
Abstract
The study of medications among pediatric patients has increased worldwide since 1997 in response to new legislation and regulations, but these studies have not yet adequately addressed the therapeutic needs of neonates. Additionally, extant guidance developed by regulatory agencies worldwide does not fully address the specificities of neonatal drug development, especially among extremely premature newborns who currently survive. Consequently, an international consortium from Canada, Europe, Japan, and the United States was organized by the Critical Path Institute to address the content of guidance. This group included neonatologists, neonatal nurses, parents, regulators, ethicists, clinical pharmacologists, specialists in pharmacokinetics, specialists in clinical trials and pediatricians working in the pharmaceutical industry. This group has developed a comprehensive, referenced White Paper to guide neonatal clinical trials of medicines – particularly early phase studies. Key points include: the need to base product development on neonatal physiology and pharmacology while making the most of knowledge acquired in other settings; the central role of families in research; and the value of the whole neonatal team in the design, implementation and interpretation of studies. This White Paper should facilitate successful clinical trials of medicines in neonates by informing regulators, sponsors, and the neonatal community of existing good practice.
Similar content being viewed by others

Purpose
This document is intended to assist investigators and sponsors of studies that evaluate medicinal products in neonates. Since terminology for drugs, medicines, and medicinal products varies around the world, we have simplified these distinctions by using the term medicinal product throughout this document. While the focus of the document is on studies that will contribute to applications to regulatory authorities, the same principles apply to other studies involving neonates. In addition, this document may be useful for regulators who will be reviewing these studies. This document expands upon previous draft guidances entitled “General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products: Guidelines for Industry” from the US Food and Drug Administration (FDA) dated December 2014 (1), the European Medicines Agency’s (EMA) Guideline on the Investigation of Medicinal Products in the Term and Preterm Neonate (2), the International Conference on Harmonization’s (ICH) E11 (3), and the Tri-Council Policy Statement (4) and other related documents by Health Canada. Studies of pharmacodynamics effect, clinical efficacy, safety, and/or dose-finding in neonates involve gathering clinical pharmacology information, such as information regarding a product’s pharmacokinetics (PK) and pharmacodynamics (PD) that pertain to dose selection, optimization, and individualization. This document addresses general clinical pharmacology considerations for conducting studies in term and preterm neonates. That is, studies that generate information about dosing and preliminary safety for medicinal products evaluated in neonates. One goal of this document is to support the standardization and harmonization of approaches to studies in neonates worldwide in order to facilitate global development of new and existing medicinal products for this vulnerable population.
The scope of this document is clinical pharmacology information (e.g., PK, PD, and exposure-response (E-R) relationships) that support findings of effect, efficacy, and safety and helps to identify appropriate dosages in term and preterm neonates. Since consideration of pharmaceutical quality and ethics influences many aspects of the design and implementation of clinical pharmacology studies, this document also discusses pharmaceutical quality, ethics, and participant welfare during studies. This document also describes the use of quantitative approaches (i.e., pharmacometrics) to employ knowledge about disease and E-R from relevant prior clinical studies to design and evaluate future studies in term and preterm neonates. The document does not describe (i) standards for regulatory approval of medicinal products in the neonatal population (ii) criteria to allow a determination that the course of a disease and the effects of a medicinal product on that disease are similar in adults and older pediatric populations.
While this document focuses on clinical pharmacology, it is important to remember that clinical pharmacology is embedded in a broader medicinal product development strategy. For example, it may be necessary/possible to include assessments of efficacy in the same studies that assess dosage. Clinical pharmacology studies may inform decisions by sponsors or regulators about proceeding with development or be designed to reduce commercial and regulatory risk.
Background
During the last two decades, recognition of the need for study of medicinal products in neonates has increased worldwide (5,6,7,8,9,10). To address this need, a group of individuals with a broad range of expertise has been assembled to describe the issues that need to be addressed in the study of medicinal products in the neonate. This group includes regulators, neonatologists, developmental pharmacologists, parents, clinical trialists, pharmacometricians, and senior members of the pharmaceutical industry with experience in the conduct of these studies. References are included to provide more in depth information than is possible in this document.
Worldwide efforts during the past two to three decades have increased the study of medicinal products in children, but not in the neonate. A review of the results of United States efforts illustrates the omission of neonates from these studies. Laughon et al. reviewed studies submitted to the FDA between 1997 and 2010 in response to legislation to increase pediatric labeling (11). From a total of 428 medicinal products studied in pediatric patients, only 28 (7%) were studied in the neonate. More importantly, the authors reviewed the actual use of those 28 medicinal products in the Newborn Intensive Care Unit (NICU) using a database comprised of 290 NICUs and 445,335 patients. Of the 28 medicinal products, 13 (46%) were never used in the NICU and 8 were used in <0.013% of patients. Even though 428 medicinal products were studied in pediatric patients, only 7 (1.6%) were actually used routinely in most NICUs. So, despite significant gains in understanding and improving medicinal product therapy for older children, neonates in the United States remain a population for whom therapy with off label medicinal products continues with minimal study (12). This off label use of medicinal products for neonates is a universal problem.
The reasons for the limited study of medicinal products in the neonate are complex. The immaturity, small size and rapid developmental changes in this high risk and vulnerable pediatric population complicate the measurement of beneficial effects as well as adverse effects of medications. Increasing survival of neonates at 23 wk gestation (just over half of a full term 40 wk gestation) requires evaluation of medical products in these extremely immature neonates, even if such studies were previously carried out in more mature neonates. Birth weights of neonates can range from <500–5,000 g and there are few validated endpoints to capture important effects of a variety of medications. Very large physiologic changes occur during this period of neonatal life, such as expression and maturation of enzymes, receptors, transporters, and neurotransmitters. Even the organs and tissues to be treated may respond differently or require different medicinal product concentrations to achieve comparable responses to those in older children or adults. Efforts to characterize maturation by age or size are still challenged by a broad range of biologic responsiveness that defies a simple description.
Additional features of the neonate make clinical trials challenging in this population. While some features of neonates are unique to this age group, others are simply more prominent than in other age groups. Many of these specific features are described in subsequent sections. The net effect of these specificities is that all neonatal medicinal product development programs are unique and require dedicated study designs to fit each individual medicinal product (13). Transposing a development plan from other age groups is unlikely to be successful. Neonatal medicinal product development plans require the integration of a number of perspectives including science, feasibility, welfare and ethics. This integration requires careful planning within study teams and across regulatory jurisdictions. Neonatal medicinal product development benefits from careful choices made early in the development of a molecule because information needed for optimal development for neonates may be captured best during preclinical and early phase clinical studies.
Definitions of the Neonate
Defining and classifying neonates is complex because different terms are used to reflect maturations or clinical metrics such as birth weight. Postconceptional age (PCA) is difficult to determine, so postmenstrual age (PMA) has traditionally been more frequently used clinically, dating the gestation from the mother’s known or reported last menstrual period (14). When available, the best Obstetric Estimate (OE) of gestational age provides the best measure of gestation with a combination of the first day of last menstrual period, physical examination of the mother, prenatal ultrasonography, and history of assisted reproduction (15). Later ultrasound(s) and direct examination of the neonate are less accurate. Large discrepancies (>2 wk) in gestation between the OE and the neonate’s examination by an experienced clinician should rely upon the examination (14). The important issue about terminology is to be consistent within a study as well as between studies. Prior to the increased survival of premature neonates that began over 40 y ago, the neonatal period was traditionally defined as the first 27 d after birth, which was followed by infancy (2). As neonatal survival at earlier stages of gestation increased, this convenient definition became increasingly inappropriate for a complete description of the neonatal population. A neonate born at 28 wk gestation would be classified as an infant when 32 wk PMA was reached and was still quite immature in organ development and responses. As has been adopted by the EMA, the optimal definition of a neonate is up to 44 completed weeks PMA. Infancy is from 44 completed weeks PMA to 1 y after the expected date of delivery (which is synonymous with 1 y corrected gestational age—corrected gestational age (CGA)—usually considered the postnatal age (PNA) added to the weeks of gestation at the time of birth) (2).
Neonates have been grouped by birth weight (which is feasible to obtain in all clinical settings) and by developmental age (which should be based on an assessment in early pregnancy and the neonate’s examination at birth). Term neonates are defined as 37–41 wk gestational age, (gestational age was used historically, but this is also postmenstrual age in newer terminology), with more recent definitions further delineating early term (37–38 wk), full term (39–40 wk), and later term (41 wk), recognizing stratification of outcomes within term gestations (16). Post-term refers to ≥42 wk gestational age. Preterm neonates are defined as any neonate born <37 completed weeks of gestation, (i.e., ≤36 wk + 6 d of gestation). A clinically-based classification for preterm neonates is as follows:
-
1. Moderate to late preterm infant: 32 to <37 wk gestational age
-
2. Very preterm infant: 28 to <32 wk gestational age
-
3. Extremely preterm infant: 24 to <28 wk gestational age
-
4. Preterm infants at the border of viability: 22 to <24 wk gestational age
Other definitions classify neonates by birth weight:
-
1. Low birth weight (LBW) infants: <2,500 g birth weight
-
2. Very low birth weight (VLBW) infants: <1,500 g birth weight
-
3. Extremely low birth weight infants (ELBW) infants: <1,000 g birth weight
-
4. Preterm infants at the border of viability: <600 g
Older age groups can be classified as follows:
-
Neonates: birth up to 1 mo after expected date of delivery (44 wk PMA)
-
Infants: 1 mo after expected date of delivery up to 1 y
-
Children: 1 y up to 12 y
-
Adolescents: 12 y up to 16 y or 18 y in some jurisdictions (some jurisdictions would extend this period to 21 y of age).
Specific cutoffs for PNA are best developed using knowledge of the product under study and expected maturational changes of neonates in organ/tissue responsiveness and the disposition of the medicinal product.
Term and preterm neonates are a high-risk cohort of children because their physiology changes dramatically with both gestational age (i.e., from 22–42 wk) and PNA. These changes can substantially affect medicinal product disposition and organ/tissue responsiveness. The measurement or prediction of a medicinal product’s PK (exposure) and PD (response) is essential to the clinical pharmacology assessment. It is important to describe as fully as possible the E-R relationship of a medicinal product across the developmental gestational and postnatal spectrum for neonates that are expected to be treated with the medication. In addition, knowledge of pharmacogenetics, organ/tissue function/dysfunction, and concomitant medications, all of which can affect a product’s exposure and response, may also be required. Attention to the timing of the expression of pertinent proteins (e.g., transporters and receptors) should be considered when assessing the disposition and PD of a medicinal product in developmentally immature infants.
A further complexity is that growth abnormalities, either large for gestational age (LGA, weight >90th centile) or small for gestational age or growth restricted (SGA, weight <10th centile), can affect neonatal developmental physiology and pharmacology (17,18). Trials that exclusively use birth weight as an inclusion criterion generally include a larger proportion of growth-restricted, more mature neonates compared with those that use gestational age. Trials should specify whether or not neonates born outside the normal growth range will be included. The assessment of growth should be based on growth charts or standards such as standard deviation scores that have been validated for the population under study. This means that a given study may have to use different growth charts or standards in different locations and for different populations within a single study.
Some trials are conducted in health care settings that do not provide reliable dating of gestation in early pregnancy. Such trials rely on birth weight to classify neonates. The design and analysis of these trials need to consider that interindividual variation may reflect both development and growth. The number of days after birth (age) is referred to as PNA. The disposition of some medicinal products (and hence dosage regimen optimization) varies with PNA and/or with PMA. PNA needs to be accounted for in clinical studies.
During long-term follow-up it is important to account for expected developmental status. This can be done using CGA. During studies that are conducted over relatively short time periods (i.e., up to 3 mo after the expected date of delivery), it may be more convenient to refer to PMA than CGA.
Inclusion of specific age groups should reflect the aims of the study and may benefit from a narrow population to generate a clear signal of efficacy (or lack thereof). In contrast, detailed understanding of PK and PD will benefit by recruiting a broader population, since the thorough assessment of PK/PD relationships hinges on the identification and quantification of sources of variability. As more neonates born at 22 or 23 wk gestation are treated in some centers, it will be important to include such neonates in all relevant studies. The age-stratifications cited here will help ensure that studies of clinical pharmacology sample all relevant stages of development. It should be noted that the results of clinical pharmacology studies may suggest that other age-stratifications for dosage are recommended in prescribing information (e.g., the label) or the summary of a medical product’s characteristics.
Clinical Pharmacology Considerations
General Considerations
Several clinical pharmacology issues must be considered when designing and undertaking clinical trials in the neonatal population. Neonatal studies require detailed planning that should start with a thorough assessment of existing knowledge. The team should then develop a clear concept of the treatment goal, identify knowledge gaps required to attain the treatment goal, design studies to fill the information gaps, conduct the studies and review the information continuously. When possible, information gaps should be filled from other sources, or at least the possibilities should be narrowed before clinical studies are conducted.
Early in the planning for a neonatal clinical trial, it is wise to involve neonatal nurses. They can provide valuable advice about the practicality of a study design and are pivotal in communication with families. Other relevant NICU caregivers, including respiratory therapists, social workers, pharmacists, and nutritionists, should be involved where appropriate. Another important aspect of planning clinical trials involves input from parents/guardians and children (potentially former preterm neonates) (19). They often provide valuable input not previously considered by investigators and their perspective on acceptable levels of risk may differ from that of investigators or even Ethics Committees (19,20,21,22). Efforts to include the perspective of NICU family advisory groups, parent support groups, and the individual family participants in the design and conduct of clinical trials should be strongly encouraged.
Neonatal specificities of the design of clinical trials arise because existing knowledge is likely to be incomplete and because studies will need designs that are specifically tailored to this population to account for the practical, ethical and welfare features of research in this population. Modeling and simulation through pharmacometrics, physiologically-based PK/PD modeling, as well as systems pharmacology modeling provide an ideal framework for knowledge synthesis, study design (including trial simulation) and analysis. Pharmacometric methods can inform decisions about the number of participants, times of sample collection, covariates, phenotypic analyses, and population analyses. Physiologically-based PKPD modeling provides a valuable knowledge synthesis for the accurate prediction of results from first-in-human studies. Depending on the state of knowledge regarding etiology and pathophysiology of a specific indication in the neonatal population, systems pharmacology can provide a useful (yet complex) platform to evaluate unexplored scenarios for future clinical trials. Given the incomplete knowledge about a medicinal product and ontogeny and the likelihood that data collection will be selective, study teams need to review their understanding of the data frequently. Models, assumptions, and study plans need to evolve as knowledge increases.
Traditionally, there is an expectation that each application for licensing/marketing authorization contains a self-sufficient body of data about the product ranging from preclinical data through a sequence of phases or exploratory/confirmatory studies. This approach is difficult to apply to neonates because of the issues identified in the Background. Instead, information about a medicinal product may come from a number of sources. Preclinical animal models that are comparable to the conditions associated with preterm neonates must be chosen carefully to correspond to the developmental stages of the organ to be studied (23,24,25). Some whole animal models do not survive long enough for adequate long-term neurodevelopmental assessment while others mature more rapidly than humans. Nonhuman primates have been used to study hypoxic-ischemic encephalopathy with findings that are similar to those seen in humans (26). Despite their small size, newborn mice have been used to study exposure to fluoroquinolone antibiotics as well as potential musculoskeletal toxicity (27).
The timing of clinical studies in neonates should be appropriate to the condition and the medicinal product. The traditional approach has been to wait until phase III clinical trials have been completed in adults. This approach is not appropriate in most situations. As outlined by Roth-Cline and Nelson, and as increasingly required by regulatory authorities, pediatric medicinal product development should proceed as soon as “proof of concept” for the likelihood of direct benefit for neonates is established in adult studies, unless the disease is unique to newborns (28). The prospect of direct benefit to the neonate would depend on the disease and its severity, availability of alternate treatments, and the absence of a major or significant safety concern based on adult data or existing neonatal data when similar medicinal products or excipients have been previously studied. This can lead to concurrent development in pediatric patients, including neonates, and adults. Delay in the initiation of neonatal studies may unduly deprive them of a new and more beneficial therapy. It may also contribute to off-label treatment without the guidance of evidence-based dosing, safety, and efficacy data.
The identification of the appropriate ages to study and decisions on how to stratify data by age are medicinal product-specific and require scientific justification, taking into consideration developmental biology and pharmacology as well as the neonatal populations that are likely to be treated. Final dosing regimens and therapeutic recommendations may use different age strata from the strata used in trials.
Pharmacokinetics
In the neonate, size (e.g., body weight) and maturation (gestational age at birth and PNA) are important determinants of medicinal product absorption, distribution, metabolism, and excretion (ADME) and explain a substantial amount of exposure variability and changes in PK parameters. Maturation is usually accounted for by using surrogate markers such as GA, PMA, and PNA. In addition, other factors such as physiologic derangements (e.g., organ dysfunction and body cooling); concomitant or prior medication exposure; feeding status and type of feedings; and pharmacogenomics (among others) can influence ADME in term and preterm neonates. Therefore, the PK of a medicinal product is typically evaluated over a wide gestational age and PNA spectrum in which the agents will be used (29,30,31). Areas that should be considered in planning neonatal PK studies are discussed in the following paragraphs. Not all of these areas are relevant to all medicinal products but they are listed so that investigators can make a comprehensive assessment of potential influences on dosing regimens.
Absorption. There are multiple developmental changes in neonates that can affect absorption including:
-
1. effects on gastric acidity
-
2. rates of gastric and intestinal emptying
-
3. surface area of the absorption site
-
4. gastrointestinal metabolizing enzyme systems
-
5. gastrointestinal permeability
-
6. biliary function
-
7. transporter expression
-
8. mode of administration (e.g., sublingual, through a nasogastric tube)
-
9. type of feeding (e.g., breast milk vs. formula)
-
10. cutaneous maturation
Similarly, developmental changes in skin, muscle, and fat, including changes in water content and degree of vascularization can affect absorption patterns of medicinal products delivered by intramuscular, subcutaneous, or percutaneous routes (32).
Distribution. Distribution of a medicinal product can be affected by changes in body composition, such as changes in total body water and adipose tissue, which are not necessarily proportional to changes in total body weight. At birth, neonates are born with a higher amount of total body water, which is mostly extracellular. The lower the gestational age of the neonate, the higher the total body water (as a percentage of body weight) (33,34). After birth, term neonates generally lose up to 10–15% of their total body water (higher for lower gestational age infants) in the first postnatal week, followed by a return to birth weight by 10–14 postnatal days (longer for lower gestational age infants). Plasma protein binding and tissue binding changes arising from changes in body composition with postnatal growth and development may also influence distribution. Both the amount of protein and the nature of its binding may be reduced in preterm neonates (35). Differences in blood flow to an organ/tissue (e.g., brain and liver) between term and preterm neonates and older children and adults can also affect the distribution of a medicinal product in the body.
The implication of these findings is that clinical pharmacology studies should control for these effects (which requires detailed collection of data informative about distribution, age, protein, and intercurrent illness) and whenever possible samples should be obtained with appropriate frequency in order to capture variation arising from changes in body composition.
Protein binding. Protein binding to a medicinal product or its metabolites may change with age and concomitant illness. An understanding of protein binding may be needed to interpret the data from a serum measurement and to determine appropriate dose adjustments (35,36,37). In term and preterm neonates, protein concentrations are lower than in older children and adults, which can influence unbound medicinal product concentrations that dictate a pharmacologic effect as well as adverse effects at concentrations that are well tolerated by adults. In addition, nutrition is a major concern for the sickest neonates and protein concentrations may remain low for weeks. In vitro plasma protein binding studies can determine the extent of binding of the parent and the major active metabolite(s) and identify specific binding proteins such as albumin and α-1 acid glycoprotein. As demonstrated for bilirubin, competition for protein binding by different endogenous and exogenous chemicals may require careful evaluation of unbound medicinal product concentrations along with measurement of total circulating medicinal product concentrations. Albumin is the primary binding site for bilirubin with a 1: 1 molar ratio for the high affinity binding site (38,39,40). Based on its stoichiometry, an albumin concentration of 2.5 mg/dl can bind ~20 mg/dl of bilirubin in the absence of other chemicals that compete for these binding sites. Because of the risk of kernicterus, displacement of bilirubin from its albumin binding site should be assessed as described by Robertson et al. if the medicinal product (e.g., ceftriaxone) is likely to bind to albumin and be administered to neonates (41,42). The implication is that studies need to account for protein binding, particularly if the product will be used in the weeks after birth when changes are most rapid and hyperbilirubinemia is common.
Metabolism. Medicinal product metabolism commonly occurs in the liver, but may also occur in many other organs, including the blood, gastrointestinal tract, kidney, lung, and skin. Developmental changes in metabolizing capacity can affect both bioavailability and elimination, depending on the degree to which intestinal and hepatic metabolic processes are involved (43). Although developmental changes are known, information on metabolism of specific medicinal products in neonates is limited. Each metabolic (or isoform) pathway, such as CYP enzymes, has unique ontogenic properties that need to be considered during study design because it will influence type and timing of study assessments (44).
A thorough review should be conducted of published studies of the most important metabolic developmental changes in the pathway for a specific medicinal product to potentially guide which developmental ages of neonates need to be targeted. In addition, some metabolizing enzymes (or their isoforms) may be present at birth in term and preterm neonates that are not present in older children and adults. Some of these isoforms may have similar or different affinity for substrates and activity relative to adult enzymes which needs to be considered during medicinal product development. Both rates of metabolite formation and the principal metabolic pathway can be significantly different in term and preterm neonates compared with adults and older children. The failure to appreciate differences in enzyme activity between age groups leads to inappropriate dosing regimens and safety problems in neonates (e.g., gray baby syndrome with chloramphenicol). In vitro studies performed early in medicinal product development may be useful in focusing attention on metabolic pathways that have already been studied in both adults and children. If specific information about relevant pathways is not available, or if it is not known which pathways are relevant, then studies should allow for unanticipated variability.
Microdosing can be used to define metabolic pathways. Neonatal microdosing is feasible and acceptable to parents/guardians, professionals and Ethics Committees and (45,46,47) may be helpful in some circumstances (e.g., complex metabolic pathways that are different in neonates compared with adults or animal models) (46).
Excretion. The preterm neonate has an extremely low glomerular filtration rate at birth, especially prior to 32–34 wk PMA (48,49). Postnatally, glomerular filtration rate increases rapidly, so PNA and gestational age can both affect the systemic exposure of medicinal products when renal excretion is a dominant pathway of elimination. Studies should be designed to evaluate those changes if renal excretion is an important pathway of elimination. The maturation of other excretory pathways, including biliary and pulmonary routes of excretion, is also important (50). Some medicinal products undergo active transport out of the liver, gastrointestinal tract, and brain which can prevent absorption (51,52). Others undergo active transport, especially into the liver and/or kidney depending on their chemical structures. The expression and maturation of transporters have not been thoroughly studied in developing humans. Additional developmental studies may be required to define the PK for medicinal products that are substrates for transporters. These data may need to be supplemented by basic science studies and organized to help define maturation of these pathways to assist with studies of medicinal products with similar pathways of elimination. Publication of studies for medicinal product approval can supplement basic science studies and add to our understanding of developmental pharmacology.
Clearance of medicinal products as a function of age (gestational and postnatal) is generally a valuable parameter for determining the dose for each age group in neonates. Medicinal product clearance has provided a valuable tool in the assessment of pediatric clinical pharmacology studies (53). Clearance may be widely variable depending on the gestational age of the infant and may change rapidly based on PNA. Clearance in target organs/tissues (e.g., brain) may also differ between neonates and older children/adults, so sampling compartments that are informative about target organs (e.g., cerebrospinal fluid) is advised whenever feasible.
Variability. Growth and developmental changes in the term and preterm neonate create substantial changes in ADME, between individuals and within individuals. This leads to variability that may lead to substantial overdosing or underdosing or may require therapeutic drug monitoring. PK measurements and parameters for a medicinal product may need to be described as a function of both gestational age and PNA and be related to body size and body weight (36,54,55). The maturational changes in systems affecting ADME, such as membrane transporters and metabolizing enzymes should be taken into consideration in choosing appropriate gestational and PNA groups and doses to study. Developmental changes in PK parameters are quite variable among metabolic pathways and according to maturation measured by PMA or PNA. The rate of change in PK should be analyzed thoroughly to determine the best description of the rates of change that can be used to guide appropriate dosing after birth.
Analysis of PK by age (PMA and PNA). Changes in the rates of clearance can be analyzed relative to PMA at the time of study or relative to PNA because birth signals the onset of large changes in physiology, such as a cortisol surge and a change in cardiovascular hemodynamics. Data regarding the changes in PK related to Cytochrome P450 (CYP) 2C19, the major pathway for clearance of pantoprazole, illustrate these differences (31). As can be seen in Supplementary Figure 1 online, clearance did not change relative to PMA, but increased significantly related to PNA.
These findings are consistent with what has been reported by Koukouritaki et al., using enzyme measurements from liver samples from a range of gestational ages showing low rates of CYP2C19 expression throughout gestation until birth when it starts to increase (Supplementary Figure 2 online) (56).
These data illustrate why the influence of birth on developmental pharmacology should be included in the analysis of medicinal products in neonates.
Variations among different phase I enzymatic pathways should be expected and analyzed for individual medicinal products. This variation with age is illustrated in Supplementary Figure 3 online where CYP activity varies widely among individual phase I enzymatic pathways (56,57,58,59). Some reach maximum activity during the first few months after birth and then decrease as adults (60), while CYP2D6 steadily increases to maximum activity in adults (58). This developmental variation in the activity of specific CYP’s illustrates why pK for individual drugs must be studied at the developmental ages when the drug is likely to be used in this developing population of neonates.
Physiologically based pharmacokinetics. When data are available to describe the developmental changes in pathways of medicinal product disposition, physiologically based PK (PBPK) may be a useful approach for integration of developmental changes in specific processes that determine ADME. Study design can benefit from a better understanding of the components of PBPK such as changes with PMA and PNA in: maturation of phase I enzymes; maturation of conjugation pathways by phase II enzymes; changes in glomerular filtration rates; renal tubular and hepatic transporter expression and maturation; and transcutaneous medicinal product or excipient absorption. Unanticipated differences in medicinal product clearance in neonates must be identified based on a careful PK study. For example, the clearance of daptomycin as well as micafungin is faster in the neonate than in older infants and adults, thus requiring higher dosages (61,62). Although this is unusual in neonates, it emphasizes how inadequate PK studies could lead to under-dosing of these antibiotics in the neonate which could limit their efficacy.
Pharmacodynamics
Investigators should collect and analyze both PK and whenever possible PD data in neonatal studies to determine how the two are linked (i.e., the PK-PD or E-R relationship). PD may include the effect of the medicinal product on biomarkers or clinical endpoints for both safety and effectiveness as long as they are validated. These measurements may allow a better understanding of whether the PK-PD relationships of the medicinal product in neonates are similar to those observed in older children or adults and may aid in deriving rational dosing strategies. Whenever possible, studies may assess E-R relationships to demonstrate the “effect” of a given medicinal product using direct measurements (i.e., physiologic parameters) or suitable biomarkers. It is important to determine whether the organ to be affected by the medicinal product in the neonate (e.g., heart) has the same capacity to respond as in older children or adults. Differences in capacity to respond can alter the E-R relationship significantly despite similar exposures. In some situations those questions may need to be studied through independent studies separate from the clinical trial. Interpretation of the E-R response in neonates is facilitated when there is an E-R response in other age groups. The ability to extrapolate may depend on a good E-R curve, which may be most efficiently obtained in the early phases of adult studies. This is one example of the importance of adequate planning for neonatal studies throughout the development of a medicinal product.
If the clinical endpoint cannot be measured directly because the effect is delayed or rare, then the selection of an appropriate biomarker to substitute for the clinical efficacy or toxicity endpoint is essential.
Biomarkers. In general, the careful application and assessment of the right biomarker in the right populations is pivotal in medicinal product development. Biomarkers can support the diagnosis, prognosis, initiation of treatment, and the response to treatment. Biomarkers can be used in an individual neonatal medicinal product development program or qualified for a particular context of use across multiple medicinal product development programs (63,64).
A large number of neonatal biomarkers and clinically important outcomes have been described in several systematic reviews (65,66). Overall, biomarkers may have some utility in clinical practice, but insufficient high-quality data are available to support their use in neonatal medicinal product development. In many cases, biomarkers are first evaluated in adult and/or non-neonatal pediatric population(s). The use of a biomarker in a neonate requires evidence to support a neonatal use. This is possible if the disease pathophysiology and pharmacologic response in pediatric patients are sufficiently similar to adults. Sufficient similarities are not always present, as exemplified by low blood pressure. For instance, low blood pressure is a useful biomarker in adults for systemic under-perfusion and shock, while it has not been a useful biomarker in neonates for evaluation of organ perfusion. In neonates, clinically meaningful surrogate outcomes or biochemical biomarkers borrowed from older age groups or therapeutic contexts may not reflect biological events (the combination of disease and ontogeny) with sufficient precision to predict a lasting effect for a medicinal product. If sufficient evidence to support the use of a biomarker in neonates is not available, then the medicinal product development program should include work to develop evidence that supports the use of that biomarker in neonates.
The pathophysiology of many neonatal conditions frequently involves multiple organ systems, so analysis of a single biomarker may not be sufficient. Despite these limitations, biomarkers can have utility in medicinal product development (e.g., population enrichment strategies). Biomarkers that are specific for age, organ, and condition may be more useful as PD biomarkers. Novel techniques and biomarkers (metabolomics, genomics, microRNA, etc.) offer great potential, but will also require a detailed and structured development program to establish validity.
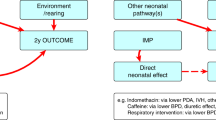
Short and long term outcomes. When selecting outcomes, it is important to note that neonates may be uniquely susceptible to medicinal products that cross the blood brain barrier and to other physiologic changes that may impact neurologic development (e.g., hypoxemia and/or acidosis). In addition, the immaturity of organ systems in neonates mean that safety signals may manifest a considerable time after the product is administered. It is necessary to include the assessment of safety in the study objectives and it is usually necessary to follow neonates beyond the period of safety surveillance of 30–90 d that is typically used in adults. Neonates may need to be followed up to 2 y CGA or older (when speech and language can be assessed). While longer-term surveillance of safety and efficacy may help define more accurate endpoints, surrogate outcomes such as reduced length of hospital stay, the incidence of comorbidities, biomarkers, and health care costs should be important factors in neonatal medicinal product development. This would remain true even if longer-term outcomes cannot consistently demonstrate benefits between experimental treatment and placebo groups. There are a number of issues to consider when selecting long-term outcomes and deciding how they best fit into a neonatal medicinal product development program.
Long-term outcome studies currently pose major challenges due to problems with patient dropout and relocation, quality control, diagnostic accuracy, interpretation of the measures themselves, underlying medical conditions, and potential environmental effects postdischarge. Parental socioeconomic status and education must be measured in the assessment of developmental outcomes.
Short-term outcomes do not always correlate with long-term outcomes. This discordance may arise because other determinants of health are dominant in the years after initial hospital discharge (e.g., social and environmental factors such as maternal education and socioeconomic status) or because short-term outcomes are not sensitive to harm or benefit.
It may be necessary to move away from a global scale of neurodevelopment or composite outcomes (including mortality) to scales where researchers can focus on those measures that might be the most important for the particular intervention being evaluated. It may be possible to use global scales as markers of safety rather than efficacy.
There is a definite need to examine the effects of gender, ethnicity, race, social status, environmental influences, etc. in clinical studies since these factors can also impact short and more importantly long-term outcomes.
There is a significant need for investigators, regulators, government, funding agencies, and industry to develop and then accept a standardized and specific set of short- and long-term endpoints for a variety of medical conditions and disease phenotypes in neonates (with proper validation of long-term assessment tools, especially with respect to neurodevelopmental outcome as well as other organ functions).
Long-term outcomes studies may be best conducted as part of a postmarketing risk management plan rather than as part of the initial dossier that leads to the availability of the product on the market. For some disorders it may be appropriate to conduct studies up to the expected date of delivery in the approval studies and to include subsequent follow-up for safety and efficacy in a postmarketing risk management plan. For some disorders (e.g., bronchopulmonary dysplasia), longer periods of follow-up are needed to accurately evaluate the effects on morbidity and even mortality.
Pharmacogenetics
Genetic differences that clinically affect both exposure and response have been increasingly documented, but the relationship between genomic profiles and developmentally regulated gene expression has not been extensively studied in neonates. Some of the difficulties in obtaining specific pharmacogenetic information in pediatric patients, including neonates have been reviewed (43). Nevertheless, if medicinal product exposure in a neonatal clinical pharmacology study is dependent on a well-known pharmacogenetic biomarker (e.g., CYP2D6) (67), obtaining DNA may provide additional information for the interpretation of the PK and PD results. In particular, if there are important pharmacogenetic differences affecting PK, efficacy and safety of a medicinal product in the adult population, pharmacogenetic analysis of the target genes is recommended in neonates, given that the relationship between phenotype and genotype may be completely different in the neonate compared with other patient groups (68,69). DNA collection may be performed on scavenged blood samples after PK analyses are performed or on buccal swabs.
Points to Consider for Neonatal Clinical Trials
A sponsor who will be submitting an application for a medicinal product that includes a new active ingredient, indication, dosage, dosing regimen, or route of administration must submit an initial pediatric study or investigation plan (70). This pediatric plan should outline the neonatal study or studies that the applicant plans to conduct unless a waiver is granted. The submission of the initial neonatal plan is intended to encourage sponsors and investigators to consider neonatal studies early in product development and begin planning for these studies when appropriate. The initial plan must include the following: (i) an outline of the neonatal study or studies that the applicant plans to conduct (including, to the extent practicable, study objectives and design, age groups, relevant endpoints, and statistical approach); (ii) any request for a deferral, partial waiver, or waiver if applicable, along with any supporting information; and (iii) other information specified in an individual country’s regulations.
When designing neonatal clinical studies, investigators and sponsors should be mindful that modeling and simulation as well as developmental pharmacologic considerations are often critical for the successful completion of a study. Modeling and simulation using all of the available information should be an integral part of all neonatal development programs. The use of modeling and simulation is still developing and lessons from individual studies should be shared in order to facilitate the development of the field.
The neonatal plan is a living document and it is expected that the plan will evolve with time. The plan is useful for directing the process for the sponsor as well as meeting a regulatory requirement. Early and frequent discussion of the neonatal plan between sponsors and regulators is extremely valuable and highly recommended. Investigators and networks can add considerable value at all stages of the development and implementation of a plan.
Approaches to Neonatal Studies
Clinical pharmacology studies assess PK (i.e., medicinal product exposure), PD (i.e., effect on biomarker or clinical endpoint), and E-R relationships. It is essential to study these topics in neonates because neonates may differ from adults. In addition, a medicinal product development program that includes neonates should consider the specific end-organ/tissue responsiveness, metabolic enzyme(s), excretory systems, and transporters that may also be specific to neonates. This is best achieved by characterizing the PK of the medicinal product and the responses across the appropriate gestational and PNA ranges of neonates who are likely to be treated with the medicinal product. Adequate numbers of neonates across age-strata should be enrolled to provide accurate guidance for dosages for the developmental ages of patients who are likely to be treated with the medication. It is not unusual to make incorrect estimates of key parameters such as clearance. For this reason it is important to include early assessments of clearance after 5 or 10 patients are studied in each neonatal age group. An early assessment of clearance is particularly important if the data will be used for the purpose of extrapolation of indications/uses because an extrapolation approach will generate a relatively small set of data. If an inaccurate initial estimate of clearance is not identified early in the study then conclusions of the study may be erroneous.
Extrapolation
Extrapolation is a well-recognized approach to providing sufficient evidence to support the safe and effective use of medicinal products in pediatric populations. As described by Dunne and associates (71) and the EMA CHMP (72) extrapolation of efficacy findings from studies in older populations can be successful. This means that a development program can be devised for neonates that minimizes the burden of the research on the participants and allows development of medicinal products for rare conditions. Note that minimizing the burden on the neonates does not make the development program “easier” for the Sponsor.
The principles of extrapolation are common across jurisdictions but the terminology can differ. Accordingly it is important to seek a globally integrated approach to extrapolation. The primary rationale for extrapolation is to avoid unnecessary studies in the target population for ethical reasons, for efficiency, and to allocate resources to areas where studies are the most needed. Many diseases in the preterm and term newborn infant are unique or have unique manifestations precluding extrapolation of efficacy from older pediatric patients and call for novel methods of outcome assessment. Alternatively, in situations where the feasibility of studies is restricted, extrapolation principles may be applied for rational interpretation of the limited evidence in the target population in the context of data from other sources (28).
The prerequisite for extrapolation is a well-justified case that reasonable similarity can be assumed between source and target population of both disease progression and response to intervention. As noted above, data from other age-groups and other conditions can inform the design of a development program based on extrapolation. It is important to use extant data systematically although regulatory agencies have different ways to structure the case for extrapolation.
The EMA Concept Paper suggests that a staged approach is used to develop a framework for reduction of the required evidence should extrapolation be justified. The stages are: examine clinical context in order to justify why extrapolation may be appropriate instead of a complete set of prospective studies; develop an extrapolation concept based on biological and pharmacological rationale with quantitative predictions on the degree of similarity in the target population (modeling and simulation can help quantify available information); develop an extrapolation plan by proposing a reduced set of supportive studies in the target population in accordance with the extrapolation concept. In general, data generated in the target population should validate the extrapolation concept and complement those data that may be extrapolated from the source population. Studies should focus on those complementary areas, e.g., age subsets, where the largest differences to the source population are expected.
The extrapolation concept should be validated using data obtained from studies included in the extrapolation plan; draw conclusions about extrapolation while taking account of risks and uncertainties (28). If the data do not confirm the extrapolation concept, the concept needs to be updated by the emerging data regarding the true extent of similarity and, hence ability to extrapolate. Consequently, the need to generate more data in the target population should be assessed and the extrapolation plan adjusted.
The FDA has published a decision tree that leads to the selection of an approach to extrapolation (71,73). In any case, if a study will be used to seek regulatory approval the assumptions will have to be clearly demonstrated, quantitatively, if possible. The approaches suggested by FDA, which may also be acceptable to the EMA are:
If Extrapolation of E-R is possible: Study Dose-Exposure. This approach is appropriate when it is reasonable to assume that neonates, when compared with adults or older children, have (i) a similar progression of disease; (ii) a similar response of the disease to treatment; (iii) a similar E-R or concentration-response relationship; and(iv) the medicinal product (or active metabolite) concentration is measureable and predictive of the clinical response. Support for concluding that there is a similar disease course and medicinal product effect in adult and pediatric populations (including term and preterm neonates) includes evidence of common pathophysiology and natural history of the disease in the adult and pediatric populations, similar concentration-response relationships in each population, and experience with the medicinal product, or other medicinal products in its therapeutic class, in the disease or condition or related diseases or conditions. This approach generally involves E-R knowledge from studies in adults. An example of this approach is fluconazole for the prevention and/or treatment of invasive candidiasis (74,75,76).
The condition is similar but extrapolation of exposure response is not possible although E-R is well characterized in other age groups: Study Dose-E-R (including studies of PD and efficacy). If the disease and intervention are believed to behave similarly in neonates and adults, but the E-R in term and preterm neonates is either inadequately defined or thought not to be sufficiently similar, a reduced set of studies in the neonates could be sufficient to demonstrate efficacy To use this approach, the E-R relationship in adults and older children should be well-characterized. The goal of such an approach is to characterize and compare the E-R relationship in adults and in the term and preterm neonatal population with the appropriate term and preterm neonatal doses based on the E-R relationships seen in term and preterm infant patients. Although clinical measures (e.g., symptoms, signs, and outcomes) can be used to select doses, an appropriate biomarker that can be considered to be related to such an endpoint should also be studied, if available. If there is uncertainty about whether extrapolation of efficacy is appropriate, an adequately powered study using a clinical endpoint may be necessary to provide an answer to the scientific question of interest. An example of this approach is the use of meropenem for the treatment of complicated intra-abdominal infections (77,78).
No extrapolation of E-R is possible: Develop a full neonatal program. If the disease progression is unique to neonates or its progression and/or response to intervention is undefined or dissimilar to that in adults or older children, then the neonatal plan should provide substantial evidence of the safety and efficacy of the medicinal product in one or more clinical studies, usually evaluating more than one dose. The study objectives are to provide evidence of safety and efficacy and to characterize the PK and E-R relationships to aid in optimizing neonatal dosing strategies. A population PK analysis can be conducted concurrently using PK data from the efficacy study to confirm PK estimates in the gestational age and PNA subgroups. If possible and appropriate, PD endpoints should also be collected in such studies to increase knowledge on the disease and the pharmacological target and to ensure sound dose finding/selection. An example of a disease limited to term and preterm neonates would be respiratory distress syndrome (79,80,81).
When response data is collected (efficacy or PD) it should be used to validate and confirm the initial assumptions related to pharmacology, disease history and clinical response to the new compound. Response or PD data may include biomarkers or clinical endpoints for both safety and effectiveness. The specific endpoints for an E-R evaluation for each medicinal product should be discussed with regulators (preferably in a globally coordinated manner). A dedicated PK study is nearly always required for neonates regardless of the approach taken if the investigational medicinal product is likely to be used in this population. Safety data must always be collected.
Sampling Procedures
Conventional PK studies with intensive blood sampling are rarely performed in neonates because of the limited circulating blood volume (82). There is a lack of consensus on permissible blood volumes and there is limited information about the tolerability of blood sampling in neonates (83). Parents are unlikely to consent if the sampling is perceived to be burdensome. Conducting PK sampling during times of routine laboratory sampling (opportunistic sampling) is an approach that reduces the number of blood draws for PK sampling only. This approach can yield similar PK models to study-specific samples taken at optimized time points (84). In any case the total amount of blood taken during a study must be meticulously recorded including blood removed for the study but not used in analysis (e.g., while flushing dead spaces). Cerebrospinal fluid may also be collected for PK sampling during times of routine clinical laboratory evaluations. Scavenged sampling, by using blood or other fluid leftover in the laboratory after clinical studies have been completed, is another noninvasive approach as is the use of residual dried blood spots. Chemical stability of the medicinal product in the storage conditions of the laboratory should be confirmed. Careful consideration to the timing of dose, time of collecting the samples (to optimize the value of the information to the PK model and fit with clinical and laboratory routines) and the integrity of the samples is important for these approaches. Urine and saliva may be collected noninvasively, but the interpretation of medicinal product analyses can be challenging. The use of salivary diagnostics and genomics has grown in importance and accuracy in correlating with blood levels of various proteins and genes (85,86). Within the next several years, saliva may be used routinely for PK or other laboratory studies if issues relating to transport into saliva and binding proteins can be resolved.
Neonatal Dose Selection for Clinical Pharmacology Studies
This section presents some general principles, acknowledging that experts in this field must be included in the study team. Since there may be limited information on the safety of the dose of medicinal product to be administered, the dose range in first-in-age group studies requires careful consideration. In general, the potential risk of the treatment must be commensurate with the prospect of direct benefit to the patient or without the prospect of direct benefit, but likely to yield generalizable knowledge about the patient’s disorder (87). Factors for consideration include (i) similarity of the disease and E-R in adults or older children; (ii) for enteral preparations, the relative bioavailability and potential differences with types of feeds; (iii) the gestational age, PNA, and developmental stage of the population; (iv) any recognized pharmacogenetic influences on disposition of the medicinal product; (v) the toxicity of the medicinal product; and (vi) PK data from other pediatric populations. Initial doses are nearly always normalized to body size (mg/kg) or some other form of scaling, based on appropriate justification. When uncertainty about the dose is high, cautious approaches will often be appropriate, including initial titration of an intentional low dose or use of therapeutic drug monitoring during the trial.
When extrapolation of efficacy is considered, so that only PK studies are conducted, the PK studies in the neonate should determine how to achieve the same level of systemic exposure in adults or older children. This requires that the neonatal organ responsiveness is similar to that of adults or older children. Dosing, safety, and/or efficacy may be affected by gestational age and PNA because neonates often have wide differences in interpatient variability in PK measures and parameters. In these instances, the sponsor should specify the criteria by which exposure matching would be acceptable. For example, one approach would be to use simulations to select the appropriate dosing strategy to estimate the appropriate range of exposures (e.g., 5th to 95th percentile) shown to be safe and effective in adults or older children.
As science and technology continue to advance, in silico and other alternative modeling methods may be developed that can provide preliminary data to inform the design and conduct of PK/PD studies for investigational medicinal products in neonates. For example, the development of a physiologically-based PK (PBPK) in silico model that integrates medicinal product-dependent parameters (e.g., renal clearance and metabolic pathways) and system-dependent parameters (e.g., nonmedicinal product parameters such as blood flow, protein binding, and enzyme and transporter activities) is one possible approach. PBPK has been used in pediatric medicinal product development programs for (i) planning for a first-in-pediatric PK study, (ii) optimizing the study design, (iii) verifying the model in specific gestational and/or PNA groups, (iv) recommending starting doses, (v) informing enzyme ontogeny using a benchmark medicinal product, and (vi) facilitating covariate analysis for the effects of organ dysfunction or medicinal product interactions in pediatric patients (88). The model selected should incorporate in vivo PK/PD data obtained in adults and older children as well as human volunteer studies, as appropriate. It is important to evaluate the reliability of the (PBPK/pop-PK/PD) model and its predictions with regards to the prior knowledge and to check the validity of major assumptions.
Clinical trial simulations can be performed to integrate PK, PD, disease progression, and study design considerations to help guide a neonatal medicinal product development program. In neonates, due to constraints related to enrollment and blood sampling, clinical trial simulations can be particularly helpful to assess sample size considerations and design a trial that’s both feasible and can adequately evaluate medicinal product exposure, safety, and effectiveness. An estimate of the E-R relationship across a range of body-size doses (dose/kg) may be important. For the “Dose-Exposure-Response” approaches discussed above, investigation of a range of doses and exposures should allow assessment of those relationships, development of rational dosing instructions and validation/confirmation of the initial assumptions.
Where PK/PD studies are designed, the dose range should account for observed differences in response between older children and adults with the neonatal population, both in terms of exposure and response (89). For example, there is evidence that pediatric populations are on average less sensitive to antihypertensive medicinal products than the adult population. Therefore, neonatal studies may include exposures greater than the highest medicinal product exposure associated with the approved adult dose, provided that prior data about the E-R relationship and safety information justify such an exposure. Studies of distinctly different ranges of exposure are desirable to provide sufficient information for the calculation of an optimal dose.
Neonatal Dosage Forms and Formulations
There are challenges associated with any route of administration in neonates, as recently summarized by Linakis et al., and the EMA Committee for Medicinal Products for Human Use (2,90). Formulations that permit accurate dosing and enhance adherence or accuracy in dosing (i.e., dose accuracy without manipulation) in neonates are a crucial part of clinical pharmacology studies and subsequent pharmacotherapy. If there is a neonatal indication, an age-appropriate dosage formulation must be made available on the market (91). The route of administration mainly depends on the characteristics of the compound. Although oral or intravenous administration is most common, other routes (e.g., inhalational, intraocular, transcutaneous, intramuscular, subcutaneous or rectal) can be considered when appropriate. Some stages of product development can be done using a product that is not ideal followed by bridging studies when the commercially available preparation becomes available. The appropriateness of bridging results from bioavailability studies in adults to children should be justified, as there could be rare examples of differences in sensitivity toward formulations.
One way to fulfill this requirement is to develop and validate a neonatal formulation, and seek approval for that formulation with respect to the available guidance on how to evaluate such a formulation. Besides concentration and dose flexibility that should result in a reasonable volume to insure accuracy of dosing, excipients are of specific relevance and importance in neonates (92). Excipients with known toxicity in neonates should be avoided (e.g., ethanol, propylene glycol or benzyl alcohol (93,94)), if possible. There has recently been a concerted effort between different stakeholders to build a dataset to cluster the available knowledge—including safety information—on excipients in neonates (95). This has been organized in the Safety and Toxicity of Excipients for Pediatrics (STEP) database at http://www.eupfi.org/step-database/ for which anyone can register (96). It is possible to measure excipients in microsamples and to perform population PK studies in neonates (97,98). It is important to consider exposure to excipients as well as to active ingredients; that is to assess the circulating concentration-time profile rather than rely on quantitative information about the excipient content of the product. Studies of product use in neonates have shown that many products thought to require excipients can be made without excipients (99). It is possible to use clinical trials to gather data about excipients and there are opportunities for precompetitive collaboration about excipient safety. It can be useful to compare exposures to excipients associated with new products to those in existing products known to be safe, however careful attention should be put on relevance of age, dose, route of administration and disease. Concern about excipients is not a valid reason to defer or avoid the development of a medicinal product for neonates.
If the sponsor demonstrates that all reasonable attempts to develop a stable, specific and safe formulation have failed, the sponsor should develop and validate an age-appropriate formulation that can be prepared by a pharmacist in a licensed pharmacy using an approved medicinal product and commercially available ingredients. If the sponsor conducts the neonatal studies using such a formulation, the following information should be provided in the study report:
-
A statement on how the selected final concentration was optimized to help ensure that the doses can be accurately measured with commercially available dosing devices;
-
A statement that the volume to be prepared is appropriate to be dispensed for a course of therapy for one neonate, unless there are safety factors that necessitate decreasing the volume to be prepared;
-
A listing of all excipients, including diluents, suspending and stabilizing agents, sweeteners and flavoring and coloring agents (quality and quantity);
-
Information on containers (designated containers should be readily and commercially available to retail pharmacies) and storage requirements (if possible the most user friendly storage condition (room temperature) should be evaluated and or studied);
-
Testing results on formulation stability, not to exceed the expiration date of the original medicinal product lot from which the pediatric formulation is derived.
The bioavailability of any formulation used in neonatal studies should be characterized in relation to the adult formulation. If needed, a relative bioavailability study comparing the age-appropriate formulation to the approved medicinal product should be conducted in adults. Potential medicinal product-food (including milk) or vehicle interactions should be considered, such as those that have been reported with apple juice (100). In addition, preterm infants <34 wk rarely feed orally, and are most often fed through a nasogastric or orogastric tube, sometimes using an infusion pump. The influences of mixing the product with feeds and rate of administration should be taken into consideration. Binding to the plastic in the most frequently used feeding tubes should be tested. Extended-release dosage forms or combination products produced for adults should be made available for neonates as an age-appropriate formulation when it is appropriate to do so (although prolonged clearance means that there is less need for extended release formulations in the neonatal population). Such formulations should not be limited to drops or liquids, since more novel approaches like uncoated minitablets have been reported (101).
Finally, intravenous administration has specific issues (e.g., dead space, flow rate, flush volume, and medicinal product volume) as recently summarized by Sherwin et al. (102). The general guidelines on formulation development of intravenous administration apply, with some aspects that are more specific for neonates. Study medicinal products should be administered separately from other medicinal products if possible. If the medicinal product is administered as a prolonged, continuous infusion then coadministration with other products will be necessary when the product is used in clinical practice, due to limited vascular access. Prior to coadministration, compatibility of the medicinal product with total parenteral nutrition and other relevant intravenous medicinal products needs to be examined. Other aspects of the dosage form should be justified including the intravenous volume (single dose and daily dose) to be administered. Data on the range of potential final strengths (concentration) may be clinically important because it influences the volume administered and dose flexibility. Issues related to electrolyte and excipient exposure, as well as choice and flexibility in diluents, should also be explored and reported.
Study Design including Sample Size
For clinical investigators working to improve the treatment of neonatal diseases with low incidence rates (e.g., rare diseases), traditional sample size requirements for clinical research may impede the conduct of the trial. Regulatory guidance on trials in small populations advises that alternative approaches to conducting such trials might be suitable if they can improve the interpretability of trial results. Innovative trial designs that have been used in rare disease populations may be applicable to neonates. Algorithms for application of innovative trial designs to neonates have not yet been developed. However, integrating aspects of innovative trial designs into more ‘‘standard’’ approaches may include adaptive designs, defined as ‘‘a study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses. This should be based on analysis of data (usually interim data) from subjects in the study (103).
Examples of such study modifications could include: (i) changes in power calculations leading to enrollment modification and early termination criteria stemming from a futility analysis; (ii)use of pragmatic trials which include patients in routine clinical practice settings, typically incorporating comparative effectiveness research comparing the safety and effectiveness of diagnostic, therapeutic, or delivery system options; (iii) incorporation of additional nontrial neonatal data from electronic health records and other auditable sources on medicinal product dose and response; and (iv) incorporation of prior information through Bayesian techniques may make adaptive designs even more practical and powerful.
Number of Patients. The precision of PK and E-R parameters in the sample size calculation are critical for neonatal studies. Prior knowledge of the disease, exposure, and response from adult and other relevant pediatric data, such as that related to variability, can be used to derive sample size for ensuring precise parameter estimation. The sponsor should account for potential sources of variability, including intersubject and intrasubject variability, and differences between adults and older children in the final selection of the sample size for each age group.
The sponsor should discuss and justify the distribution of the number of participants across each age range and the appropriateness of the ranges with regulators, because this will be medicinal product-specific. For example, one approach would be to prospectively target a 95% confidence interval within 60% and 140% of the geometric mean estimates of clearance and volume of distribution for the medicinal product in each subgroup with at least 80% power. In general, noncompartmental modeling with rich sampling has not been used in neonates. Instead, population PK modeling analysis based on sparse PK sampling, or other scientifically justified methods can be applied to achieve this precision standard (104). Conceivably, certain disease states might not allow for recruitment of an adequate number of participants to meet the standard, but practical considerations should be taken into account in determining the sample size.
Number of Samples per Patient. The number of blood samples collected in a clinical pharmacology study, to estimate PK measures and parameters for each patient in the study should be carefully considered. The number of samples is often very limited in neonates (for more on collection of blood or plasma samples, see section below). Using times of routine blood draws (e.g., clinically indicated laboratory testing) for opportunistic sampling can reduce the number of blood draws for research purposes only. Clinical trial simulations or optimal sampling techniques may be recommended to justify the proposed sampling scheme. Blood volume limitations for PK sampling will vary by gestational and PNA and this can affect the number of PK samples for medicinal products requiring >0.5 ml of whole blood per sample. Newer microsampling techniques can provide measurements of multiple analytes (e.g., electrolytes, blood glucose, and blood gases) on a single 0.3 ml sample of blood. Accelerator mass spectrometry can use samples as small as 7 µl. Additional sampling for medicinal product or metabolite concentrations is also recommended when an adverse event occurs.
Sample Collection. Blood or plasma concentrations of medicinal product or metabolites have been used as supporting evidence of effect or dose selection through E-R analyses in children. However, the volume and frequency of blood sampling are often of concern in neonatal studies (105). Blood samples can be obtained by direct venipuncture or through the use of an indwelling intravascular catheter, especially with the need for repeated samples. Heel sticks, commonly used in this population, can also be used to collect blood samples. Unless there are data that indicate a difference between arterial, venous, and capillary blood concentrations, all sources of blood should be allowed. In order to validate the microanalytic approaches in neonatal PK studies, this approach needs to be incorporated into adult development programs by inclusion of capillary samples when possible. The volume and frequency of blood sampling can be minimized by using microvolume medicinal product assays, dried blood spots, sparse-sampling techniques, and population PK approaches. These types of assays and analyses are especially relevant when studying neonates (106). Modern assay techniques allow small sample volumes to be used to determine medicinal product concentration, but data quality may be affected if the sample volume is insufficient to allow for reanalysis when necessary (107). Blood samples for analysis should be collected from the circulating blood volume and not from reservoir dead space created by catheters or other devices. Sampling technique is critical when using available neonatal indwelling intravenous catheters. The time of sample collection, proper sample transportation and storage, and sample handling techniques should be documented. Whenever possible, PK samples should be collected from a separate site from the one used to administer the medicinal product. The collection of fluids including blood, cerebral spinal fluid, or bronchial fluids for medicinal product concentration measurements may be beneficial when samples are being obtained for clinical purposes. Noninvasive sampling procedures, such as urine and saliva collection, may be adequate if correlated with outcomes or if the correlation with blood or plasma levels has been documented.
Given the difficulty in collecting blood samples in neonates, special approaches to allow optimal times of sample collection may be useful (108,109,110). Sampling windows may need to be wider than is typical for an adult study to account for difficulty in sampling. The sampling scheme should be planned carefully to obtain the maximum information using the minimum number of samples. If possible, additional PK samples should be collected when adverse events occur in order to better understand the relationship between medicinal product exposure and potential toxicity. Samples for DNA should be collected when appropriate, as discussed previously.
Participant welfare is of paramount importance during clinical trials. Trial planning needs to account for the availability of experienced staff, techniques for analgesia (e.g., topical anesthetics, pacifiers or oral sucrose) and applicability across different units. During the first week or two after birth, sick neonates may have umbilical catheters, which may be an optimal site for blood sampling. The small caliber of other vascular access devices in neonates may limit direct sampling (e.g., venous cannulae or long lines). Neonates are susceptible to infections so multiple samples from surgically-placed (Broviac/Hickman) or other central lines may need to be limited (111). The pros and cons of sampling routes have been summarized (112).
Covariates and Phenotype Data. The sponsor should obtain, at a minimum, the following covariates for each neonate: gestational age at birth, birth weight and head circumference, PNA, current weight, race, ethnicity, sex, diagnoses, concomitant and recent medications or intravenous fluids (including blood transfusions), and relevant laboratory tests that reflect the function of the organs responsible for medicinal product elimination. Occasionally the weight that dosing is based on may differ from the actual weight (e.g., treatment of neonatal abstinence syndrome with opioids). Investigators are encouraged to collect DNA samples in neonatal PK studies under the circumstances described above, along with appropriate phenotypic information to optimize the interpretation of pharmacogenetic influences on PK and PD parameters. For example, when genetic information is obtained for a cytochrome P450 enzyme, the sponsor should examine the influence of genetic mutations on PK, PD, and/or dose-response to determine whether genetically defined subsets of patients may need special dosing considerations (113).
The sponsor should examine the relationship between the covariates and the PK of the medicinal product of interest. The contribution of size (e.g., body weight), maturation (e.g., gestational age at birth, PNA, PMA), and other covariates to PK variability should be assessed. If factors affecting the PK of the medicinal product are to be studied (e.g., the effect of a concomitant medication or the presence or absence of a disease), a justification for the numbers of patients with and without those factors in the study should be included.
Sample Analysis. An accurate, precise, sensitive, chemically-specific, and reproducible analytical method to quantify the medicinal product and metabolites in the biologic fluids of interest is essential. A method that is readily adaptable and that uses only minimum sample volumes should be chosen.
Data Analysis
Two basic approaches for performing the PK analysis in neonates can be used; a noncompartmental PK approach and a population PK approach.
Noncompartmental Analysis. The noncompartmental analysis PK approach involves administering either single or multiple doses of a medicinal product to a relatively small group of patients with relatively frequent blood and urine sample collection. Samples are collected over specified time intervals chosen on the basis of absorption and disposition half-lives (predicted from other studies with modification based on known maturation of the route(s) of disposition), and subsequently assayed for either total or unbound concentrations of medicinal product and relevant metabolites. Noncompartmental analysis can be used to establish PK parameters such as area under the curve (AUC), maximum (peak) concentration (Cmax), clearance (CL), volume of distribution, and half-life which are descriptive of the concentration of medicinal product or metabolite(s) over time. Data are usually expressed as the means of the relevant measure or parameter and interindividual variances. This approach should include a sufficient number of neonates to give a precise estimate of the mean: standard approaches can be applied to neonates. If medicinal product administration and sampling are repeated in a PK study, some understanding of intraindividual variability in PK parameters can be obtained. A noncompartmental approach is often not feasible in neonates.
Population Analysis. An alternative approach for analysis in pediatric clinical pharmacology studies is the population approach to PK analysis. Population PK accommodates infrequent (sparse), but informative, sampling of blood or plasma from a larger patient population than would be used in a compartmental or noncompartmental analysis PK approach to determine PK parameters. Sparse sampling of blood or plasma is considered more acceptable for neonatal studies, because the total volume of blood sampled can be minimized. Sampling can often be performed concurrently with clinically necessary blood or urine sampling. Because relatively large numbers of patients are studied and samples can be collected at various times of the day and repeated over time in a given patient, estimates of both population and individual means, as well as estimates of intra and intersubject variability, can be obtained if the population PK study is properly designed.
E-R analyses predominantly employ a population analysis approach. Individual analysis is generally not recommended unless responses from a wide range of doses from each neonate are available. Simultaneous modeling of data across all enrolled neonates provides the best opportunity to describe the E-R relationship.
Adverse and Serious Adverse Event Reporting
It is crucial to capture safety data in all neonatal clinical pharmacology studies. This need is particularly acute in neonates because of the limited number of participants in clinical studies—particularly if an extrapolation approach is used. Safety must be included in the objectives of all studies of medicinal products in neonates. Maturing organs may be damaged in the neonatal period, but that damage may only become manifest as the child develops. It is often difficult to distinguish whether morbidity and mortality observed in a critically ill neonate is from a possible effect of the medicinal product being studied or the underlying clinical condition (especially in neonates at the limits of viability). Neonatal pharmacovigilance has been described (114). Du et al. have outlined and tested a method for evaluation of the relation of an adverse event to the administration of a medication (115). They developed a classification of the relation to the medicinal product as definite, probable, possible or unlikely based on the following 13 criteria:
-
1. Timing of the event relative to the administration of the medication
-
2. Whether the event was a well-documented complication of the medication
-
3. Whether there are published reports of the event related to the suspected medicinal product in neonates
-
4. Whether the event was a change in a pre-existing condition
-
5. Whether alternative explanations might explain the event
-
6. Whether any alternative explanations are able to be confirmed
-
7. Whether the event improved after the medicinal product was discontinued
-
8. Whether a reduction in dosage reduced the severity of the event
-
9. Whether the event improved after administration of a specific antagonist
-
10. Whether the event improved or disappeared while the patient continued to receive the suspected medication
-
11. Whether the event reappeared or worsened when the suspected medication was reintroduced
-
12. Whether the suspected medication was detected in blood or other body fluids in toxic concentrations
-
13. Whether there was unequivocal evidence that the dose was an overdose
14. Adverse events represent any change in a condition or appearance of a new problem that was not present before the beginning of the study medication. Such changes in condition are quite common in all premature neonates who are not involved in a study making it difficult to distinguish medicinal product-related changes and potential interactions between medicinal products. A placebo treated group can be quite important in separating out the effects of a study medication from changes related to the underlying condition (e.g., prematurity). Appropriate reporting of serious adverse events and adverse events to designated Institutional Review Boards, Data Safety Monitoring Committees, and Regulatory Agencies should occur based on regulations in each country where the research is being conducted. It is worth noting that in some countries, serious adverse events that are anticipated as part of neonatal intensive care and are thought to be unrelated to the study medicinal products or procedures may be reported over the same timelines as nonserious adverse events.
Ethical Considerations
During the last 50 y, medical treatment of sick neonates has progressed from antibiotics to mechanical ventilation to prolonged artificial heart-lung support with extracorporeal membrane oxygenators. Numerous medicinal products accompany these medically complex treatments, but most are prescribed off-label or not licensed for the treatment of neonates (11,116). Even the few that are labeled for the neonate are unlikely to have received adequate study to support prescribing for the extremely premature neonate. Treatment of neonates with medical products without the benefit of comparable evidence provided for adults through appropriate clinical trials is usually unethical (117,118).
Not only do clinical trials in neonates present special medical and pharmacologic challenges that have been outlined above, they also present ethical challenges (119). Neonates are a vulnerable population for a variety of reasons such as the inability to comprehend the risks of a study, to express their views about those risks or to choose whether or not to participate (28,120). As noted at several points in this document, there are significant uncertainties about the effects of medicinal products, the optimal dose and the long-term consequences of administering medicinal products to neonates. These factors make a benefit-risk balance more difficult than in other settings. Sick neonates face significant burdens during routine care. The additional burdens of participation in clinical research need careful justification. Nevertheless it is more important to protect neonates from poor or missing medicinal products by conducting well-planned and justified research than it is to protect neonates from research.
In order to provide adequate protection for children in clinical trials, many countries have developed guidelines about how the studies should be conducted (3,6,22,105,121,122,123). As clinical trials have become international in scope, it is essential to adhere to the pediatric policies within each country and those of the responsible Ethics Committees while adapting to the needs of neonates. However, variations among nations and among local Institutional Review Board’s (IRB’s) within countries in the interpretation and implementation of guidelines for studies in neonates can present challenges to any international clinical trial.
Certain ethical principles that are found in most international regulations related to clinical trials apply to neonates (22). International guidelines such as ICH E6 state that “before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks” (22). In general, most, if not all investigators, regulators, regulations and ethicists recognize that neonates are a vulnerable population that should only be exposed to low risk interventions. Some national guidelines manage the process of weighing risks against benefits of a trial by classifying the risks, for example as minimal risk (4,124). Some countries recognize a category of trials that represent a minor increase over minimal risk (125). The use of risk categories adds to the complexity of assessing risk because the interpretation of which interventions are low or minimal risk varies between countries and Ethics Committees. Some Ethics Committees would classify a blood draw as minimal risk while others regard this as a minor increase over minimal risk (126). For studies classified as a minor increase over minimal risk that provide the prospect of direct benefit for the subject, the ceiling on risk is determined by whether it is proportional to the probability and magnitude of benefit (127). According to most regulatory guidelines, a clinical trial without the prospect of direct benefit to the individual neonate is possible with appropriate parental consent, but only if the trial will provide generalizable knowledge about the underlying condition being studied (87). Parents/guardians, children and neonatal care providers can provide important advice about acceptable levels of risk relative to the potential benefits and should be included early in the planning process for all clinical trials involving children, especially neonates (14,22,23,24). If available data do not indicate whether a specific treatment is beneficial for neonates, then participation in a study about the treatment is deemed to provide a prospect of direct benefit for those infants receiving the investigational product. When a placebo arm is involved, risk to those infants must be low (128). In any case, therapeutic trials are usually conducted in neonates with a condition or disorder for which the medicinal product is indicated. It is essential to recognize that neonates face significant risks because of exposure to medicinal products that have not been adequately evaluated (129,130). Once new, improved medicinal products reach the market, they will be used in neonates even if they have not been studied adequately in this population. Furthermore, sick neonates are routinely exposed to a number of interventions as part of routine clinical care, many of which are painful (131). In this context, the risks and potential harms arising from research studies may not add substantively to the burdens borne by neonates. The risks, burdens and benefits of each research study should be identified. For each study the recognition of acceptable risks and burdens should be based on an explicit balance with the benefits of research in a way that takes into account the views of families.
Before initiation of a clinical trial, a duly appointed, independent Ethics Committee must approve the proposed trial. The Ethics Committee must have access to sufficient expertise in neonatal research. Given the limited number of relevant experts and the need for consistent decision-making arrangements, federated Ethics Committees or a single Ethics Committee for each country will work particularly well for neonatal clinical trials (similar to current models for cancer trials) as long as it includes appropriate expertise.
An Independent Data, Safety and Monitoring Board (separate from the local Ethics Committee) may be needed to oversee the trial (122,132). This committee should be comprised of Pediatricians and/or Neonatologists, Biostatisticians, community members, and other appropriate personnel with sufficient expertise to be able to examine the safety and efficacy of the trial and stop a trial if there are significant concerns about safety or if it becomes apparent (through interim analyses) that the medicinal product is not efficacious.
Permission for the participation of a neonate in a clinical trial can be challenging. This permission must be free of undue influence and coercion, although the parents or legally appointed guardian(s) of a sick neonate are likely to be in a state of heightened anxiety. This heightened anxiety may make the possibility of benefit from a therapeutic intervention seem particularly attractive or the uncertainty about therapeutic options and outcomes in a randomized trial particularly stressful. The person obtaining parental permission must remain neutral and maintain equipoise about whether either arm of the study is better or worse. Parents must be provided ample time to consider the study and to formulate questions about their child’s participation. The definition of ample time will depend on the context of the study. Families may visit sick neonates irregularly and consent procedures need to account for this. When it is appropriate for the study design, the process of continuous consent allows families to extend the decision-making process (133).
Ethical considerations may be interpreted differently in different jurisdictions. Sponsors and investigators need to be mindful of the variation in ethical approaches while aiming to develop a consistent global approach and avoid locating studies in outside countries where the regulatory agencies will be most flexible and provide limited oversight. This will be become increasingly complex with an increased number of clinical trials in neonates being conducted in multiple countries simultaneously, requiring standardizing and harmonizing clinical and ethical approaches globally. Despite the ethical challenges of conducting clinical trials in neonates, they do deserve the benefit of drug therapy grounded in thorough studies conducted in patients with similar degrees of immaturity.
Conclusions
Neonatal clinical pharmacology studies are unique. There is an ethical imperative to minimize the number of participants in neonatal clinical studies and a need to study new and existing medicinal products as efficiently as possible. These drivers promote an approach to clinical pharmacology that starts with a broad search for existing knowledge and uses pharmacometric tools to integrate existing knowledge in order to plan and analyze clinical studies. The interpretation of new and existing knowledge is an iterative process with multiple review steps.
It is important to plan neonatal studies early in the medicinal product development process. Important data to support the application of a medicinal product to neonates may need to be gathered during the adult phases of clinical medicinal product development. Preclinical studies may need to include juvenile animal studies.
Multiple stakeholders must work well together to insure the successful development and regulation of neonatal medicinal products, and the International Neonatal Consortium (INC) was established to help promote that collaborative process. Establishing and maintaining relationships among the key stakeholders of a neonatal development plan need special attention. Teams including regulators need to be open-minded about study design and focused on filling information gaps using the most appropriate approach. Sponsors and investigators need to develop better ways to share information relevant to all neonatal studies in a precompetitive way. For example, pooling adverse event rates in the placebo arms of neonatal trials will inform all clinical development programs by identifying the rates of anticipated adverse events in neonates who have not been exposed to novel treatments. Information about how surrogate outcomes relate to clinically important outcomes will improve the design of clinical programs by allowing the refinement of surrogate outcome measures. It is essential that both positive and negative trials be published so that all relevant information concerning the outcomes of a trial is available.
Many neonatal conditions have a major public health impact but involve rare diseases and have relatively few patients. This means that a global development pathway is needed for most medicinal products used in neonates. While sponsors need to work closely with multiple regulatory agencies and investigators, regulators need to develop processes to reach agreement as often as possible during the development and implementation of neonatal programs to develop medicinal products.
Statement of Financial Support
The International Neonatal Consortium (INC) is supported in part by grant number U18FD005320-01 from the US Food and Drug Administration (FDA) to the Critical Path Institute (http://c-path.org) and through annual dues of member companies.
Disclosure
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article. Two coauthors are employees of a pharmaceutical company, as noted in the list of affiliations. No products are discussed in this article. The consortium aims to improve methods that can be applied to evaluating the safety and effectiveness of any medical product for neonates and is consequently drug agnostic.
WORKSHOP
On 23 October 2015, the Clinical Pharmacology Workgroup of INC held a workshop in Washington, D.C. to draft this document. Cochaired by Robert Ward, Jeffrey Barrett, and Karel Allegaert, participants at the workshop and INC members who contributed to drafting and finalizing the document included the following: Dina Apele Freimane, Pauls Stradins Clinical University Hospital, Latvia; Jack Aranda, University Hospital of Brooklyn; Ronald Ariagno, Stanford University/US Food and Drug Administration; Daniel Benjamin, Duke University; Raafat Bishai, AstraZeneca; Edmund Capparelli, University of California, San Diego; Edress Darsey, Pfizer; Jonathan M. Davis, Tufts Medical Center and the Tufts Clinical and Translational Science Institute; Walter Kraft, Thomas Jefferson University; Irja Lutsar, University of Tartu, Estonia; Jeffrey Ming, Sanofi Pharmaceuticals; Yeruk Mulugeta, US Food and Drug Administration; Min-Soo Park, Yonsei University College of Medicine; Ronald Portman, Novartis Pharmaceuticals; Randy Prescilla, Eli Lilly and Company/Boston’s Children Hospital; Catherine Sherwin, University of Utah; Vikram Sinha, US Food and Drug Administration; Ine Skottheim Rusten, Norwegian Medicines Agency; Adina Tocoian, Shire; Mark Turner, University of Liverpool; John van den Anker, Children’s National Health System, University of Basel Children’s Hospital, and Sophia Children’s Hospital; Alexander Vinks, Cincinnati Children’s Hospital Medical Center; Kelly Wade, Children’s Hospital of Philadelphia; Siri Wang, Norwegian Medicines Agency; Anne Zajicek, National Institutes of Health.
DISCLAIMERS
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency (EMA) or one of its committees or working parties or the position of the US FDA.
References
General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products. Draft Guidance for Industry. 2014. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm425885.pdf.
Guideline on the Investigation of Medicinal Products in the Term and Preterm Neonate. 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003750.pdf.
International Conference on Harmonization: Clinical Investigation of Medicinal Products in the Pediatric Population E11. 2000. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E11/ICH_E11_R1_Step_2_25Aug2016_Final.pdf.
Tri-Council policy statement 2: ethical conduct for research involving humans. 2014. http://www.pre.ethics.gc.ca/pdf/eng/tcps2-2014/TCPS_2_FINAL_Web.pdf. http://www.pre.ethics.gc.ca/pdf/eng/tcps2-2014/Highlights_of_Changes_EN.pdf.
Survey of Current Guidance for Child Health Clinical Trials. The StaR Child Health Project: Standards for Research with Children. 2009. http://www.who.int/childmedicines/publications/GUIDANCECHILDHEALTH.pdf.
REGULATION (EC) No 1901/2006 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. 2006. http://ec.europa.eu/health/files/eudralex/vol-1/reg_2006_1901/reg_2006_1901_en.pdf.
US Food and Drug Administration Modernization Act of 1997. 1997. http://www.fda.gov/RegulatoryInformation/Legislation/SignificantAmendmentstotheFDCAct/FDAMA/FullTextofFDAMAlaw/default.htm.
Best Pharmaceuticals for Children Act Page. 2002. http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=107_cong_public_laws&docid=f:publ109.107.
Pediatric Research Equity Act of 2003. 2003. http://www.gpo.gov/fdsys/pkg/PLAW-108publ155/pdf/PLAW-108publ155.pdf.
US Food and Drug Administration Safety and Innovation Act. 2012. http://www.gpo.gov/fdsys/pkg/PLAW-112publ144/pdf/PLAW-112publ144.pdf.
Laughon MM, Avant D, Tripathi N, et al. Drug labeling and exposure in neonates. JAMA Pediatr 2014;168:130–6.
Stiers JL, Ward RM. Newborns, one of the last therapeutic orphans to be adopted. JAMA Pediatr 2014;168:106–8.
Turner MA. Clinical trials of medicines in neonates: the influence of ethical and practical issues on design and conduct. Br J Clin Pharmacol 2015;79:370–8.
Engle WA. A recommendation for the definition of “late preterm” (near-term) and the birth weight-gestational age classification system. Semin Perinatol 2006;30:2–7.
Engle WA ; American Academy of Pediatrics Committee on Fetus and Newborn. Age terminology during the perinatal period. Pediatrics 2004;114:1362–4.
Sengupta S, Carrion V, Shelton J, et al. Adverse neonatal outcomes associated with early-term birth. JAMA Pediatr 2013;167:1053–9.
Allegaert K, Anderson BJ, van den Anker JN, Vanhaesebrouck S, de Zegher F. Renal drug clearance in preterm neonates: relation to prenatal growth. Ther Drug Monit 2007;29:284–91.
Calkins K, Devaskar SU. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care 2011;41:158–76.
Thompson H, Frederico N, Smith SR, Chowdhury M, Dicks P, Preston J, Thompson C. iCAN: Providing a voice for children and families in pediatric research. Therapeutic Innovation & Regulatory Science 2015;49:673–679.
Concept paper on the involvement of children and young people at the Paediatric Committee (PDCO). 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/09/WC500132555.pdf.
International Children’s Advisory Network. 2015. http://www.icanresearch.org/.
Guidance for industry: E6 Good clinical practice. Consolidated guidance. 1996. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073122.pdf.
Gauda EB. Knowledge gained from animal studies of the fetus and newborn: Application to the human premature infant. ILAR J 2006;47:1–4.
Nathanielsz PW. Animal models that elucidate basic principles of the developmental origins of adult diseases. ILAR J 2006;47:73–82.
Festing MF. Design and statistical methods in studies using animal models of development. ILAR J 2006;47:5–14.
Inder T, Neil J, Yoder B, Rees S. Patterns of cerebral injury in a primate model of preterm birth and neonatal intensive care. J Child Neurol 2005;20:965–7.
Bourgeois T, Delezoide AL, Zhao W, et al. Safety study of Ciprofloxacin in newborn mice. Regul Toxicol Pharmacol 2016;74:161–9.
Roth-Cline M, Nelson RM. Ethical considerations in conducting pediatric and neonatal research in clinical pharmacology. Curr Pharm Des 2015;21:5619–35.
Kauffman RE, Kearns GL. Pharmacokinetic studies in paediatric patients. Clinical and ethical considerations. Clin Pharmacokinet 1992;23:10–29.
Kearns GL. Impact of developmental pharmacology on pediatric study design: overcoming the challenges. J Allergy Clin Immunol 2000;106(3 Suppl):S128–38.
Ward RM, Tammara B, Sullivan SE, et al. Single-dose, multiple-dose, and population pharmacokinetics of pantoprazole in neonates and preterm infants with a clinical diagnosis of gastroesophageal reflux disease (GERD). Eur J Clin Pharmacol 2010;66:555–61.
Anderson BJ, Woollard GA, Holford NH. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol 2000;50:125–34.
Friis-Hansen B. Body water compartments in children: changes during growth and related changes in body composition. Pediatrics 1961;28:169–81.
Friis-Hansen B. Body composition during growth. In vivo measurements and biochemical data correlated to differential anatomical growth. Pediatrics 1971;47:Suppl 2:264–274.
Aranda JV, Sitar DS, Parsons WD, Loughnan PM, Neims AH. Pharmacokinetic aspects of theophylline in premature newborns. N Engl J Med 1976;295:413–6.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med 2003;349:1157–67.
McNamara PJ, Alcorn J. Protein binding predictions in infants. AAPS PharmSci 2002;4:E4.
Brodersen R, Honoré B. Drug binding properties of neonatal albumin. Acta Paediatr Scand 1989;78:342–6.
Odell GB. The distribution and toxicity of bilirubin. E. Mead Johnson address 1969. Pediatrics 1970;46:16–24.
Wennberg RP, Ahlfors CE, Rasmussen LF. The pathochemistry of kernicterus. Early Hum Dev 1979;3:353–72.
Robertson A, Karp W, Brodersen R. Bilirubin displacing effect of drugs used in neonatology. Acta Paediatr Scand 1991;80:1119–27.
Fink S, Karp W, Robertson A. Ceftriaxone effect on bilirubin-albumin binding. Pediatrics 1987;80:873–5.
Leeder JS. Translating pharmacogenetics and pharmacogenomics into drug development for clinical pediatrics and beyond. Drug Discov Today 2004;9:567–73.
Leeder JS, Kearns GL, Spielberg SP, van den Anker J. Understanding the relative roles of pharmacogenetics and ontogeny in pediatric drug development and regulatory science. J Clin Pharmacol 2010;50:1377–87.
Garner CR, Park KB, French NS, et al. Observational infant exploratory [(14)C]-paracetamol pharmacokinetic microdose/therapeutic dose study with accelerator mass spectrometry bioanalysis. Br J Clin Pharmacol 2015;80:157–67.
Turner MA, Mooij MG, Vaes WH, et al. Pediatric microdose and microtracer studies using 14C in Europe. Clin Pharmacol Ther 2015;98:234–7.
Roth-Cline M, Nelson RM. Microdosing studies in children: A US regulatory perspective. Clin Pharmacol Ther 2015;98:232–3.
Aperia A, Broberger O, Elinder G, Herin P, Zetterström R. Postnatal development of renal function in pre-term and full-term infants. Acta Paediatr Scand 1981;70:183–7.
Engle WD, Arant BS Jr . Renal handling of beta-2-microglobulin in the human neonate. Kidney Int 1983;24:358–63.
Rhodin MM, Anderson BJ, Peters AM, et al. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol 2009;24:67–76.
Daood M, Tsai C, Ahdab-Barmada M, Watchko JF. ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics 2008;39:211–8.
Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res 2015;56:1085–99.
Rodriguez W, Selen A, Avant D, et al. Improving pediatric dosing through pediatric initiatives: what we have learned. Pediatrics 2008;121:530–9.
Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 2008;48:303–32.
Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 2009;24:25–36.
Koukouritaki SB, Manro JR, Marsh SA, et al. Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther 2004;308:965–74.
Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver–evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem 1997;247:625–34.
Allegaert K, van Schaik RH, Vermeersch S, et al. Postmenstrual age and CYP2D6 polymorphisms determine tramadol o-demethylation in critically ill neonates and infants. Pediatr Res 2008;63:674–9.
Blake MJ, Castro L, Leeder JS, Kearns GL. Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med 2005;10:123–38.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med 2003;349:1157–67.
Cohen-Wolkowiez M, Watt KM, Hornik CP, Benjamin DK Jr, Smith PB. Pharmacokinetics and tolerability of single-dose daptomycin in young infants. Pediatr Infect Dis J 2012;31:935–7.
Yanni SB, Smith PB, Benjamin DK Jr, Augustijns PF, Thakker DR, Annaert PP. Higher clearance of micafungin in neonates compared with adults: role of age-dependent micafungin serum binding. Biopharm Drug Dispos 2011;32:222–32.
Qualification of novel methodologies for medicine development. 2016. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000319.jsp&mid=WC0b01ac0580022bb0.
Biomarker Qualification Program. 2016. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/default.htm.
Amur S, Frueh FW, Lesko LJ, Huang SM. Integration and use of biomarkers in drug development, regulation and clinical practice: a US regulatory perspective. Biomark Med 2008;2:305–11.
Bai JP, Barrett JS, Burckart GJ, Meibohm B, Sachs HC, Yao L. Strategic biomarkers for drug development in treating rare diseases and diseases in neonates and infants. AAPS J 2013;15:447–54.
Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J 2005;5:6–13.
Leeder JS. Developmental pharmacogenetics: a general paradigm for application to neonatal pharmacology and toxicology. Clin Pharmacol Ther 2009;86:678–82.
Guideline on key aspects for the use of pharmacogenomic methodologies in the pharmacovigilance evaluation of medicinal products. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/01/WC500160232.pdf.
European Medicines Agency: Paediatric investigation plans. 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000293.jsp&mid=WC0b01ac0580025b91.
Dunne J, Rodriguez WJ, Murphy MD, et al. Extrapolation of adult data and other data in pediatric drug-development programs. Pediatrics 2011;128:e1242–9.
Concept paper on extrapolation of efficacy and safety in medicine development. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/04/WC500142358.pdf.
Pediatric Research and Science Activities. 2016. http://www.fda.gov/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/ucm106614.htm.
Wade KC, Wu D, Kaufman DA, et al.; National Institute of Child Health and Development Pediatric Pharmacology Research Unit Network. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother 2008;52:4043–9.
Ascher SB, Smith PB, Watt K, et al. Antifungal therapy and outcomes in infants with invasive Candida infections. Pediatr Infect Dis J 2012;31:439–43.
Benjamin DK Jr, Hudak ML, Duara S, et al.; Fluconazole Prophylaxis Study Team. Effect of fluconazole prophylaxis on candidiasis and mortality in premature infants: a randomized clinical trial. JAMA 2014;311:1742–9.
Smith PB, Cohen-Wolkowiez M, Castro LM, et al.; Meropenem Study Team. Population pharmacokinetics of meropenem in plasma and cerebrospinal fluid of infants with suspected or complicated intra-abdominal infections. Pediatr Infect Dis J 2011;30:844–9.
van den Anker JN, Pokorna P, Kinzig-Schippers M, et al. Meropenem pharmacokinetics in the newborn. Antimicrob Agents Chemother 2009;53:3871–9.
Hudak BB, Egan EA. Impact of lung surfactant therapy on chronic lung diseases in premature infants. Clin Perinatol 1992;19:591–602.
Kwong MS, Egan EA, Notter RH, Shapiro DL. Double-blind clinical trial of calf lung surfactant extract for the prevention of hyaline membrane disease in extremely premature infants. Pediatrics 1985;76:585–92.
Notter RH, Egan EA, Kwong MS, Holm BA, Shapiro DL. Lung surfactant replacement in premature lambs with extracted lipids from bovine lung lavage: effects of dose, dispersion technique, and gestational age. Pediatr Res 1985;19:569–77.
Howie SR. Blood sample volumes in child health research: review of safe limits. Bull World Health Organ 2011;89:46–53.
Heidmets LT, Metsvaht T, Ilmoja ML, Pisarev H, Oselin K, Lutsar I. Blood loss related to participation in pharmacokinetic study in preterm neonates. Neonatology 2011;100:111–5.
Leroux S, Turner MA, Guellec CB, et al.; TINN (Treat Infections in NeoNates) and GRiP (Global Research in Paediatrics) Consortiums. Pharmacokinetic studies in neonates: The utility of an opportunistic sampling design. Clin Pharmacokinet 2015;54:1273–85.
Castagnola M, Inzitari R, Fanali C, et al. The surprising composition of the salivary proteome of preterm human newborn. Mol Cell Proteomics 2011;10:M110.003467.
Lee TC, Charles BG, Steer PA, Flenady VJ. Saliva as a valid alternative to serum in monitoring intravenous caffeine treatment for apnea of prematurity. Ther Drug Monit 1996;18:288–93.
21 CFR Sec. 50.53 Clinical investigations involving greater than minimal risk and no prospect of direct benefit to individual subjects, but likely to yield generalizable knowledge about the subjects’ disorder or condition. 2015. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=50&showFR=1&subpartNode=21:1.0.1.1.20.4.
Leong R, Vieira ML, Zhao P, et al. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther 2012;91:926–31.
Benjamin DK Jr, Smith PB, Jadhav P, et al. Pediatric antihypertensive trial failures: analysis of end points and dose range. Hypertension 2008;51:834–40.
Linakis MW, Roberts JK, Lala AC AC, et al. Challenges associated with route of administration in neonatal drug delivery. Clin Pharmacokinet 2016;55:185–196.
Guideline on pharmaceutical development of medicines for paediatric use. August 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/07/WC500147002.pdf.
Turner MA, Shah U. Why are excipients important to neonates? Curr Pharm Des 2015;21:5680–7.
Brown WJ, Buist NR, Gipson HT, Huston RK, Kennaway NG. Fatal benzyl alcohol poisoning in a neonatal intensive care unit. Lancet 1982;1:1250.
Gershanik J, Boecler B, Ensley H, McCloskey S, George W. The gasping syndrome and benzyl alcohol poisoning. N Engl J Med 1982;307:1384–8.
Excipients labelling EMA. 2003. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003412.pdf.
Salunke S, Brandys B, Giacoia G, Tuleu C. The STEP (Safety and toxicity of excipients for paediatrics) database: part 2 - the pilot version. Int J Pharm 2013;457:310–22.
Yakkundi S, McElnay J, Millership J, et al. Use of dried blood spots to study excipient kinetics in neonates. Bioanalysis 2011;3:2691–3.
Mulla H, Yakkundi S, McElnay J, et al. An observational study of blood concentrations and kinetics of methyl- and propyl-parabens in neonates. Pharm Res 2015;32:1084–93.
Nellis G, Metsvaht T, Varendi H, et al.; ESNEE consortium. Potentially harmful excipients in neonatal medicines: a pan-European observational study. Arch Dis Child 2015;100:694–9.
Abdel-Rahman SM, Reed MD, Wells TG, Kearns GL. Considerations in the rational design and conduct of phase I/II pediatric clinical trials: avoiding the problems and pitfalls. Clin Pharmacol Ther 2007;81:483–94.
Klingmann V, Seitz A, Meissner T, Breitkreutz J, Moeltner A, Bosse HM. Acceptability of uncoated mini-tablets in neonates–a randomized controlled trial. J Pediatr 2015;167:893–896.e2.
Sherwin CM, Medlicott NJ, Reith DM, Broadbent RS. Intravenous drug delivery in neonates: lessons learnt. Arch Dis Child 2014;99:590–4.
Adaptive Design Clinical Trials for Drugs and Biologics. Guidance for Industry. 2010. http://www.fda.gov/downloads/Drugs/Guidances/ucm201790.pdf.
Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clin Pharmacol 2012;52:1601–6.
Ethical Considerations for Clinical Trials on Medicinal Products conducted with the Paediatric Population 2008. http://ec.europa.eu/health/sites/health/files/files/eudralex/vol-10/ethical_considerations_en.pdf.
Long D, Koren G, James A. Ethics of drug studies in infants: how many samples are required for accurate estimation of pharmacokinetic parameters in neonates? J Pediatr 1987;111(6 Pt 1):918–21.
Kauffman RE, Kearns GL. Pharmacokinetic studies in paediatric patients. Clinical and ethical considerations. Clin Pharmacokinet 1992;23:10–29.
Retout S, Comets E, Samson A, Mentré F. Design in nonlinear mixed effects models: optimization using the Fedorov-Wynn algorithm and power of the Wald test for binary covariates. Stat Med 2007;26:5162–79.
Retout S, Duffull S, Mentré F. Development and implementation of the population Fisher information matrix for the evaluation of population pharmacokinetic designs. Comput Methods Programs Biomed 2001;65:141–51.
Retout S, Mentré F. Optimization of individual and population designs using Splus. J Pharmacokinet Pharmacodyn 2003;30:417–43.
Perlman SE, Saiman L, Larson EL. Risk factors for late-onset health care-associated bloodstream infections in patients in neonatal intensive care units. Am J Infect Control 2007;35:177–82.
Hawcutt DB, Rose AC, Fuerst-Recktenwald S, Nunn T, Turner MA. Points to consider when planning the collection of blood or tissue samples in clinical trials of investigational medicinal products in children, infants and neonates. In: Rose K, van den Anker JN, eds. Guide to Paediatric Drug Development and Clinical Research. Karger: Basel, 2009:99–110.
Ward RM, Kearns GL. Proton pump inhibitors in pediatrics: mechanism of action, pharmacokinetics, pharmacogenetics, and pharmacodynamics. Paediatr Drugs 2013;15:119–31.
Turner MA, Hill H. Pharmacovigilance in neonatal intensive care. In: Mimouni FB, van den Anker JN, eds. Neonatal Pharmacology. Karger: Basel, 2014;28–40.
Du W, Lehr VT, Lieh-Lai M, et al. An algorithm to detect adverse drug reactions in the neonatal intensive care unit. J Clin Pharmacol 2013;53:87–95.
Conroy S, McIntyre J. The use of unlicensed and off-label medicines in the neonate. Semin Fetal Neonatal Med 2005;10:115–22.
American Academy of Pediatrics Committee on Drugs. Guidelines for the ethical conduct of studies to evaluate drugs in pediatric populations. Committee on Drugs, American Academy of Pediatrics. Pediatrics 1995;95:286–294.
Shaddy RE, Denne SC ; Committee on Drugs and Committee on Pediatric Research. Clinical report–guidelines for the ethical conduct of studies to evaluate drugs in pediatric populations. Pediatrics 2010;125:850–60.
Ward RM, Sherwin CM. Ethics of drug studies in the newborn. Paediatr Drugs 2015;17:37–42.
Kipnis K. Seven vulnerabilities in the pediatric research subject. Theor Med Bioeth 2003;24:107–20.
Release and Adoption of Health Canada Addendum to the ICH1 Guidance E11: Clinical Investigation of Medicinal Products in the Pediatric Population. 2003. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/clini/e11_addendum-eng.pdf.
EMEA Committee for medicinal products for human use: Guideline on data monitoring committees. 2005. http://osp.od.nih.gov/sites/default/files/resources/WC500003635.pdf.
2016 Edition of the International Compilation of Human Research Standards. 2016. https://www.hhs.gov/ohrp/international/compilation-human-research-standards/index.html.
REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. 2014. http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf.
Protection of Human Subjects. Subpart D. Additional Protections for Children Involved as Subjects in Research. 48 FR 9818. Section 46.401-46.409. 1991. http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html#subpartd.
Shah S, Whittle A, Wilfond B, Gensler G, Wendler D. How do institutional review boards apply the federal risk and benefit standards for pediatric research? JAMA 2004;291:476–82.
Office for Human Research Protections (OHRP). Secretary’s Advisory Committee on Human Research Protections (SACHRP). Appendix B. 2005. https://www.hhs.gov/ohrp/sachrp-committee/recommendations/2005-july-28-letter-appendix-b/index.html.
21 CFR sec. 50.52 Clinical investigations involving greater than minimal risk but presenting the prospect of direct benefit to individual subjects. 2015. http://www.ecfr.gov/cgi-bin/text-idx?SID=0f4329b2e4045a0a62b37bf42590c837&mc=true&node=se21.1.50_152&rgn=div8.
Ward RM, Daniel CH, Kendig JW, Wood MA. Oliguria and tolazoline pharmacokinetics in the newborn. Pediatrics 1986;77:307–15.
Turner S, Nunn AJ, Fielding K, Choonara I. Adverse drug reactions to unlicensed and off-label drugs on paediatric wards: a prospective study. Acta Paediatr 1999;88:965–8.
Simons SH, van Dijk M, Anand KS, Roofthooft D, van Lingen RA, Tibboel D. Do we still hurt newborn babies? A prospective study of procedural pain and analgesia in neonates. Arch Pediatr Adolesc Med 2003;157:1058–64.
Guidance for Clinical Trial Sponsors. Establishment and Operation of Clinical Trial Data Monitoring Committees. 2006. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm127073.pdf.
Allmark P, Mason S. Improving the quality of consent to randomised controlled trials by using continuous consent and clinician training in the consent process. J Med Ethics 2006;32:439–43.
Acknowledgements
The authors gratefully acknowledge the review by Ralph Bax (EMA), Skip Nelson (FDA), Agnes Klein (Health Canada), Christine Gleason (March of Dimes), and Nick Hall (Graham’s Foundation).
Author information
Authors and Affiliations
Consortia
Corresponding author
Supplementary information
Supplementary Information
(ZIP 1051 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ward, R., Benjamin, D., Barrett, J. et al. Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr Res 81, 692–711 (2017). https://doi.org/10.1038/pr.2016.221
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.221
This article is cited by
-
Why population pharmacokinetic studies are instrumental to boost clinical research in neonates, and suggestions on how clinicians should assess these papers
Pediatric Research (2024)
-
Challenges and opportunities for improving access to approved neonatal drugs and devices
Journal of Perinatology (2022)
-
The conect4children (c4c) Consortium: Potential for Improving European Clinical Research into Medicines for Children
Pharmaceutical Medicine (2021)
-
How to introduce MSC-based therapy for the developing lung safely into clinical care?
Pediatric Research (2020)
-
Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population
Clinical Pharmacokinetics (2020)
