
Abstract
Background:
Necrotizing enterocolitis (NEC) is an inflammatory bowel necrosis of premature infants. Based on our recent findings of increased Smad7 expression in surgically resected bowel affected by NEC, we hypothesized that NEC macrophages undergo inflammatory activation because increased Smad7 expression renders these cells resistant to normal, gut-specific, transforming growth factor (TGF)-β-mediated suppression of inflammatory pathways.
Methods:
We used surgically resected human NEC tissue, murine models of NEC-like injury, bone marrow-derived and intestinal macrophages, and RAW264.7 cells. Smad7 and IκB kinase-beta (IKK-β) were measured by quantitative PCR, western blots, and immunohistochemistry. Promoter activation was confirmed in luciferase reporter and chromatin immunoprecipitation assays.
Results:
NEC macrophages showed increased Smad7 expression, particularly in areas with severe tissue damage and high bacterial load. Lipopolysaccharide-induced Smad7 expression suppressed TGF-β signaling and augmented nuclear factor-kappa B (NF-κB) activation and cytokine production in macrophages. Smad7-mediated NF-κB activation was likely mediated via increased expression of IKK-β, which, further increased Smad7 expression in a feed-forward loop. We show that Smad7 induced IKK-β expression through direct binding to the IKK-β promoter and its transcriptional activation.
Conclusion:
Smad7 expression in NEC macrophages interrupts TGF-β signaling and promotes NF-κB-mediated inflammatory signaling in these cells through increased expression of IKK-β.
Similar content being viewed by others

Main
Necrotizing enterocolitis (NEC) continues to be a leading cause of death among neonates born before 32 wks of gestation (1). The etiology of NEC is complex and not well understood, although current evidence suggests that NEC may occur in the preterm intestine when an altered/disrupted epithelial barrier allows bacterial translocation from the lumen into the bowel wall, where these bacteria and their products trigger an exaggerated and damaging inflammatory reaction (2).
The cellular inflammatory response in NEC lesions is marked by macrophage-rich infiltrates (3,4,5). In both the inflamed and noninflamed intestinal mucosa, macrophage populations are maintained through recruitment of circulating monocytes to the lamina propria (6,7). In the healthy mucosa, newly recruited macrophage precursors undergo inflammatory downregulation under the influence of transforming growth factor-beta (TGF-β) present in the local extracellular matrix. Intestinal macrophages display avid phagocytic and bacteriocidal activity, but do not produce cytokines upon exposure to bacterial products (8). This “inflammatory anergy” of macrophages in the uninflamed mucosa contrasts with macrophages in NEC lesions, which express a variety of cytokines and other inflammatory mediators (4). We recently showed that surgically resected bowel with NEC shows increased expression of Smad7, which is a negative regulator of TGF-β signaling (9). Based on this information, we now hypothesized that the increased Smad7 expression seen in NEC promotes inflammatory activation of NEC macrophages by rendering these cells resistant to normal TGF-β-mediated suppression of inflammatory pathways. In the following sections, we present data from human intestinal tissue samples of NEC, our previously described neonatal murine model of 2, 4, 6-trinitrobenzene sulfonic acid (TNBS)-mediated intestinal injury, which shows mucosal inflammation similar to NEC (4), and from native and genetically modified murine macrophages.
Results
Macrophages in NEC Lesions Express Smad7, Particularly in Areas With Severe Tissue Damage and High Bacterial Load
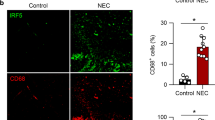
We first compared macrophages in surgically resected tissue samples of NEC (n = 8; postmenstrual age 29.1 ± 2.6 wks) with uninflamed premature intestine resected for reasons other than NEC (n = 5; postmenstrual age 27.2 ± 3 wks). Macrophages in NEC lesions, but not in the uninflamed mucosa, showed strong cytoplasmic and nuclear immunoreactivity for Smad7 ( Figure 1a ). Smad7 immunoreactivity in macrophages was most prominent in areas with severe tissue damage, which, on Brown and Brenn staining were also noted to contain the highest number of bacteria/high-power field ( Figure 1b ).

Macrophages in NEC lesions express Smad7, particularly in areas with severe tissue damage and high bacterial load. (a) Top: Uninflamed premature intestine (ileum) showing intact crypt-villus histoarchitecture (hematoxylin & eosin, magnification 25×). Fluorescence photomicrographs on the right (650×) show macrophages (red; HAM56+) with no immunoreactivity for Smad7 (green). Bottom: NEC lesion with extensive necrosis and inflammation. Fluorescence photomicrographs on the right show macrophages with strong Smad7 immunoreactivity (green). Bar = 50 μm. Data represent five controls and eight infants with NEC; (b) representative photomicrographs (650×) show Brown and Brenn staining of uninflamed premature intestine, NEC with mild tissue damage, and NEC with severe tissue damage. Bacteria were seen in NEC lesions (arrows) but not in the uninflamed mucosa. Bar = 50 μm. N = 8 infants with NEC; box-whisker plots below show (left) increasing fluorescence intensity of Smad7 staining in macrophages with severity of tissue damage; (middle) greater bacterial load in areas with severe tissue injury than in regions with no or mild tissue damage; and (right) fluorescence intensity of Smad7 staining in macrophages increased with increasing number of bacteria per high-power field. Groups compared by Kruskal–Wallis H test; *P < 0.01. NEC, necrotizing enterocolitis.
Smad7 Expression in RAW264.7 Cells and in Neonatal Gut Macrophages in NEC-Like Injury
Based on the observations in surgically resected human NEC tissue, we postulated that bacterial products such as lipopolysaccharide (LPS) drive Smad7 expression during NEC. Consistent with this hypothesis, LPS treatment of RAW264.7 murine macrophages induced Smad7 expression in these cells ( Figure 2a , b ). Similar results were obtained when RAW264.7 cells were stimulated with flagellin (Salmonella typhimurium; not depicted). We also detected increased Smad7 expression in intestinal tissue from pups with TNBS-induced enterocolitis ( Figure 2c , d ) and in macrophages isolated from affected bowel by immunomagnetic separation ( Figure 2e ). Smad7 expression in gut macrophages in TNBS enterocolitis was further confirmed by immunohistochemistry ( Figure 2f ). TNBS-mediated intestinal injury occurs only in the presence of gut microbial flora and not in germ-free mice (4), and therefore, in conjunction with those from human NEC specimens and the in-vitro models, these findings were interpreted as evidence that bacterial products induce Smad7 expression in neonatal gut macrophages during NEC. We further confirmed these findings in another murine model where NEC-like injury was induced in formula-fed mice exposed to hypoxia and hypothermia. NEC-like injury increased Smad7 expression by 2.8 ± 0.3 folds over control (P < 0.05; not depicted).

Smad7 expression in LPS-treated RAW264.7 cells and in gut macrophages in NEC-like injury: (a) LPS increasedSmad7 expression in RAW264.7 macrophages. Bar-diagram (mean values ± SEM) shows fold change in Smad7 mRNA after treatment with 0.5 μg LPS/ml × 12 h. Data represent three separate experiments; (b) western blots confirm increased Smad7 expression in LPS-treated RAW264.7 cells at the protein level. Bar diagram (mean values ± SEM) summarizes densitometric data normalized against β-actin. Data represent three separate experiments; (c) neonatal TNBSenterocolitis increased tissue expression of Smad7 mRNA. Bar diagram shows fold change in Smad7 mRNA in proximal colon from control vs. TNBS mice. N = 5 animals/group; (d) western blots confirmed increased Smad7 protein expression in TNBS colon. Bar diagram (mean values ± SEM) shows densitometric data normalized against β-actin. Data represent three separate experiments in different litters; (e) increased Smad7 expression in macrophages isolated from ileocecal region of pups with TNBS-enterocolitis. Bar diagram (mean values ± SEM) show fold change in Smad7 mRNA. N = 5 animals/group; (f) fluorescence photomicrographs (100×) show localize Smad7 immunoreactivity in F4/80+ macrophages in TNBS enterocolitis, but not in control. Bar = 50 μm. N=5 animals/group. Groups compared by Mann–Whitney U-test; *P < 0.05. LPS, lipopolysaccharide; NEC, necrotizing enterocolitis; TNBS, 2, 4, 6-trinitrobenzene sulfonic acid.
Developmental Differences in Macrophage Smad7 Expression
We have previously shown that the preterm intestine is developmentally deficient in the Ski-like protein, which is a physiological repressor of the Smad7 promoter (9,10). Therefore, we now asked whether these developmental differences extend to hematopoietic cells such as macrophages and compared Smad7 expression in BMDMs from 10-d-old mouse pups and adult animals. Unlike macrophages from adult mice, neonatal macrophages were able to upregulate Smad7 expression upon exposure to LPS ( Figure 3a ). There was no difference in constitutive Smad7 expression (not depicted). Consistent with our earlier findings, neonatal macrophages expressed less Ski-like mRNA than macrophages from adult mice ( Figure 3b ).

Developmental differences in macrophage Smad7 expression: (a) LPS induced Smad7 in BMDMs from 10-d-old mouse pups but not in macrophages isolated from adult animals. Bar diagram (mean values ± SEM) shows fold change in Smad7 mRNA expression. (b) Macrophages from pups showed lower expression of Ski-like mRNA than adults. Bar diagram shows fold changes in mRNA. Data represent three separate experiments; groups compared by the Kruskal–Wallis H test with pairwise comparisons; *P < 0.05. LPS, lipopolysaccharide.
LPS-Mediated Suppression of TGF-β Signaling in Macrophages is Mediated via Smad7
In the healthy intestinal mucosa, macrophages undergo inflammatory downregulation under the influence of TGF-β isoforms TGF-μ2 and TGF-β1 (8,11). We asked whether bacterial products present in NEC lesions could interrupt this normal, TGF-β-mediated non-inflammatory differentiation of gut macrophages. Consistent with this hypothesis, LPS treatment of RAW264.7 cells blocked TGF-β2-induced Smad2 phosphorylation ( Figure 4a ). We next used Smad7 knockdown to investigate whether these LPS effects are indeed mediated via Smad7. In these experiments, we used the SRE-reporter RAW264.7 cells, which express a TGF-β-responsive luciferase reporter construct (12) and performed transient transfection with a Smad7 shRNA plasmid to achieve Smad7 knockdown in these cells. After overnight treatment with LPS, these cells were stimulated with TGF-β2 × 1h and the relative strength of the TGF-β signal was measured as luciferase activity. Smad7 knockdown reversed LPS-mediated suppression of TGF-β signaling in macrophages ( Figure 4b ). Finally, we investigated the contribution of Smad7 to the inflammatory activation of macrophages by comparing LPS-induced cytokine expression in control and Smad7-overexpressing RAW264.7 cells. As shown in Figure 4c , cells overexpressing Smad7 produced more interleukin (IL)-1β, IL-6, tumor necrosis factor, and granulocyte macrophage-colony stimulating factor than control.

LPS-mediated suppression of TGF-β signaling in macrophages is mediated via Smad7: (a) LPS treatment inhibited TGF-β-induced Smad2 phosphorylation in RAW264.7 cells. Western blots show phospho-Smad2 and β-actin expression in RAW264.7 macrophages treated with LPS (0.5 μg/ml overnight), TGF-β2 (500 pg/ml × 30 min), and LPS (0.5 μg/ml overnight) followed by TGF-β2 (500 pg/ml, 30 min). Bar diagram (mean values ± SEM) summarizes densitometric data from three separate experiments. *P < 0.05 vs. media alone, ‡P < 0.05 vs. TGF-β2-treated cells; (b) Smad7 knockdown prior to LPS blocked LPS-mediated suppression of TGF-β signaling in macrophages. Bar diagram (mean values ± SEM) shows luciferase activity in SRE-reporter cells transfected with either control or Smad7-specific shRNA. These cells were treated overnight with LPS (or mock). Finally, we added rTGF-β2 × 1h and then measured TGF-β signaling. In the absence of LPS, Smad7 shRNA did not affect TGF-β signaling (not depicted). Data represent three separate experiments; inset: western blots confirmed Smad7 knockdown at protein level; (c) Smad7 overexpression increased LPS-induced cytokine expression in RAW264.7 cells. Bar diagrams (mean values ± SEM) show cytokine concentrations in supernatants: top left: IL-1β; top right: IL-6; bottom left: TNF; and bottom right: GM-CSF. Data represent three separate experiments. Groups compared by the Kruskal–Wallis H test with pairwise comparisons; *P < 0.05, **P < 0.01 and †P < 0.001 vs. media alone, ‡P < 0.05 vs. LPS-treated control cells. GM-CSF, granulocyte macrophage-colony stimulating factor; LPS, lipopolysaccharide; rTGF-β2, recombinant TGF-β2; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor.
Smad7 Augments LPS-Induced NF-κB Activation in Macrophages by Inducing IKK-β Expression in These Cells
Because TGF-β2 normally suppresses nuclear factor-kappa B (NF-κB)-mediated inflammatory signaling in gut macrophages (11), we next asked whether increased Smad7 expression during NEC would augment NF-κB activation in these cells. To test this hypothesis, we used the NF-κB/SEAP reporter RAW264.7 cells, which express secreted embryonic alkaline phosphatase (SEAP) activity upon NF-κB activation. These cells were transiently transfected to overexpress Smad7. Compared with control, Smad7-overexpressing macrophages showed a significant increase in LPS-induced NF-κB activation ( Figure 5a ).

Smad7 augments LPS-induced NF-κB activation in macrophages by inducing IKK-β expression in these cells: (a) Smad7 promoted LPS-mediated NF-κB activation in macrophages. Bar diagram (mean values ± SEM) shows relative NF-κB activity in control vs. Smad7-overexpressing NF-κB/SEAP reporter cells. Inset: fold change (mean ± SEM) in Smad7 expression in macrophages following transfection with pCMV-Smad7; data represent three experiments; *P < 0.05, **P < 0.01 and †P < 0.05 vs. pCMV-Smad7-transfected cells treated with media alone; (b) Smad7 overexpression in RAW264.7 macrophages induced IKK-β expression in these cells. Bar diagram (mean values ± SEM) shows fold change in mRNA expression of major NF-κB pathway genes. (c) Smad7-overexpression did not increase the expression of genes involved in TLR4-activated signaling; (d) Smad7 overexpression increased IKK-β protein expression in macrophages. Immunoblots show IKK-β expression in control and Smad7-overexpressing RAW264.7 cells. Bar diagram (mean values ± SEM) summarizes densitometric data, normalized against β-actin, from three experiments; (e) TNBS enterocolitis increased tissue expression of IKK-β. Bar diagram (mean values ± SEM) shows fold change in IKK-β mRNA in the proximal colon in TNBS-treated pups. N = 5 pups per group. Inset: increased IKK-β expression in intestinal macrophages isolated from the ileocecal region of TNBS pups; (f) Smad7 activated the IKK-β gene promoter. Bar diagram (mean values ± SEM) shows luciferase activity in control and Smad7-overexpressing RAW264.7 cells, transfected with a luciferase reporter carrying the IKK-β promoter. Data were normalized for total protein in the cell lysates; (g) rTGF-β2 suppression of LPS-induced IKK-β expression is not complete. Bar diagrams (mean values ± SEM) shows fold change in IKK-β mRNA in RAW264.7 cells in media alone, treated with LPS (0.5 μg/ml × 1 h and then cultured overnight), rTGF-β2 (500 ng/ml, overnight), or LPS stimulation × 1 h followed by rTGF-β2 overnight. *P < 0.05, **P<0.01 vs. control; †P < 0.05 vs. LPS-treated control cells. Two-group comparisons performed by the Mann–Whitney U-test; Kruskal–Wallis H test (with pairwise comparisons) used for multiple groups. IKK-β, IκB kinase-beta; LPS, lipopolysaccharide; NF-κB, nuclear factor-kappa B; SEAP, secreted embryonic alkaline phosphatase; TLR, toll-like receptor; TNBS, 2, 4, 6-trinitrobenzene sulfonic acid.
To identify the specific signaling mediator(s) through which Smad7 augments LPS-induced NF-κB activation, we used reverse transcriptase-quantitative PCR to compare control and Smad7-overexpressing RAW264.7 cells for mRNA expression of key NF-κB pathway genes. Out of 14 genes included in this panel, IKK-β was the only one to show significant induction in Smad7-overexpressing cells ( Figure 5b ). We also examined a few, well-established mediators in the toll-like receptor-4-activated signaling pathway but did not detect increased expression for genes included in this panel ( Figure 5c ). Smad7-overexpressing cells showed decreased expression of TIR-domain-containing adapter-inducing interferon-β, which is a key adaptor involved in toll-like receptor-associated delayed signaling cascades. Data from TIR-domain-containing adapter-inducing interferon-β knockout mice (13) suggest that the loss of TIR-domain-containing adapter-inducing interferon-β expression alone cannot explain increased tumor necrosis factor, IL-6, and IL-1β production by Smad7-overexpressing macrophages, and therefore, we decided to focus on IKK-β as our mediator of interest in this study.
We next confirmed increased IKK-β expression in Smad7-overexpressing RAW264.7 cells at the protein level ( Figure 5d ). In support of our findings, we detected increased IKK-β expression in intestinal tissue from pups with TNBS-induced enterocolitis and in macrophages isolated from affected bowel by immunomagnetic separation ( Figure 5e ).
To elucidate the mechanisms by which Smad7 overexpression increased IKK-β expression, we cotransfected RAW264.7 cells to overexpress Smad7 and also express a luciferase reporter driven by the IKK-β promoter. Compared with control, Smad7-overexpressing cells showed significantly greater activation of the IKK-β promoter ( Figure 5f ). Finally, to determine whether Smad7 effects on IKK-β expression were direct and specific, or were secondary to interrupted TGF-β signaling as noted in Figure 4a , b (which would presumably increase NK-κB–mediated transcription), we investigated whether TGF-β played a role in IKK-β gene expression. Treatment of RAW264.7 cells with rTGF-β2 did not suppress IKK-β expression to levels below the constitutive state. In LPS-stimulated cells, rTGF-β2 showed a significant, but partial suppression ( Figure 5g ). These findings did not convincingly exclude direct effects of Smad7 on IKK-β transcription, and therefore, we next investigated whether Smad7 could directly activate the IKK-β gene promoter. These experiments were also warranted because the onset of NEC may accentuate the developmental deficiency of TGF-β in the preterm intestine (9,11). In this low TGF-β environment, direct, non-TGF-β-mediated inflammatory effects of Smad7 remained plausible.
Smad7 Induces IKK-β Expression in Macrophages by Directly Activating its Promoter
To investigate whether the IKK-β promoter displays Smad-binding element(s), we first aligned human and murine IKK-β promoter sequences in silico using the application MULAN (14) to identify conserved regions, and then used its multi-transcription factor scan to identify Smad-binding elements. This exercise predicted at least 2 Smad-binding elements in a conserved region within 350 base pairs of the transcription start site ( Figure 6a ). Using anti-Smad7 antibody and specific primers designed to detect a 150-bp amplicon that included these two Smad-binding elements ( Table 1 ) in a ChIP assay, we demonstrated Smad7 binding to the IKK-β promoter in RAW264.7 cells ( Figure 6b ). To confirm these findings in vivo, we repeated the ChIP on intestinal tissue with TNBS-induced enterocolitis and showed Smad7 binding to the IKK-β promoter in control intestine and further enrichment of Smad7 in TNBS-induced NEC-like injury ( Figure 6c ).

Smad7 induces IKK-β expression in macrophages by directly activating its promoter: (a) In-silico analysis predicted two Smad-binding elements in human and murine IKK-β promoters; (b) Smad7 bound the IKK-β promoter in RAW264.7 macrophages. Bar diagram (mean values ± SEM) shows fold enrichment of IKK-β promoter in LPS-treated native and Smad7-overexpressing RAW264.7 cells. Data represent three experiments; (c) Smad7 binding to IKK-β promoter was confirmed in tissue samples of TNBS-enterocolitis. N = 5 animals/group; (d) Smad7 binding to the IKK-β promoter increased H4K12 acetylation on IKK-β nucleosome. Bar diagram (mean values ± SEM) show fold enrichment of IKK-β promoter in ChIP using antiacetyl-H4K12; (e) increased H4K12 acetylation of the IKK-β promoter was confirmed in tissue samples of TNBS-enterocolitis. N = 5 animals/group. Groups compared by the Mann–Whitney U-test; *P < 0.05. IKK-β, IκB kinase-beta; TNBS, 2, 4, 6-trinitrobenzene sulfonic acid.
To determine whether Smad7 binding to the IKK-β promoter actually resulted in transcriptional activation, we sought epigenetic marker(s) of euchromatin on the IKK-β nucleosome. Because Smad7 augments LPS-induced NF-κB activation in macrophages, we measured the acetylation of Lys12 residue on histone H4 (H4K12; an epigenetic modification characteristic of LPS-induced transcriptional activation) on the IKK-β nucleosome (15). We repeated the ChIP assay using antiacetyl-H4K12 and the primers for the IKK-β promoter sequence described above, which confirmed that Smad7 promoted the development of acetyl-H4K12 marks on the IKK-β nucleosome ( Figure 6d ). These findings were then confirmed in vivo by repeating the ChIP assay on intestine from pups with TNBS enterocolitis ( Figure 6e ).
IKK-β Promotes Smad7 Expression in Macrophages and Forms a Positive Feedback Loop of Inflammatory Activation
In chondrocytes, NF-κB activation can induce Smad7 expression (16). Therefore, we asked if increased IKK-β expression in inflammatory macrophages could promote Smad7 expression and thereby complete a feed-forward activation loop. Consistent with this hypothesis, transient transfection of RAW264.7 cells to overexpress IKK-β increased Smad7 at both mRNA and protein levels ( Figure 7a , b ).

IKK-β promotes Smad7 expression in macrophages and forms a positive feedback loop of inflammatory activation: (a) IKK-β overexpression induced Smad7 in macrophages. Bar diagram (mean values ± SEM) shows fold change in Smad7 mRNA in control vs. pCR-IKK-β-transfected RAW264.7 cells. Inset: fold change (mean ± SEM) in IKK-β expression in macrophages transfected with pCR-IKK-β; (b) immunoblots show Smad7 expression in LPS-treated control and IKK-β-overexpressing RAW264.7 cells. Bar diagram (mean values ± SEM) summarizes densitometric data, normalized against β-actin, from three experiments. Groups compared by the Mann–Whitney U-test; *P < 0.05. (c) Schematic showing the proposed signaling mechanisms for Smad7 effects on macrophages. IKK-β, IκB kinase-beta.
Discussion
We present a detailed investigation into the role of Smad7 in inflammatory activation of NEC macrophages. We show that bacterial products induce Smad7 expression in neonatal macrophages, which can interrupt TGF-β-mediated inflammatory downregulation of macrophages that normally occurs following recruitment to the noninflamed mucosa. Smad7 sensitizes macrophages to bacterial products and promotes NF-κB activation and cytokine production in these cells. To our knowledge, this is the first study to show that Smad7 can induce IKK-β expression in macrophages through direct binding and transcriptional activation of the IKK-β promoter. We also show that increased IKK-β expression can promote Smad7 expression, thereby setting up a feed-forward loop in the inflammatory activation of macrophages ( Figure 7c ).
In this study, we used our previously described model of TNBS-induced murine neonatal acute necrotizing enterocolitis to study NEC (4). This model was originally conceived because (i) the incidence of NEC peaks at 32 wks corrected gestational age (17); and (ii) clinical antecedents of NEC are diverse and often difficult to connect by a unifying mechanism of injury. Emphasizing the developmental predilection (over specific insults) in the pathogenesis of NEC, we postulated that the pathoanatomy of NEC represents a generic tissue injury response of the developing intestine that could be evoked nonspecifically by a variety of antecedent insults (4). In this context, we used TNBS as a nonspecific, but predictable, immunological insult to cause bowel injury in neonatal mice. The mechanisms by which TNBS induces inflammation in the neonatal intestine are not clear and likely differ from those postulated in adult animals. In adult mice, TNBS is believed to activate Th1–Th17 lymphocytes in the mucosa (18), whereas TNBS enterocolitis in pups is marked by macrophage-rich infiltrates with very few lymphocytes (4). Also, TNBS causes severe mucosal injury in C57BL/6 pups, even though adult animals of this strain are relatively resistant to TNBS (4). TNBS does not cause injury in germ-free mice, indicating that the mucosal damage requires the presence of bacteria and is not caused by direct chemical/corrosive action. As a model of NEC, TNBS-induced neonatal enterocolitis offers some important advantages: (i) the cellular inflammatory response resembles surgically resected tissue samples of NEC (4); (ii) similar to NEC (19), TNBS enterocolitis requires the presence of gut microflora (4); (iii) unlike the hypothermia-hypoxia model where individual animals develop bowel injury at different times during the NEC protocol, the temporal evolution of bowel injury can be accurately mapped from the time of TNBS administration; (iv) administration of weight-normalized doses of TNBS in mouse pups and adults can facilitate the study of developmental aspects of NEC. Infectious agents such as Enterobacter sakazakii (20) can also allow age-based comparisons, but the dose of the inciting agent (luminal Enterobacter populations) is difficult to control due to ongoing bacterial proliferation; (v) unlike models where bowel injury is induced by platelet-activating factor or splanchnic ischemia–reperfusion and the animals need to be killed within a few hours (21,22), mice subjected to TNBS enterocolitis can be observed for several days to study sequential inflammatory changes; (vi) because all pups are affected predictably, fewer animals are needed (vs. the hypoxia-hypothermia model where only 40–70% animals develop injury); and (vii) the use of 10-d-old pups allows time to test prophylactic interventions; it was convenient that the rodent intestine at birth resembles the preterm human intestine and takes 3 wks to undergo structural and functional maturation seen in the term human neonate (23,24).
Unlike macrophages from adult mice, neonatal macrophages were able to upregulate Smad7 expression upon exposure to LPS. These data are consistent with our earlier report that the gut epithelium in the premature intestine expresses Smad7 at high levels because of the developmental deficiency of the ski-like protein, which normally binds the Smad7 promoter and maintains it in state of repression (10). Ski-like protein is generally expressed at very low levels in the fetus, except for specific developmental epochs (25).
Smad7 is an important negative regulator of TGF-β signaling in the gastrointestinal tract (26). We have previously demonstrated high Smad7 expression both in the uninflamed preterm intestine and during NEC, where it blocks normal autocrine induction of TGF-β2 in epithelial cells (9). Smad7 can suppress TGF-β signaling by competing with activating Smads, interfering with TGF-β receptor function and increasing its degradation, or through epigenetic mechanisms by interacting with histone deacetylases (14,27,28). In this study, we show that Smad7 can also promote inflammation through direct activation of the IKK-β promoter in macrophages. IKK-β is an essential catalytic subunit of the IKK complex, which includes another catalytic subunit, IKK-α, and a regulatory subunit, IKK-γ. During inflammation, cytokines and bacterial products promote phosphorylation of IKK-β, which in turn, activates the IKK complex. Activated IKK phosphorylates the IκBs, triggering their degradation and thereby releasing the NF-κB dimers for nuclear translocation. In our study, Smad7 activation of the IKK-β promoter was associated with increased H4K12 acetylation, which is an epigenetic marker of euchromatin (29), on the IKK-β nucleosome. H4K12 acetylation presumably neutralizes its electrical charge, leading to structural changes that promote DNA accessibility and interactions with the H2A-H2B dimer in neighboring histones (30). In addition to Smad7 effects on IKK-β expression, we have also shown that increased IKK-β forms a feed-forward activation loop and upregulates Smad7 expression. Although we did not investigate the mechanisms for IKK-β-mediated Smad7 expression in macrophages, our findings are consistent with existing data in other cell lineages where IKK-β mediates LPS-induced phosphorylation of Rel A/p65 (31), which, in turn, can trigger Smad7 expression (16).
In conclusion, we have shown that Smad7 promotes the inflammatory activation of NEC macrophages and is a potential therapeutic target. We acknowledge that rodent models, important for proof-of-concept studies, may not always simulate pathological changes in a natural disease process. There is a need for further corroboration of these findings in clinical studies, which can provide complementary information on the role(s) of comorbidities and the clinical course, feeding experience, and the microbial flora.
Methods
Human NEC and Controls
Deidentified formalin-fixed paraffin-embedded archived human intestinal tissues were used at University of Illinois at Chicago. The Institutional Review Board at the University of Illinois at Chicago deemed this study as not involving human subjects. Sections of NEC (n = 8) were compared with healthy tissue margins resected for indications other than NEC (intestinal obstruction or spontaneous intestinal perforation (n = 5).
Mice
Animal studies were approved by the Institutional Animal Care and Use Committees at the University of Illinois at Chicago, University of South Florida, and Baylor College of Medicine. As described previously (4), TNBS was administered by gavage and rectal instillation (50 mg/kg each) to induce enterocolitis in 10-d-old C57BL/6 mice (n = 15). Animals were followed for up to 48 h and killed using CO2 inhalation followed by cervical dislocation. Control animals received vehicle alone. NEC-like injury was confirmed by histopathological analysis of ileocolic region using criteria described previously (4).
Increased Smad7 expression was confirmed in a second model of NEC-like injury (32). Briefly, newborn mice pups delivered at 18.5 d of gestation (by cesarean section) were gavage fed with infant formula (Similac PM60/40 200 mL/kg/d, Abbott Nutrition, Columbus, OH) every 3 h × 72 h. Pups were exposed to hypoxia (99% N2) × 1 min in a hypoxic chamber, followed by hypothermia (4 °C) × 10 min twice daily for 3 d. The animals were monitored every 3h and killed if they developed signs of illness or at 72 h. NEC-like injury was confirmed by histopathological analysis and tissues were used for mRNA isolation and PCR.
Cell Culture and Reagents
RAW264.7 cells (ATCC, Manassas, VA) were grown in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY) with 10% fetal calf serum. In some experiments, we added recombinant TGF-β2 (rTGF-β2; 0.5–1 ng/mL; R&D, Minneapolis, MN) or E. coli O111:B4 LPS (0.5 µg/mL; Sigma).
Plasmids
pCMV-Smad7 and pCR-IKK-β have been described previously (33,34). Smad7 shRNA plasmid sc-36508-SH (Santa Cruz Biotechnology, Santa Cruz, CA) and IKK-β promoter pEZX-PG02 luciferase reporter (GeneCopoeia, Rockville, MD) are commercially available. RAW264.7 cells were transfected using the Fugene-6 reagent (Roche, Branchburg, NJ) and used at 24 h post-transfection (predetermined optimum).
Brown and Brenn Staining and Immunohistochemistry
Bacteria were identified in intestinal tissue sections using the Brown and Brenn stain. We used our previously described immunofluorescence protocol to stain human/murine tissues for macrophage markers and Smad7 (9). After antigen retrieval (EZ-AR solution, Biogenex, San Remon, CA), tissue sections were blocked (SuperBlock, Thermo Scientific, Rockford, IL) and incubated overnight at 4 °C with goat polyclonal anti-Smad7 IgG (Santa Cruz) along with either mouse monoclonal anti-HAM56 IgM (Ebioscience, Santa Cruz, CA) or rat monoclonal anti-mouse F4/80 IgG2a (Ebioscience), Secondary staining was performed with Alexa Flour-conjugated antibodies (Invitrogen, San Diego, CA). Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI, Sigma). Imaging was performed using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Thornwood, NY).
Reverse Transcriptase-Quantitative PCR
Primers ( Table 1 ) were designed using the Beacon Design software (Bio-Rad, Hercules, CA). We used a standard reverse transcriptase reaction and a SYBR green-based method to measure mRNA expression (35). Data were normalized against 18S ribosomal RNA.
Western Blots
Smad7, phospho-Smad2 (Thr220), and IKK-β expression (antibodies from Santa Cruz) were measured by western blots (9). Data were normalized against β-actin.
Cytokine Concentrations
A magnetic bead-based 4-plex assay (Life Technologies) was used to measure IL-1β, IL-6, tumor necrosis factor, and granulocyte macrophage-colony stimulating factor.
Intestinal and Bone Marrow-Derived Macrophages
Murine intestinal macrophages were isolated by immunomagnetic cells separation. Briefly, intestinal tissue was digested with HBSS containing 1 mM collagenase type IV (Sigma) × 2 h at 37 °C. Macrophages were first stained with phycoerythrin-conjugated anti-Ly6C and anti-F4/80 antibodies and then separated using antiphycoerythrin-conjugated ferromagnetic beads (Miltenyi Biotec, San Diego, CA). More than 90% of these cells were confirmed as CD11b+ F4/80+ CD11cint macrophages by FACS.
BMDMs were prepared (36) by culturing bone marrow cells in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum and 40 ng/ml monocyte-colony stimulating factor (PeproTech, Rocky Hill, NJ). After 5 d, BMDMs were collected by scraping and cultured in RPMI with 10% fetal calf serum.
TGF-β Signal Transduction
SRE-reporter cells (RAW264.7 cells stably transduced to express a Smad-response element) (12) were transfected to express Smad7 shRNA (above). Smad7-knocked down SRE cells were first treated overnight with E. coli LPS (or mock) and then stimulated with TGF-β2 (500 ng/ml) × 1h. Luciferase activity was measured using a commercially available kit (GloMax, Promega, Madison, WI).
NF-κB Reporter Cells
NF-κB activation was measured using NF-κB/SEAP reporter RAW264.7 cells (Imgenex, San Diego, CA), which show NF-κB-driven expression of SEAP (37). These reporter cells were transfected with pCMV-Smad7 to overexpress Smad7. Cells were stimulated with LPS × 1h after which the media were replaced and SEAP expression was measured after 18 h per manufacturer’s protocol.
In-silico Promoter Analysis
Murine and human IKK-β promoter sequences were retrieved from the transcriptional regulatory element database (38) and were aligned using the online application MULAN to identify conserved regions and Smad-binding elements (14,39).
Chromatin Immunoprecipitation (ChIP)
Chromatin was harvested from RAW246.7 cells or tissue samples using the Magnify ChIP system (Invitrogen). Cross-linked, sheared chromatin was used for immunoprecipitation with protein A/G dynabeads coupled with anti-Smad7, rabbit antiacetyl-histone H4 Lys12 (H4K12Ac) IgG (EMD Millipore, Billerica, MA) or isotype control from a different species (rabbit). Precipitated DNA was reverse crosslinked and amplified by PCR using primers specific for IKK-β promoter ( Table 1 ). Data were expressed as fold change after normalization for background levels (9).
Statistical Methods
Two-tailed parametric and nonparametric tests were applied using SPSS for Windows (version 17.0, SPSS, Chicago, IL). A p value of <0.05 was accepted as significant.
Statement of Financial Support
Eunice Kennedy Shriver National Institutes of Child Health and Human Development, Bethesda, MD, award R01HD059142 (to A.M.) and National Heart, Lung, and Blood Institute, Bethesda, MD, award R01HL102171 (to S.M.T.).
Disclosure
The authors disclose no conflicts.
References
Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med 2011;364:255–64.
Maheshwari A. Immunologic and hematological abnormalities in necrotizing enterocolitis. Clin Perinatol 2015;42:567–85.
Remon JI, Amin SC, Mehendale SR, et al. Depth of bacterial invasion in resected intestinal tissue predicts mortality in surgical necrotizing enterocolitis. J Perinatol 2015;35:755–62.
MohanKumar K, Kaza N, Jagadeeswaran R, et al. Gut mucosal injury in neonates is marked by macrophage infiltration in contrast to pleomorphic infiltrates in adult: evidence from an animal model. Am J Physiol Gastrointest Liver Physiol 2012;303:G93–102.
Ballance WA, Dahms BB, Shenker N, Kliegman RM. Pathology of neonatal necrotizing enterocolitis: a ten-year experience. J Pediatr 1990;117:S6–13.
Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 2014;15:929–37.
Smythies LE, Maheshwari A, Clements R, et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol 2006;80:492–9.
Smythies LE, Sellers M, Clements RH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 2005;115:66–75.
Namachivayam K, Blanco CL, MohanKumar K, et al. Smad7 inhibits autocrine expression of TGF-β2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2013;304:G167–80.
Briones-Orta MA, Sosa-Garrocho M, Moreno-Alvarez P, Fonseca-Sánchez MA, Macías-Silva M. SnoN co-repressor binds and represses smad7 gene promoter. Biochem Biophys Res Commun 2006;341:889–94.
Maheshwari A, Kelly DR, Nicola T, et al. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011;140:242–53.
Namachivayam K, Blanco CL, Frost BL, et al. Preterm human milk contains a large pool of latent TGF-β, which can be activated by exogenous neuraminidase. Am J Physiol Gastrointest Liver Physiol 2013;304:G1055–65.
Power MR, Li B, Yamamoto M, Akira S, Lin TJ. A role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol 2007;178:3170–6.
Ovcharenko I, Loots GG, Giardine BM, et al. Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res 2005;15:184–94.
Aung HT, Schroder K, Himes SR, et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J 2006;20:1315–27.
Baugé C, Attia J, Leclercq S, Pujol JP, Galéra P, Boumédiene K. Interleukin-1beta up-regulation of Smad7 via NF-kappaB activation in human chondrocytes. Arthritis Rheum 2008;58:221–6.
Sharma R, Hudak ML, Tepas JJ 3rd, et al. Impact of gestational age on the clinical presentation and surgical outcome of necrotizing enterocolitis. J Perinatol 2006;26:342–7.
Alex P, Zachos NC, Nguyen T, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis 2009;15:341–52.
Hsueh W, Caplan MS, Tan X, MacKendrick W, Gonzalez-Crussi F. Necrotizing enterocolitis of the newborn: pathogenetic concepts in perspective. Pediatr Dev Pathol 1998;1:2–16.
Hunter CJ, Singamsetty VK, Chokshi NK, et al. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis 2008;198:586–93.
Liu SX, Tian R, Baskind H, Hsueh W, De Plaen IG. Platelet-activating factor induces the processing of nuclear factor-kappaB p105 into p50, which mediates acute bowel injury in mice. Am J Physiol Gastrointest Liver Physiol 2009;297:G76–81.
Maheshwari A, Christensen RD, Calhoun DA, Dimmitt RA, Lacson A. Circulating CXC-chemokine concentrations in a murine intestinal ischemia-reperfusion model. Fetal Pediatr Pathol 2004;23:145–57.
Walthall K, Cappon GD, Hurtt ME, Zoetis T. Postnatal development of the gastrointestinal system: a species comparison. Birth Defects Res B Dev Reprod Toxicol 2005;74:132–56.
Nanthakumar NN, Dai D, Meng D, Chaudry N, Newburg DS, Walker WA. Regulation of intestinal ontogeny: effect of glucocorticoids and luminal microbes on galactosyltransferase and trehalase induction in mice. Glycobiology 2005;15:221–32.
Jahchan NS, Luo K. SnoN in mammalian development, function and diseases. Curr Opin Pharmacol 2010;10:670–5.
Monteleone G, Pallone F, MacDonald TT. Smad7 in TGF-beta-mediated negative regulation of gut inflammation. Trends Immunol 2004;25:513–7.
Yan X, Lin Z, Chen F, et al. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J Biol Chem 2009;284:30097–104.
Zhang S, Fei T, Zhang L, et al. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol 2007;27:4488–99.
Sasaki K, Ito T, Nishino N, Khochbin S, Yoshida M. Real-time imaging of histone H4 hyperacetylation in living cells. Proc Natl Acad Sci USA 2009;106:16257–62.
Ausio J, van Holde KE. Histone hyperacetylation: its effects on nucleosome conformation and stability. Biochemistry 1986;25:1421–8.
Yang F, Tang E, Guan K, Wang CY. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 2003;170:5630–5.
Premkumar MH, Sule G, Nagamani SC, et al. Argininosuccinate lyase in enterocytes protects from development of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2014;307:G347–54.
Eliseev RA, Schwarz EM, Zuscik MJ, O’Keefe RJ, Drissi H, Rosier RN. Smad7 mediates inhibition of Saos2 osteosarcoma cell differentiation by NFkappaB. Exp Cell Res 2006;312:40–50.
Nakano H, Shindo M, Sakon S, et al. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci USA 1998;95:3537–42.
Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat Protoc 2006;1:1559–82.
Xu D, Wang S, Liu W, Liu J, Feng X. A novel receptor activator of NF-kappaB (RANK) cytoplasmic motif plays an essential role in osteoclastogenesis by committing macrophages to the osteoclast lineage. J Biol Chem 2006;281:4678–90.
Namachivayam K, Coffing HP, Sankaranarayanan NV, et al. Transforming growth factor-β2 is sequestered in preterm human milk by chondroitin sulfate proteoglycans. Am J Physiol Gastrointest Liver Physiol 2015;309:G171–80.
Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 2007;35:D137–40.
Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev 2005;19:2783–810.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
MohanKumar, K., Namachivayam, K., Chapalamadugu, K. et al. Smad7 interrupts TGF-β signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr Res 79, 951–961 (2016). https://doi.org/10.1038/pr.2016.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.18
This article is cited by
-
Bench to bedside — new insights into the pathogenesis of necrotizing enterocolitis
Nature Reviews Gastroenterology & Hepatology (2022)
-
Colonization of fecal microbiota from patients with neonatal necrotizing enterocolitis exacerbates intestinal injury in germfree mice subjected to necrotizing enterocolitis-induction protocol via alterations in butyrate and regulatory T cells
Journal of Translational Medicine (2021)
-
Role of platelets in neonatal necrotizing enterocolitis
Pediatric Research (2021)
-
Role of macrophages in fetal development and perinatal disorders
Pediatric Research (2021)
-
Intestinal dysbiosis and necrotizing enterocolitis: assessment for causality using Bradford Hill criteria
Pediatric Research (2020)
