
Abstract
Background:
rhIGF-1/rhIGFBP-3 is being investigated for prevention of retinopathy of prematurity in extremely preterm infants.
Methods:
A population pharmacokinetic model was developed using data from phase I/II (Sections A–C) trials of rhIGF-1/rhIGFBP-3 and additional studies in preterm infants to predict optimal dosing to establish/maintain serum IGF-1 within physiological intrauterine levels. In Section D of the phase II study, infants (gestational age (GA) (wk+d) 23+0 to 27+6) were randomized to rhIGF-1/rhIGFBP-3, administered at the model-predicted dose of 250 µg/kg/d continuous i.v. infusion up to postmenstrual age (PMA) 29 wk+6 d or standard of care. An interim pharmacokinetic analysis was performed for the first 10 treated infants to verify dosing.
Results:
Serum IGF-1 data were reviewed for 10 treated/9 control infants. Duration of therapy in treated infants ranged 1–34.5 d. At baseline (before infusion and <24 h from birth), mean (SD) IGF-1 was 19.2 (8.0) μg/l (treated) and 15.4 (4.7) μg/l (controls). Mean (SD) IGF-1 increased to 45.9 (19.6) μg/l at 12 h in treated infants, and remained within target levels for all subsequent timepoints. For treated infants, 88.8% of the IGF-1 measurements were within target levels (controls, 11.1%).
Conclusion:
Through the reported work, we determined appropriate rhIGF-1/rhIGFBP-3 dosing to achieve physiological intrauterine serum IGF-1 levels in extremely preterm infants.
Similar content being viewed by others


Main
Retinopathy of prematurity (ROP), characterized by abnormal postnatal retinal vascular growth, is a common comorbidity of extreme prematurity. Despite improvements in disease management, infants with ROP remain at risk of life-long visual impairments, and there is an unmet need for a preventive therapy (1).
rhIGF-1/rhIGFBP-3 (recombinant human IGF-1 complexed with its binding protein recombinant human IGFBP-3) is being investigated as an IGF-1 replacement therapy for the prevention of ROP in extremely preterm infants. This approach is based on the hypothesis that increasing early postnatal serum IGF-1 concentrations in preterm infants to corresponding intrauterine levels could promote normal retinal vascularization and prevent development of ROP. This hypothesis is supported by the following literature and clinical observations: (i) IGF-1 is an important regulator of fetal growth and development, with fetal levels of IGF-1 increasing with gestational age (GA), particularly in the second and third trimesters of pregnancy (2,3); (ii) preclinical studies suggest that IGF-1 plays a key role in retinal vascular development (4,5); (iii) extremely preterm birth is associated with a rapid fall in serum levels of IGF-1 (6,7), which then rise more slowly postnatally in preterm infants than in term infants (8); and (iv) longitudinal studies have shown that lower IGF-1 levels are associated with an increased risk of ROP (9,10,11), with severity of ROP reported to correlate with duration of low IGF-1 (9).
Historically, pharmacokinetic (PK) analyses have not been routinely conducted in newborns, with dosing typically extrapolated from PK data obtained in older children and adults (12,13). However, newborns undergo rapid developmental changes, which often alter the PK properties of a drug (14), and we believe that PK analyses specific to the neonatal population are essential from an ethical perspective in order to establish optimal dosing, and to ensure the greatest potential therapeutic benefit and safety of drug usage in these infants. Early clinical studies have been conducted that aimed (among other objectives) to evaluate PK and optimize dosing of rhIGF-1/rhIGFBP-3 in extremely preterm infants. An initial phase I study showed an increase in serum IGF-1 with a 3-h infusion of rhIGF-1/rhIGFBP-3 and highlighted a need for continuous i.v. infusion to maintain adequate IGF-1 levels due to a much shorter than anticipated half-life in preterm infants (15). A phase II study was subsequently initiated to assess dosing of rhIGF-1/rhIGFBP-3 administered as a continuous i.v. infusion and to evaluate efficacy and safety for the prevention of ROP. rhIGF-1/rhIGFBP-3 dosing in this study (initially designed in three sections (A–C), data on file) was individualized and adjusted via intensive therapeutic drug monitoring to achieve target levels (2). However, intensive monitoring requires frequent blood sampling, and therefore, it was considered necessary to standardize the rhIGF-1/rhIGFBP-3 dose regimen to alleviate the burden this imposes on the neonatal population, as well as to simplify the infusion protocol. Further, a review of the data for serum IGF-1 levels in Sections A–C showed that a substantial number of measurements fell below the target range. This prompted reconsideration of the literature (2,16,17) that had been used to establish IGF-1 target levels, which reported intrauterine IGF-1 levels from pregnancies undergoing diagnostic cordocentesis due to various abnormalities/anomalies, and which may not have been representative of a normal population. For these reasons, enrollment in Section C was paused to re-evaluate target levels and optimize the dosing regimen for a fourth section of the study (Section D).
We report the additional work undertaken in order to: (i) establish the normal range of intrauterine fetal serum IGF-1 and the range of serum IGF-1 levels in infants born preterm; (ii) predict the optimal dose of rhIGF-1/rhIGFBP-3 for Section D of the phase II study through population PK modeling and clinical trial simulations; and (iii) verify the model-predicted dose of rhIGF-1/rhIGFBP-3 in an interim PK analysis among 10 preterm infants who received the redesigned and standardized dose regimen.
Methods
Estimation of Physiological Intrauterine Serum IGF-1 Levels
A meta-analysis of the published literature was performed to establish normal physiological intrauterine levels of IGF-1. Two published studies were selected for inclusion (16,17) (number of infants, n = 174; Table 1 ). Graphical data from the source publications were digitized using a plot digitizer, combined, and then log-transformed and subjected to nonlinear regression analysis. A centered second-order polynomial curve fitting was used. The resulting data were converted back to a linear (anti-log) scale to obtain the final plot and estimate of normal physiological IGF-1 levels based on mean (5– 95% prediction range) between GA 23–28 wk ( Figure 1 ).

Intrauterine and preterm infant serum IGF-1 levels. Open black circles represent IGF-1 in normal in utero fetus. Closed pink circles represent IGF-1 in preterm infants. Solid black line represents mean predicted physiological levels of corresponding intrauterine levels (R2 = 0.64, N = 174). Solid pink line represents mean predicted values of preterm infants (R2 = 0.15, N = 137). Dashed black line represents upper prediction interval (95th). Dotted black line represents lower prediction interval (95th). From Hellström et al. (3), Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Copyright © 2016 Acta Paediatrica. Reproduced with permission of Blackwell Publishing Ltd.
Population PK Modeling
Data sources in preterm infants. A population PK model was developed incorporating data for rhIGF-1/rhIGFBP-3‒treated infants from the phase I and II studies (n = 19), and for untreated preterm infants from the phase II study Sections B/C (n = 8) and additional studies in untreated preterm infants (n = 137; Table 1 ). From these data sources, a total of 812 PK sample records (collected at specified time intervals per the protocol for each study) from 156 infants were included in the dataset for initial PK modeling. The model was updated after incorporation of interim analysis data on the first 10 infants treated in Section D.
For all samples, analysis of serum IGF-1 levels (measuring total unbound and bound IGF-1 circulating in blood) was performed by an accredited laboratory using a validated IGFBP-blocked radioimmunoassay (RIA) (Mediagnost GmbH, Tübingen, Germany) (17,18). This technique enabled IGF-1 to be separated from its binding protein before measurement (direct measurements of IGF-1 in serum samples without pretreatment result in inaccurate values because of the slow dissociation of the IGF-1/IGFBP-3 complex during assay incubation).
Model development. Model development was conducted in five steps: (i) exploratory graphical analysis of IGF-1 concentration‒time data; (ii) base structural model development; (iii) exploratory covariate analysis; (iv) model refinement; and (v) final model verification.
The model was developed using an explicit equation estimating baseline IGF-1, a first-order production rate constant for IGF-1, and a lag time to the start of endogenous production to describe the untreated controls. An explicit equation describing a short-term infusion with mono-exponential decline was used to capture the time course of the IGF-1 serum concentrations following i.v. infusion (see Supplementary Methods online). Total serum IGF-1 was, therefore, modeled as the sum of endogenous IGF-1 and IGF-1 from the short-term infusion.
Covariate analysis was performed to evaluate the effect of patient cofactors as potential predictors of interindividual variability in the PK of the drug. Covariates included in the exploratory analysis were demographic factors (postmenstrual age (PMA), sex, weight) and blood chemistries (e.g., aspartate aminotransferase, alanine aminotransferase, glucose). PMA and weight were excluded from the final model because they were not found to be statistically significant or to improve the predictive power. Although there were slight trends to suggest a relationship between PMA and weight with PK parameters of rhIGF-1/rhIGFBP-3, model diagnostics did not support the inclusion of either covariate to explain the interindividual variability in PK of rhIGF-1/rhIGFBP-3 (in a comparison between simulated concentrations from the visual predictive check model and the observed IGF-1 concentrations stratified by PMA and weight, minimal differences were observed up to a PMA of 37 wk and a weight range of 0.5‒3 kg).
The initial model was optimized by inclusion of significant covariates and tested by a visual predictive check comparing IGF-1 observations with simulated concentrations in order to predict optimal dosing for Section D (described further below). Additional verification was carried out through incorporation of the interim PK data from 10 infants treated with the model-predicted dose in Section D, and comparison of the final vs. original models. Further information on the development and refinement of the model is reported in the online supplementary material (see Supplementary Methods online).
Trial simulations and dose prediction. The initial population PK model was used to simulate IGF-1 concentration‒time profiles following continuous i.v. infusion at incremental doses ranging 100–750 µg/kg/24 h (assuming linear kinetics) for a period of 2, 4, 6, and 8 wk in 500 randomly sampled patients. The 25th, 50th, and 75th prediction intervals were plotted and overlaid with physiological intrauterine IGF-1 levels (determined from the literature meta-analysis) to assess the effect of dose and dosing frequency on IGF-1 concentration levels and to predict the dose required to achieve and maintain physiological intrauterine IGF-1 levels. Data were analyzed to quantify the percentage of simulations within target levels for each dose relative to the percentages over and under target.
Population PK and statistical analysis software. Population PK modeling was performed using NONMEM software version 7.2, level 1.1, NM-TRAN version III, level 1.0, and PREDPP version IV, level 1.0 (ICON plc, Ellicott City, MD) at KinderPharm/PKPD Bioscience (Exton, PA). The first-order conditional estimation with interaction and first-order minimization methods were implemented. Statistical analyses were performed using SAS software Version 9.3 (SAS Institute, Cary, NC) (16,17,19) and PDxPop version 5.1 (SAS Institute, version 9.1).
Clinical Dose Verification
Section D study design overview. The model-predicted dose was evaluated in Section D of the phase II study (completed March 2016), which was multicenter, randomized, and assessor masked, and intended to evaluate the efficacy and safety of rhIGF-1/rhIGFBP-3 to prevent ROP in extremely preterm infants. Follow up evaluations were performed to 40 wk PMA. The study was conducted at clinical sites in the United States and Europe. Approval was obtained from the relevant institutional review boards and independent ethics committees before study initiation, and the study adhered to International Conference on Harmonisation Good Clinical Practice guidelines and the tenets of the Declaration of Helsinki. All parents/guardians of the study infants provided written informed consent. A clinical study monitor ensured that the investigation was conducted according to protocol design and regulatory requirements by frequent site visits and communications. Infant safety was monitored on a continuous basis until the last subject completed his or her last scheduled study visit/assessment, and quarterly safety review meetings were held throughout study Section D. Safety data collected during the trial was reported annually to competent authorities in the form of a Development Safety Update Report. Additionally, an independent data monitoring committee provided an ongoing review and assessment of safety data, to safeguard the interests and safety of the infants participating in the study, and a special data monitoring committee meeting was to be convened if the safety-related stopping rules (i.e., if a death occurred that was considered possibly or probably related to the study drug) were met. The trial (Sections A–D) is registered at ClinicalTrials.gov (NCT01096784). Results for the full Section D study will be published separately.
Interim PK analysis. An interim PK analysis was conducted in the first 10 treated infants and corresponding control infants in Section D in order to evaluate PK parameters and attainment of target levels of IGF-1 (28–109 μg/l) with the model-predicted dose regimen (250 µg/kg/24 h).
Infants with GA 23 wk + 0 d to 27 wk + 6 d at birth were randomized to receive rhIGF-1/rhIGFBP-3 using the model-predicted dose regimen up to a PMA of 29 wk + 6 d or standard neonatal care. Blood samples for IGF-1 analysis in serum were taken at baseline (immediately before starting the infusion and within 24 h from birth), at 12- and 24-h postbaseline/start of infusion, then every 72 h in treated infants, and every 168 h in infants receiving standard neonatal care, up to a PMA of 29 wk + 6 d. Follow-up measurements also were taken 1 h after termination of infusion (treated only) and at a PMA of 32, 36, and 40 wk (data not included for dose verification). Serum IGF-1 levels were measured using a validated RIA at a central laboratory (17,18). All measurements below the lower limit of quantification (8.93 μg/l) were excluded from the analysis.
Results
Intrauterine and Preterm Infant Serum IGF-1 Levels
Based on the published literature (16,17), mean physiological intrauterine serum IGF-1 between GA 23–28 wk was estimated at 54 μg/l (5th‒95th prediction intervals 28‒109 μg/l). This represented the median value from eight independent analyses conducted by four different operators ( Figure 1 ). In preterm infants born between GA 23–28 wk, the mean predicted serum IGF-1 level was 13 μg/l (5th‒95th prediction intervals 4‒40 μg/l; Figure 1 ).
Trial Simulations and Predicted Dose Regimen
Dose simulations indicated that a dosing regimen of ≥250 μg/kg administered as a continuous 24-h infusion for at least 4 wk would be needed to achieve serum IGF-1 within the range of physiological intrauterine levels, considered to be 28–109 μg/l per 95% prediction intervals from the above analysis. At this dose, the simulation indicated that the majority of values (65.5%) would be predicted to be in the normal range ( Figure 2 ). Further, at this simulated dose, only 1.9% of values were above 109 μg/l (this percentage increasing at the higher simulated doses of 300, 500, and 750 μg/kg/24 h), suggesting a minimal chance for overexposure at this dose. Thus, 250 μg/kg/24 h was considered a conservative dose from a safety perspective. These predictions assumed linear PK for the production and elimination of IGF-1, an hypothesis that was tested and confirmed in the interim analysis reported herein.

Simulation of the 250 μg/kg/24 h dose over 6 wk of treatment. Blue lines represents 5th to 95th percentiles for simulated IGF-1 levels with 250 µg/kg, 24-h infusion, 6 wk treatment. Red dots represent in utero endogenous IGF-1. The black line represents the 50th percentile (~68.5 µg/l) of the targeted range of physiological intrauterine levels (28–109 µg/l).
The estimated half-life of IGF-1 with rhIGF-1/rhIGFBP-3 administration was ~2 h, supporting the conclusion from previous studies that continuous i.v. infusion is required to maintain target levels of IGF-1 (15,20).
The onset of significant endogenous IGF-1 production in preterm infants was estimated to be ~30 wk PMA, based on previous prospective studies (7,21), thus suggesting administration over this period would maintain IGF-1 throughout the period of endogenous IGF-1 production deficit.
Clinical Dose Verification
Based on the model and simulation results presented, the recommended rhIGF-1/rhIGFBP-3 dosing regimen for use in Section D was 250 μg/kg/24 h, with treatment up to a PMA of 29 wk + 6 d, in order to achieve IGF-1 target levels of 28–109 μg/l.
Patients. To verify the model predictions, serum IGF-1 data were reviewed and analyzed for the first 10 infants treated with rhIGF-1/rhIGFBP-3 in Section D, and for 9 control infants receiving standard neonatal care only. Patient demographics are summarized in Table 2 .
Duration of therapy in treated infants ranged from 1.0 to 34.5 d. One treated infant received therapy for only 1 d (this infant died due to early onset sepsis); for the nine other infants, infusion duration (defined as the total duration of infusion excluding the time for infusion interruptions) was between 13.6 and 34.5 d (mean 20.7 d). The total dose received ranged from 2.85–7.07 mg (mean 4.36 mg), excluding the infant treated for only 1 d. Infusion interruptions were documented for 5 of 10 treated infants: 1 interruption for each of infants #2, #4, and #9; 45 interruptions for infant #6; 82 interruptions for infant #7. Twelve of the total 130 infusion interruptions occurred during or very close to sampling for IGF-1 levels.
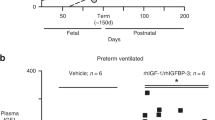
Serum IGF-1 levels. At baseline (before infusion or within 24 h from birth), mean (SD) serum IGF-1 was 19.2 (8.0) μg/l for treated and 15.4 (4.7) μg/l for control infants. After baseline, a clear separation was observed for the mean IGF-1 profiles between treated and control infants ( Figure 3 ). Mean (SD) serum IGF-1 levels increased to 45.9 (19.6) μg/l at 12 h in treated infants, and remained within target levels (28–109 µg/l) for all subsequent time points during treatment. In control infants, mean serum IGF-1 remained below target levels for all time points.

Serum IGF-1 profile (mean (SD); treated vs. control infants). Note: IGF-1 levels from treated infants on days 28, 31, and 34 were from 1 infant only (treated infant #1 in Figure 4 ), and therefore should be interpreted with caution. Closed black squares represent rhIGF-1/rhIGFBP-3 treated infants (n = 10). Closed grey circles represent control infants (n = 9).
All serum IGF-1 concentrations were included for analysis, except for one observation due to a blood sampling error, in which the sample was taken from the central venous line where study drug was infused (188 μg/l, day 25, treated patient #1 in Figure 4 ).

Attainment of target levels of serum IGF-1. (a) Individual serum IGF-1 concentrations (b) and proportions within target range. n, number of infants in the group considered; N, number of IGF-1 measurements. aBaseline (predose) values from treated infants are not included. One observation of 188 µg/l in 1 treated infant (patient #1) was excluded due to a sampling procedure error on day 25. All IGF-1 values for control infant #5 were below the lower limit of quantification (LLQ) and were not included in the population PK model (for control infants #8 and #9; only 1 IGF-1 measurement was above the LLQ). bPredose data excluded. cExcludes data from time points around infusion interruptions (N = 12 measurements).
Attainment of target levels of serum IGF-1. Over the course of the study, 88.8% of individual serum IGF-1 measurements were within target levels for treated infants, compared with 11.1% for control infants ( Figure 4 ). No infants had serum IGF-1 measurements above the upper limit (109 μg/l) of the target range.
Excluding data for samples close to or during infusion interruptions (N = 12 measurements; accuracy of IGF-1 concentrations at these time points being uncertain), the percentage of treated infants with serum IGF-1 within target levels increased to 91.2%.
PK parameters. Over the course of the study, PK parameters for average and maximum concentrations of serum IGF-1 were within the target range for the 10 infants in the treated group (mean (SD) average serum concentration (Cavg) during treatment period 47 (16) μg/l; maximum serum concentration (Cmax) observed after administration 66 (16) μg/l), and below target range in the control group (Cavg 18 (4) μg/l; Cmax 25 (9) μg/l).
Model verification. The interim data from Section D included 151 PK sample records (from 19 infants), of which 33 data points were excluded (32 below the lower limit of quantification, one sampling error). After exclusions, 118 PK sample records from 18 infants (8 control, 10 treated) were included in the dataset to verify the initial model. When data from the interim PK analysis were incorporated into the population PK model, comparison of the final model (with incorporation of interim data) with the original showed that PK parameter estimates were comparable between the two. Further detail on the verification and development of the final model are reported in the online Supplementary Material (see Supplementary Methods online).
Discussion
Population PK modeling predicted an optimal rhIGF-1/rhIGFBP-3 dose of 250 μg/kg/24 h, administered by continuous i.v. infusion, to establish and maintain physiological intrauterine serum IGF-1 levels in extremely preterm infants. PK analysis among infants treated with this dosing regimen in Section D of a phase II trial verified the population PK model and confirmed the appropriateness of the selected dose. At this dose, serum IGF-1 levels were within target levels (28–109 μg/l) for the majority of measurements (88.8%) during therapy. In contrast, the majority of measurements (88.9%) in infants receiving standard neonatal care were below targeted levels.
Since very few therapeutic indications are unique to infants, dosing is typically derived from adult dosing regimens, a common practice that results in the widespread off-label use of drugs within the neonatal population. A prospective study conducted in 70 infants (including preterm), over 13 wk in the NICU showed that 90% were given a drug that was either unlicensed or used in an off-label way (22). However, PK properties of a drug are frequently age dependent, and may be particularly variable between a neonatal population (undergoing rapid developmental changes) and an older population. As an example, half-life for IGF-1 following administration of rhIGF-1/rhIGFBP-3 in preterm infants has been shown to be significantly shorter than in older children and adults (15), highlighting the importance of PK analysis and dose optimization specifically in this population. We believe the work undertaken in this study is unique in its approach and in ensuring optimization of drug dosing at an early stage of clinical development. The study was carefully designed in several stages, first compiling information from published literature and prior clinical trials in order to estimate IGF-1 levels in utero and in preterm infants and determine IGF-1 target levels, followed by population PK modeling and dose simulations to predict optimal dosing, and finally, verification of the predicted dose regimen within a clinical trial. We would urge a similar level of study for other investigational agents in development for use in the neonatal preterm population.
A limitation of the study was the small sample size for the interim PK analysis within the phase II trial (Section D), and the variable duration of treatment among infants. However, PK is being further evaluated within the full Section D trial population, which will provide data for a larger sample (121 infants enrolled in total). PK analyses within the Section D study also will evaluate serum IGF-1 levels postinfusion in both treated and control infants to establish whether there are continuing differences between groups that may affect safety or efficacy. Although dose selection was intended to be conservative, the dose of 250 µg/kg/24 h is higher than the previously studied doses in the phase I study and the phase II Sections A–C (data on file); Section D will further serve to monitor and evaluate safety at this higher and fixed dose. Section D will also permit evaluation of the overall safety of rhIGF-1/rhIGFBP-3 administration by continuous IV infusion in a larger population than studied previously, including possible adverse events linked to IGF-1 treatment such as hypoglycemia, and any potential increase in the risk of sepsis associated with catheter infusion.
An additional limitation of the PK analysis was occurrence of a number of infusion interruptions among the included infants, and uncertainty of sampling around these time points. This served to highlight the importance of maintaining a continuous i.v. infusion for the duration of treatment (up to a PMA of 29 wk + 6 d), since the treated infants with the greatest number of infusion interruptions had the highest proportion of IGF-1 measurements outside target range. Through follow-up with trial investigators, we determined that a high proportion of infusion interruptions were due to the administration of other medications through a single lumen i.v. line. Subsequent education regarding the importance of maintaining a continuous infusion reduced interruptions during the remainder of the trial. It is clear, however, that due to limited line access in neonates, and the frequent need for multiple concomitant medications in the preterm population, maintaining continuous i.v. infusion can be challenging without coinfusion of drugs through the same line. In parallel with the Section D study, a comprehensive analysis of the physical and chemical compatibility of rhIGF-1/rhIGFBP-3 with other i.v. medications commonly administered in the NICU is, therefore, being conducted to confirm compatibility for coinfusion (results will be published separately).
Through the reported body of work, we have determined an effective dose of rhIGF-1/rhIGFBP-3 to achieve physiological intrauterine serum IGF-1 levels in extremely preterm infants. Generally, optimal dosing and PK are not well characterized before medicines are introduced to the neonatal population. We believe this work will help to establish a new standard for testing of drugs for utilization in infants.
Statement of Financial Support
This study was funded by Premacure AB (Uppsala, Sweden), a member of the Shire Group of Companies. J-K.C., A.T., and N.B. are employees of Shire PLC (Lexington, MA) and hold stock/stock options in Shire PLC. B.H. has received consulting fees from Premacure AB and Shire PLC. I.H-P. holds stock/stock options in Premalux AB, and has received consulting fees from Shire PLC. M.A.G. is an employee of KinderPharm/PKPD Bioscience Inc. (Exton, PA), who were paid consultants to Shire Human Genetic Therapies Inc. (Lexington, MA) in relation to this study. A.H. and D.L. hold stock/stock options in Premalux AB (Stockholm, Sweden), and have received consulting fees from Shire PLC. At the time of the study, G.F. was a consultant to and J.S. an employee of KinderPharm/PKPD Bioscience Inc., who were paid consultants to Shire Human Genetic Therapies Inc. in relation to this study. N.C.K. was an employee of Shire PLC at the time of this study and holds stock in Shire PLC.
Meeting presentations: Parts of these data have been presented at the International Society of Pharmacometrics 2014 Annual Meeting, 12 October 2014, Las Vegas, Nevada, USA; at the Pediatric Academic Societies 2014 Annual Meeting, 3 May 2014, Vancouver, British Columbia, Canada; and at the 1st Congress of joint European Neonatal Societies, 16 September 2015, Budapest, Hungary.
Clinical trial registry name and registration number: ClinicalTrials.gov, NCT01096784.
References
Blencowe H, Lawn JE, Vazquez T, Fielder A, Gilbert C. Preterm-associated visual impairment and estimates of retinopathy of prematurity at regional and global levels for 2010. Pediatr Res 2013;74(suppl 1):35–49.
Langford K, Nicolaides K, Miell JP. Maternal and fetal insulin-like growth factors and their binding proteins in the second and third trimesters of human pregnancy. Hum Reprod 1998;13:1389–93.
Hellström A, Ley D, Hansen-Pupp I, et al. Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr 2016;105:576–86.
Hellstrom A, Perruzzi C, Ju M, et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci USA 2001;98:5804–8.
Vanhaesebrouck S, Daniëls H, Moons L, Vanhole C, Carmeliet P, De Zegher F. Oxygen-induced retinopathy in mice: amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr Res 2009;65:307–10.
Hansen-Pupp I, Hellström-Westas L, Cilio CM, Andersson S, Fellman V, Ley D. Inflammation at birth and the insulin-like growth factor system in very preterm infants. Acta Paediatr 2007;96:830–6.
Hansen-Pupp I, Löfqvist C, Polberger S, et al. Influence of insulin-like growth factor I and nutrition during phases of postnatal growth in very preterm infants. Pediatr Res 2011;69(5 Pt 1):448–53.
Lineham JD, Smith RM, Dahlenburg GW, et al. Circulating insulin-like growth factor I levels in newborn premature and full-term infants followed longitudinally. Early Hum Dev 1986;13:37–46.
Hellström A, Engström E, Hård AL, et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics 2003;112:1016–20.
Löfqvist C, Engström E, Sigurdsson J, et al. Postnatal head growth deficit among premature infants parallels retinopathy of prematurity and insulin-like growth factor-1 deficit. Pediatrics 2006;117:1930–8.
Pérez-Muñuzuri A, Fernández-Lorenzo JR, Couce-Pico ML, Blanco-Teijeiro MJ, Fraga-Bermúdez JM. Serum levels of IGF1 are a useful predictor of retinopathy of prematurity. Acta Paediatr 2010;99:519–25.
Laughon MM, Benjamin DK Jr, Capparelli EV, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol 2011;4:643–52.
O’Hara K, Wright IM, Schneider JJ, Jones AL, Martin JH. Pharmacokinetics in neonatal prescribing: evidence base, paradigms and the future. Br J Clin Pharmacol 2015;80:1281–8.
Amin SB, McDermott MP, Shamoo AE. Clinical trials of drugs used off-label in neonates: ethical issues and alternative study designs. Account Res 2008;15:168–87.
Löfqvist C, Niklasson A, Engström E, et al. A pharmacokinetic and dosing study of intravenous insulin-like growth factor-I and IGF-binding protein-3 complex to preterm infants. Pediatr Res 2009;65:574–9.
Lassarre C, Hardouin S, Daffos F, Forestier F, Frankenne F, Binoux M. Serum insulin-like growth factors and insulin-like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res 1991;29:219–25.
Bang P, Westgren M, Schwander J, Blum WF, Rosenfeld RG, Stangenberg M. Ontogeny of insulin-like growth factor-binding protein-1, -2, and -3: quantitative measurements by radioimmunoassay in human fetal serum. Pediatr Res 1994;36:528–36.
Blum WF, Breier BH. Radioimmunoassays for IGFs and IGFBPs. Growth Regul 1994;4 (suppl 1):11–9.
Beal SL, Sheiner LB. NONMEM Users Guide. Ellicott City, MD: ICON Development Solutions, 1992.
Hansen-Pupp I, Hövel H, Hellström A, et al. Postnatal decrease in circulating insulin-like growth factor-I and low brain volumes in very preterm infants. J Clin Endocrinol Metab 2011;96:1129–35.
Ley D, Hansen-Pupp I, Niklasson A, et al. Longitudinal infusion of a complex of insulin-like growth factor-I and IGF-binding protein-3 in five preterm infants: pharmacokinetics and short-term safety. Pediatr Res 2013;73:68–74.
Conroy S, McIntyre J, Choonara I. Unlicensed and off label drug use in neonates. Arch Dis Child Fetal Neonatal Ed 1999;80:F142–4; discussion F4–5.
Acknowledgements
The authors thank Valérie Boissel, PhD, of Excel Scientific Solutions, who provided medical writing assistance funded by Shire Human Genetic Therapies Inc.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(ZIP 356 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chung, JK., Hallberg, B., Hansen-Pupp, I. et al. Development and verification of a pharmacokinetic model to optimize physiologic replacement of rhIGF-1/rhIGFBP-3 in preterm infants. Pediatr Res 81, 504–510 (2017). https://doi.org/10.1038/pr.2016.255
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.255
This article is cited by
-
An exploratory study of clinical factors associated with IGF-1 and IGFBP-3 in preterm infants
Pediatric Research (2024)
-
Pilot dose-ranging of rhIGF-1/rhIGFBP-3 in a preterm lamb model of evolving bronchopulmonary dysplasia
Pediatric Research (2023)
-
Assessment of compatibility of rhIGF-1/rhIGFBP-3 with neonatal intravenous medications
World Journal of Pediatrics (2023)
-
Metabolic-endocrine disruption due to preterm birth impacts growth, body composition, and neonatal outcome
Pediatric Research (2022)
