety were better defined, which occurs in later phase clinical trials. Enrollment of a large number of participants with acute SCI, who meet inclusion criteria, can be difficult. The challenge of informing trauma and intensive care unit-based personnel in a large number of facilities nationwide is a daunting task. In some cases physicians are reluctant to refer a patient to a Phase 2 trial, without published results. Furthermore, clinical staff may be aware of the trial, but may be reluctant to refer an acute patient to another facility. One option to address some of these issues is through the use of clinical trial networks with both trauma and acute care facilities, which would facilitate screening and choice of clinical study sites, improve dissemination of inclusion/exclusion criteria, and in turn improve subject recruitment. To conduct a trial such as described herein, multiple centers are needed, potentially even in Phase 1 trials, thus adding complexity, cost and increased variability in techniques and data collection.
Unfortunately, many of these issues are unalterable. This study necessitated the transportation of participants to participating centers affiliated with a cell center, as cell centers and personnel were limited and costly. However, the alternative of transporting product and extending stability beyond 2 h (extended to 8 h) only was able to be validated close to the end of the trial. The expanded window would have allowed the possibility of adding more centers and eliminating the inconvenience of transporting and displacing participants and their families. In future research, these considerations should be addressed in pre-clinical studies and validation.
The time window for injection and the use of autologous product significantly impacted study logistics. By using an autologous product with a 14-day window for injection, the raw materials were only available after injury and the processing was complex and time consuming. An ‘off the shelf’ allogeneic cellular-based product may allow more flexibility and simplify logistical issues. The scheduling of the macrophage injection surgery was encouraged as early in the study window as feasible, to have a variety of time points for injection, enabling analysis of day of injection as a potentially important independent variable. Owing to the complexity of enrollment procedures and use of an autologous product, the average day for macrophage injection was day 13. With the narrow window of 14 days for injection combined with a small sample size, it is difficult to draw conclusions about effectiveness at a variety of time points.
An issue impacting all acute SCI trials is the route of administration of the product. For a high risk route of administration (that is, intradural surgery), more participants may be excluded from the trial due to inherent surgical risks, even if all other criteria are met. A major surgery may also affect when participants can initiate rehabilitation. The less invasive the route of administration, the less risk to the participant, and therefore, a potentially higher rate of enrollment can be achieved. Of the 96 patients who contacted a study site or the Call Center and chose not to participate, the reason was obtainable from only 28. The most commonly stated reason for choosing not to participate was concern about undergoing a second surgery. In future studies, enrollment may be facilitated by validation of the least invasive route of administration, development of a product that can be delivered to the participant without requiring medical transport to specialized sites, and administration of the product during initial stabilization without a stand alone surgical procedure. The sponsor was, in fact, conducting pre-clinical studies using an intrathecal infusion of cells, to explore a less invasive alternative to an open surgical procedure.
A significant logistical issue was transmission of MRIs. At the outset of the study, it was assumed that all images could be sent from referring sites and study sites to the central radiologists. However, many facilities had difficulty transferring electronic images; therefore pre-screening images were sent from the referring site either as hard copies or electronically to the centralized MRI reading center. Raw Digital Imaging and Communications in Medicine (DICOM) images stored on compact disk (CD) and shipped via courier overnight became the most common method of sending MRIs. Unfortunately these methods of image transmission caused delays in central reading of MRI images in an already tight window (14 days). Use of an imaging contract research organization, specializing in electronic image transmission could improve this logistical challenge and the resulting delay in assessing preliminary eligibility.
The careful choice of inclusion/exclusion criteria is critical to ensure good study design, balanced with the need to target a population that will benefit if the product is labeled for commercial use and to ensure that the funnel effect is not prohibitive. An example is the inclusion criteria of patients with complete SCI. From a safety standpoint, this is often the optimal choice in a trial with an invasive route of administration; however, these are also the most severe injuries. With the advent of regenerative therapies, patients with an incomplete SCI, who may benefit the most from these types of interventions, may not be included until later stage trials for safety reasons. Therefore, sponsors must either have compelling safety data to include patients with incomplete SCI in early stage trials, or be prepared to accept limited efficacy in more severe injuries.
There are many potential reasons for the high rate (1/3 of participants) of screen failure. The most common reasons were a neurologically incomplete injury (29.6%) and level of injury (22.2%), together accounting for 51.8% of screen failures. Frequently, participants were examined remotely during pre-screening and, once transported and examined by study trained personnel, did not meet the trial inclusion criteria based on the ISNCSCI. In some cases, spontaneous conversions from complete to incomplete SCI may have occurred. Another reason for the discrepancy between remote and study-trained examiners may be that the ISNCSCI although detailed in a reference manual, are clinically applied with a wide degree of interpretation and are not commonly used in non-rehabilitation focused trauma and local hospitals therefore, it was often difficult to confirm with certainty that participants met criterion for completeness and level of injury. Even experienced examiners, if not trained, may not be reliable. Reliability was critical in this study, as participants frequently traveled a long distance to study sites. One mechanism used to potentially improve reliability was for the study treatment site investigator to guide a local therapist or physician through the exam, at times sending portions of the training manual and describing the exam in detail with local examiners.
Although an Investigators Meeting (IM) and Site Initiations occurred before the trial and an IM was also held mid-study, sites became active in the trial sequentially. There was a learning curve for both the sponsor and sites in terms of enrollment and minimizing screen failures. For example the criteria for neurological level of injury was amended from ‘A single spinal cord lesion with last fully preserved neurological level from C-5 to T-11’ to ‘A single spinal cord lesion between C5 motor/C4 sensory level and T11 neurological level by the ISNCSCI’ following a number of screen failures. The rationale for this change is discussed in the Supplement. Approaches the investigators used to minimize screen failures were also communicated between sites. The rate of screen failure for neurological level of injury did diminish as the trial progressed.
In this study, pre-trial training was formally conducted for ISNCSCI motor/sensory testing and AIS. Formal training for radiologists and surgeons should be considered for future trials. Clinical sites and thus surgeons were added sequentially to the trial, thus making standardization issues more challenging but, on the positive side, enabling new surgeons to learn from experiences of other sites.
Despite the small sample size, random allocation of participants to active and control arms resulted in equivalent groups in terms of demographics, pre-study surgical intervention and steroid use before study entry. The data collected may provide information about practice patterns in the population. For example, participants were recruited from a variety of centers throughout the United States (not just study centers) during the era when the results of the NASCIS II and III trials were considered controversial; however, steroids were widely used. All US participants who met the 8-h eligibility criteria defined in the NASCIS trials, had received clinical steroid treatment before referral to the study.3, 4
Additionally, it is interesting to note that the epidemiology of enrolled participants (96.0% Caucasian), varies from the epidemiology in the NSCID.13 Although an in-depth analysis is beyond the scope of this article, this disparity may be a reflection of recruitment methods, referring facilities and/or clinical sites.
Tests and procedures such as lumbar puncture (LP), which may be difficult, uncomfortable or have potential side effects may result in poor compliance. The primary reason for the low rate of CSF collection at day 7 and month 12 was that participants declined the LP. The rate of obtaining CSF was higher in participants allocated to the treatment versus control group on day 7, 48.3% of participants allocated to active arm consented to LP, whereas only 23.5% consented in the control arm. As CSF was collected for research purposes only (versus safety) and a separate informed consent was required, participants frequently declined. One reason CSF was not always obtained at the day 0 time point was that CSF was collected following the durotomy; therefore cord edema sometimes made collection difficult. In participants without a significant contraindication, a lumbar puncture performed after anesthesia but before opening of the dura for injection, may improve the collection rate.
Conclusion
Many challenging issues are illustrated when examining the ProCord trial, some of which may be alterable in future trials. Understanding which factors can be simplified, and facilitating these changes where possible, may be critical in future trials.
Some of the issues illustrated in this trial include the ‘funnel effect’, which occurs in all studies but was particularly evident in this study. Several factors likely contributed to the funnel effect in this study including: the inclusion criteria of 14 days since injury, medical transportation of participants to study sites, which necessitated relying on ISNCSCI exams from remote sites (that may have less experience with ISNCSCI examinations), and a route of administration requiring major surgery.
Data collected in this trial may reflect current practice patterns, which may impact the design of future protocols. Standardization of tests and procedures where possible, particularly those that may impact outcomes, is important in the context of current standards of care.
This trial shows that a complex cell-based intervention clinical trial protocol in this population can be implemented, despite the collective challenges of subject recruitment and trial logistics described herein.
References
Geisler FH, Coleman WP, Grieco G, Poonian D . The Sygen multicenter acute spinal cord injury study. Spine 2001; 26: S87–S98.
Bracken MB, Collins WF, Freeman DF, Shepard MJ, Wagner FW, Silten RM et al. Efficacy of methylpredisone in acute spinal cord injury. JAMA 1984; 251: 45–52.
Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS et al. A randomized, controlled trial of methylprednisone or naloxone in the treatment of acute spinal-cord injury: results of the second national acute spinal cord injury study. N Engl J Med 1990; 322: 1405–1411.
Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M et al. Administration of methylprednisone for 24 or 48 h or tirilazad mesylate for 48 h in the treatment of acute spinal cord injury: results of the third national acute spinal cord injury randomized controlled trial. JAMA 1997; 277: 1597–1604.
Knoller N, Auerbach G, Fulga V, Zelig G, Attias J, Bakimer R et al. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: Phase I study results. J Neurosurg Spine 2005; 3: 173–181.
American Spinal Injury Association. International standards for neurological classification of spinal cord injury, revised 2000, reprinted 2002. American Spinal Injury Association: Chicago, IL, 2002.
Steeves JD, Lammertse D, Curt A, Fawcett JW, Tusynski MH, Ditunno JF et al. Guidelines for the conduct of clinical trials for spinal cord injury (SCI) as developed by the ICCP panel: clinical trial outcome measures. Spinal Cord 2007; 45: 206–221.
Priebe MM, Waring WP . The interobserver reliability of the revised American Injury Association Standards for neurological classification of spinal injury patients. Am J Phys Med Rehabil 1991; 70: 1991.
Cohen ME, Sheehan TP, Herbison GJ . Content validity and reliability of the international standards for neurological classification of spinal cord injury. Topics Spinal Cord Injury Rehab 1996; 1: 15–31.
Cohen ME, Ditunno JF, Donovan WH, Maynard FH . A test of the 1992 International Standards for neurological classification and functional classification of spinal cord injury. Spinal Cord 1998; 36: 554–560.
Burns AS, Lee BS, Ditunno JF, Tessler A . Patient selection for clinical trials: the reliability of the early spinal cord injury examination. J Neurotrauma 2003; 20: 477–482.
Marino RJ, Jones L, Kirshblum S, Tal J, Dasgupta A . Reliability and repeatability of the motor and sensory examination of the international standards for neurological classification in spinal cord. J Spinal Cord Med 2008; 31: 166–170.
Jackson AB, Dijkers M, DeVivo MJ, Poczatek RB . A demographic profile of new traumatic spinal cord injuries: change and stability over 30 years. Arch Phys Med Rehabil 2004; 85: 1740–1748.
Acknowledgements
We thank the following study coordinators and site personnel for their tireless and continual efforts from remote screening to last data capture upon which this paper is based: Anousheh Behnegar, Susan B Charlifue, Ismari Clesson, Tamar Kazoula- Halabi, Steven H Koltenuk, and Leslie R VanHeil. We thank members of the Safety Committee (John F Ditunno, Michael D Walker, and other members of the Safety Committee), Steering Committee (Marcalee S Alexander, Edward C Benzel, and the Medical Monitor (David R Cornblath) for monitoring safety and providing guidance throughout the trial. We thank DP Clinical for their support in preparing this paper. This trial was supported by Proneuron Biotechnologies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Spinal Cord website
Supplementary information
Rights and permissions
About this article
Cite this article
Jones, L., Lammertse, D., Charlifue, S. et al. A phase 2 autologous cellular therapy trial in patients with acute, complete spinal cord injury: pragmatics, recruitment, and demographics. Spinal Cord 48, 798–807 (2010). https://doi.org/10.1038/sc.2010.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2010.29
Keywords
This article is cited by
-
The challenge of recruitment for neurotherapeutic clinical trials in spinal cord injury
Spinal Cord (2019)
-
Natural history of neurological improvement following complete (AIS A) thoracic spinal cord injury across three registries to guide acute clinical trial design and interpretation
Spinal Cord (2019)
-
Spinal Cord Injury Scarring and Inflammation: Therapies Targeting Glial and Inflammatory Responses
Neurotherapeutics (2018)
-
Translational considerations in injectable cell-based therapeutics for neurological applications: concepts, progress and challenges
npj Regenerative Medicine (2017)
-
International standards for neurological classification of spinal cord injury: classification skills of clinicians versus computational algorithms
Spinal Cord (2015)
