
Abstract
Background:
Endothelin-1 (ET-1) and Rho-kinase (ROCK) increase vascular tone in experimental persistent pulmonary hypertension of the newborn (PPHN). Whether ET-1 activates ROCK to decrease angiogenesis in the developing lung remains unknown.
Methods:
Proximal pulmonary artery endothelial cells (PAECs) were harvested from fetal sheep after partial ligation of the ductus arteriosus in utero (PPHN) and controls. Growth and tube formation were assessed after ET-1 treatment. The effect of ET-1 antagonism on tube formation was studied using ET-1 small interfering RNA (siRNA), ET-1 monoclonal antibodies (ET-1mAbs), BQ-123 (an ETA blocker), and bosentan (an ETA/ETB blocker). ET-1 gene and protein and ETA/ETB receptor protein expression were measured in normal and PPHN PAECs. ET-1–ROCK interactions were assessed by measuring ROCK activity after ET-1, ET-1 siRNA, and bosentan treatments, and tube formation with ET-1 and Y-27632 (ROCK inhibitor).
Results:
ET-1 did not affect growth but decreased tube formation in normal and PPHN PAECs. ET-1 protein and gene expression were increased and ETB receptor protein decreased in PPHN PAECs. ET-1 siRNA, ET-1mAbs, and bosentan, but not BQ-123, increased tube formation. ROCK activity was increased in PPHN PAECs and decreased with ET-1 siRNA and bosentan treatments. Y-27632 prevented the decrease in tube formation with ET-1.
Conclusion:
ET-1 activation of ROCK impairs angiogenesis of fetal PAECs. Disruption of ET-1–ROCK interactions may increase vascular growth in PPHN.
Similar content being viewed by others

Main
Persistent pulmonary hypertension of the newborn (PPHN) is a clinical syndrome characterized by elevated pulmonary vascular resistance (PVR) that persists after birth (1). Mechanisms contributing to increased PVR in PPHN include increased vascular tone, hypertensive remodeling, and, in the most severe cases, impaired angiogenesis (2). Decreased vascular growth often occurs in the setting of PPHN with lung hypoplasia, such as that in infants with congenital diaphragmatic hernia (2,3). In the presence of lung hypoplasia, decreased arterial number plays a prominent role in maintaining high PVR and contributes to increased mortality. Although inhaled nitric oxide is effective in treating many newborns with persistent pulmonary hypertension, PPHN with impaired angiogenesis is often refractory to nitric oxide therapy (4). Novel strategies that can stimulate vascular growth and increase arterial number may improve long-term outcomes; however, due to the lack of developmentally relevant models, mechanisms that impair angiogenesis and enhance lung vascular growth in severe PPHN remain poorly understood.
Previous studies have shown that partial ligation of the ductus arteriosus (DA) in late-gestation fetal sheep provides a useful animal model for studying the pathogenesis and treatment of PPHN (5). In this model, partial DA ligation increases pulmonary artery pressure without causing sustained elevations of pulmonary blood flow or hypoxemia (5). PVR remains elevated at birth, with extrapulmonary shunting and hypoxemia (5). Along with changes in vascular tone, DA ligation impairs lung vascular growth in vivo and causes sustained abnormalities of pulmonary artery endothelial cell (PAEC) growth and tube formation in vitro (6,7). The mechanism by which hemodynamic stress induced by hypertension disrupts normal endothelial cell signaling and function and impairs vascular growth in PPHN is unknown.
In adult models of experimental pulmonary hypertension, high endothelin-1 (ET-1) activity increases vascular tone and promotes hypertensive structural remodeling with smooth muscle proliferation (8,9,10). ET-1 is strongly expressed in the developing lung and maintains high PVR in the normal fetus (11,12). ET-1 has also been implicated in the pathogenesis of PPHN in experimental models and the clinical setting. PPHN in fetal sheep caused by DA ligation is characterized by increased lung ET-1 content and altered ET receptor expression and is prevented by treatment with a selective ETA receptor antagonist (13,14). In addition, human newborns with severe persistent pulmonary hypertension have high circulating ET-1 levels (15), and in infants who died from congenital diaphragmatic hernia, lung ET-1 levels are also markedly increased (3,16). These studies demonstrate the importance of ET-1 in the pathogenesis of severe PPHN, especially in the setting of lung hypoplasia.
Although most studies have focused on the effects of ET-1 on vascular smooth muscle, previous studies of tumor angiogenesis and human umbilical vein endothelial cells have suggested a proangiogenic effect of ET-1 on endothelial cells (17,18). ET-1 levels are markedly increased in clinical and experimental PPHN, but whether ET-1 regulates angiogenesis in the developing lung and alters angiogenesis in severe PPHN has not been previously studied.
Rho-kinase (ROCK) is a complex signaling pathway responsible for cellular proliferation, migration, barrier function, differentiation, and gene expression in diverse vascular beds (19,20). ROCK activity maintains high PVR and contributes to the myogenic response in the normal fetal lung (21,22). In experimental models of neonatal and adult pulmonary hypertension, high ROCK activity causes high vascular tone, increased myogenic reactivity, and hypertensive vascular remodeling (20,22,23,24). ET-1 is an upstream regulator of ROCK activity (25), and previous reports have suggested that activation of ROCK and excessive ROCK activity impair PAEC function in vitro (26). ET-1 levels and ROCK activity are increased in diverse models of pulmonary hypertension, but whether ET-1 activation of ROCK causes endothelial cell dysfunction and impaired angiogenesis in PPHN remains unknown.
Therefore, we hypothesized that ET-1 impairs angiogenesis in experimental PPHN and that these effects are mediated through the activation of ROCK. In this study, we report increased ET-1 production by PAECs from PPHN fetal sheep and that ET-1 activation of ROCK decreases tube formation in vitro. These findings support the hypothesis that ET-1–ROCK interactions contribute to endothelial dysfunction and decreased vascular growth in PPHN.
Results
ET-1 Decreases Three-Dimensional Tube Formation in Normal PAECs
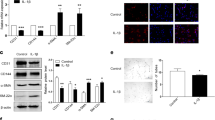
Growth and tube formation were assessed in normal fetal PAECs with and without ET-1 treatment (100 nmol/l). ET-1 treatment had no effect on cell growth ( Figure 1a ) but decreased tube formation in normal PAECs. ET-1 treatment decreased tube length by 15% (P < 0.01) and branch points per high-power field (HPF) by 32% (P < 0.01) in normal fetal PAECs ( Figure 1b ).

Decreased three-dimensional tube formation by normal PAECs after ET-1 treatment. (a) Growth and (b,c) tube formation were assessed in normal fetal PAECs with and without ET-1 treatment (100 nmol/l). (a) ET-1 treatment (black triangles, dashed line) had no effect on cell growth as compared with untreated controls (black squares, continuous line), but (b) decreased tube formation in normal PAECs (×10 original magnification). (c) ET-1 treatment decreased tube length by 15% (*P < 0.01) and branch points per HPF by 32% (*P < 0.01) in normal fetal PAECs. ET-1, endothelin-1; HPF, high-power field; PAEC, pulmonary artery endothelial cell.
ET-1 Decreases Three-Dimensional Tube Formation in PPHN PAECs
Growth and tube formation were assessed in PPHN fetal PAECs with and without ET-1 treatment. ET-1 treatment had no effect on PPHN PAEC growth ( Figure 2a ). Tube formation by PPHN PAECs was significantly decreased as compared with age-matched controls. Tube length was decreased by 27% (P < 0.01) and the number of branch points per HPF was decreased by 32% (P < 0.01) in PPHN PAECs. ET-1 treatment (100 nmol/l) further decreased tube length by 24% (P < 0.05) and the number of branch points per HPF by 28% (P < 0.05) in PPHN fetal PAECs ( Figure 2b ).

Decreased three-dimensional tube formation by PAECs from PPHN fetal sheep after ET-1 treatment. (a) Growth and (b,c) tube formation were assessed in PPHN fetal PAECs with and without ET-1 treatment. (a) ET-1 treatment (black triangles, dashed line) had no effect on PPHN PAEC growth as compared with untreated PPHN controls (black squares, continuous line). (b) Tube formation by PPHN PAECs was significantly decreased as compared with age-matched controls (×10 original magnification). (c) Tube length was decreased by 27% (*P < 0.01) and the number of branch points per HPF was decreased by 32% (*P < 0.01) in PPHN PAECs. ET-1 treatment (100 nmol/l) further decreased tube length by 24% (**P < 0.05) and number of branch points per HPF by 28% (**P < 0.05) in PPHN fetal PAECs (as shown in b,c). ET-1, endothelin-1; HPF, high-power field; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn.
Increased ET-1 Production by PPHN PAECs
As compared with controls, ET-1 protein expression in the cell supernatant increased twofold ( Figure 3a ) and ET-1 mRNA expression increased by 47% ( Figure 3b ) in PPHN PAECs.

Increased ET-1 gene and protein expression in PAECs from PPHN fetal sheep. ET-1 mRNA and protein expression were measured in PAECs from normal and PPHN fetal sheep. (a) ET-1 protein expression increased by 100% (**P < 0.05) and (b) ET-1 mRNA expression increased by 47% (*P < 0.01) in PPHN fetal PAECs. ET-1, endothelin-1; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn.
Effect of ET-1 Inhibition on Tube Formation in Normal and PPHN Fetal PAECs
Tube formation was assessed in normal and PPHN PAECs after exposure to ET-1 monoclonal antibodies (ET-1mAbs) and ET-1 small interfering RNA (siRNA) (4 µmol/l). ET-1mAbs bind to ET-1 and prevents binding to ETA and ETB receptors. ET-1mAbs increased tube length by 36% (P < 0.05 for each comparison) ( Figure 4a ) and the number of branch points per HPF by 53% and 56% (P < 0.05 for each comparison) in normal and PPHN PAECs, respectively ( Figure 4a ). ET-1 siRNA decreased ET-1 mRNA expression by 50% and 54% in normal and PPHN PAECs, respectively ( Figure 4b ). ET-1 siRNA increased tube length by 32% and 46% (both P < 0.01) ( Figure 4c ) and the number of branch points per HPF by 53% and 61% (both P < 0.01) in normal and PPHN fetal PAECs, respectively ( Figure 4d ). Both ETmAbs and ET-1 siRNA restored tube formation by PPHN fetal PAECs to similar values seen in normal untreated controls.

ET-1 inhibition increases tube formation in normal and PPHN fetal PAECs. Tube formation was assessed in normal and PPHN PAECs afterexposure to (a) ET-1mAbs and (c,d) ET-1 siRNA (4 µmol/l). ET-1mAbs bind to ET-1 and prevent binding to ETA and ETB receptors. (a) With ET-1mAb exposure, tube length increased by 36% (**P < 0.05 for each comparison) and number of branch points per HPF by 53% and 56% (**P < 0.05 for each comparison) in normal and PPHN PAECs, respectively. (b) ET-1 siRNA decreased ET-1 mRNA expression by 50% and 54% in normal and PPHN PAECs, respectively, and (c) increased tube length by 32% and 46%, respectively (*P < 0.01 for each comparison). (d) ET-1 siRNA increased the number of branch points per HPF by 53% and 61% (*P < 0.01 for each comparison) in normal and PPHN fetal PAECs, respectively. Both ET-1mAbs and ET-1 siRNA restored tube formation by PPHN fetal PAECs to values similar to those seen in normal untreated fetal PAECs. ET-1, endothelin-1; HPF, high-power field; mAb, monoclonal antibody; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn; siRNA, small-interfering RNA.
Decreased ETB Receptor Protein by PAECs From PPHN Fetal Sheep
ETB receptor protein was measured in whole-cell lysates from normal and PPHN PAECs (n = 4 clones) by western blot analysis. ETB receptor protein expression was decreased by 48% (P < 0.01) in PAECs from PPHN fetal sheep ( Figure 5a ). ETA receptor protein was detected in pulmonary artery smooth muscle cells but not in PAEC whole-cell lysates, demonstrating absence of ETA receptor expression in PAECs ( Figure 5b ). Twenty micrograms of protein was loaded per lane, and differences in β-actin protein expression were noted between pulmonary artery smooth muscle cells and PAECs.

Decreased ETB protein by PPHN PAECs and absence of ETA receptor expression in normal and PPHN fetal PAECs. ETB receptor protein was measured in whole-cell lysates from normal and PPHN PAECs (n = 4 clones) by western blot analysis. (a) ETB receptor protein expression was decreased by 48% (*P < 0.01) in PAECs from PPHN fetal sheep. (b) ETA receptor protein was detected in PASMC but not in PAEC whole-cell lysates, demonstrating absence of ETA receptor expression in PAECs. A total of 20 µg of protein was loaded per lane and differences in β-actin protein expression were noted between PASMCs and PAECs. ET, endothelin; PAEC, pulmonary artery endothelial cell; PASMC, pulmonary artery smooth muscle cell; PPHN, persistent pulmonary hypertension of the newborn.
Effect of ETA and Combined ETA/ETB Receptor Blockade on Tube Formation in Normal and PPHN Fetal PAECs
Tube formation was assessed in normal and PPHN PAECs in the presence of ETA receptor blockade (BQ-123, 1 µmol/l) and combined ETA/ETB receptor blockade (bosentan, 1 µmol/l) with and without ET-1 (100 nmol/l) treatment. BQ-123 alone had no effect on tube length ( Figure 6a ) or number of branch points per HPF in normal and PPHN PAECs ( Figure 6b ). BQ-123 did not prevent the decrease in tube length and number of branch points per HPF with ET-1 treatment. Tube length was decreased by 49% (P < 0.01) in normal PAECs and 26% (P < 0.05) in PPHN PAECs ( Figure 6a ) and branch points per HPF were decreased by 27% in normal and 34% in PPHN PAECs ( Figure 6b ) (P < 0.05 for each comparison). Bosentan alone increased tube length by 35% and 42% (P < 0.01 for each comparison) ( Figure 6c ) and increased the number of branch points per HPF by 45% and 62% (P < 0.01 for each comparison) in normal and PPHN PAECs, respectively ( Figure 6d ). Bosentan prevented the decrease in tube length and number of branch points per HPF with ET-1 treatment. When combination treatment with bosentan and ET-1 was used, tube length was increased by 42% (P < 0.01) in normal PAECs and 32% (P < 0.05) in PPHN PAECs ( Figure 6c ), and the number of branch points per HPF was increased by 48% in normal and 61% in PPHN PAECs ( Figure 6d ) (P < 0.05 for each comparison). Bosentan either alone or in combination with ET-1 treatment restored PPHN PAEC tube formation to values seen in normal untreated controls.

Effect of ETA receptor blockade and nonselective ETA/ETB receptor blockade on tube formation in normal and PPHN fetal PAECs. Tube formation was assessed in normal and PPHN PAECs (a,b) in the presence of selective ETA receptor blockade (BQ-123, 1 µmol/l) and (c,d) nonselective ETA/ETB receptor blockade (bosentan, 1 µmol/l) with and without ET-1 (100 nmol/l) treatment. BQ-123 alone had no effect on tube length (in a) and number of branch points per HPF (in b) in normal and PPHN PAECs and did not prevent the decrease in tube length (in a) and number of branch points per HPF (in b) with ET-1 treatment. Tube length was decreased by 49% (*P < 0.01) and 26% (**P < 0.05) (in a) and branch points per HPF by 27% and 34% (in b) in normal and PPHN PAECs, respectively (**P < 0.05 for each comparison). Bosentan alone increased tube length by 35% and 42% (in c) (**P < 0.01 for each comparison) and number of branch points per HPF by 45% and 62% (in d) (*P < 0.01 for each comparison) in normal and PPHN PAECs, respectively. Bosentan also prevented the decrease in tube length and number of branch points per HPF with ET-1 treatment (in c,d). With bosentan and ET-1 treatment in combination, tube length was increased by 42% (*P < 0.01) and 32% (**P < 0.05) (in c) and branch points per HPF increased by 48% and 61% (in d) in normal and PPHN PAECs, respectively (**P < 0.05 for each comparison). ET, endothelin; HPF, high-power field; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn.
Effect of ET-1 and ET-1 siRNA on ROCK Activity in Normal and PPHN Fetal PAECs
The effect of ET-1 treatment (100 nmol/l) and ET-1 siRNA (4 µmol/l) on ROCK activity was measured by MYPT-1 and phosphorylated-MYPT-1 protein by western blot analysis in normal and PPHN PAEC whole-cell lysates (n = 4 clones). ROCK activity was increased by 65% in PPHN PAECs (P < 0.01) and by 44% in normal and 55% in PPHN PAECs with ET-1 treatment ( Figure 7a ) (P < 0.01 for each comparison). ET-1 increased ROCK activity in normal PAECs to values similar to those seen in untreated PPHN PAECs. ET-1 siRNA treatment decreased phosphorylated-MYPT-1:MYPT-1 protein ratio by 35% in normal and PPHN PAECs ( Figure 7b ) (P < 0.01 for each comparison), decreasing ROCK activity to levels seen in normal untreated PAECs.

ET-1 Rho-kinase interaction in normal and PPHN fetal PAECs. (a) ET-1 increases Rho-kinase activity in normal and PPHN fetal PAECs. Effect of ET-1 treatment (100 nmol/l) on Rho-kinase activity was measured by MYPT-1 and phosphorylated-MYPT-1 (p-MYPT-1) protein by western blot analysis in normal and PPHN PAEC whole-cell lysates (n = 4 clones). Rho-kinase activity increased by 44% in normal and 55% in PPHN PAECs with ET-1 treatment (*P < 0.01 for each comparison). (b) ET-1 siRNA decreases Rho-kinase activity in normal and PPHN PAECs. The effect of ET-1 siRNA (4 µmol/l) on Rho-kinase activity was measured by MYPT-1 and p-MYPT-1 protein by western blot analysis in normal and PPHN PAEC whole-cell lysates (n = 4 clones). p-MYPT-1:MYPT-1 protein ratio decreased by 35% in normal and PPHN PAECs with ET-1 siRNA treatment (*P < 0.01 for each comparison). (c) Rho-kinase inhibition prevents the decreases in three-dimensional tube formation by normal PAECs and rescues the abnormal in vitro PPHN phenotype after ET-1 treatment. Three-dimensional tube formation was assessed in normal and PPHN PAECs in the presence and absence of ET-1 treatment (100 nmol/l) with and without Y-27632 (1 μmol/l) (Rho-kinase inhibitor). After ET-1 treatment, Y-27632 increased tube length by 25% (**P < 0.05 vs. ET-1 treatment alone) and the number of branch points per HPF by 32% (*P < 0.01 vs. ET-1 treatment alone) in normal PAECs, restoring tube formation to values seen in normal PAECs. In PPHN PAECs, Y-27632 increased tube length by 75% (*P < 0.01 vs. ET-1 treatment alone) and number of branch points per HPF by 95% (**P < 0.01 vs. ET-1 treatment alone), restoring tube formation to values seen in normal PAECs. ET-1, endothelin-1; HPF, high-power field; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn; siRNA, small-interfering RNA.
Effect of ROCK Inhibition on Tube Formation After ET-1 Treatment in Normal and PPHN PAECs
Y-27632 blocked the effect of ET-1 treatment on three-dimensional tube formation in both normal and PPHN PAECs. After ET-1 treatment, Y-27632 increased tube length by 25% (P < 0.05) ( Figure 7c ) and branch points per HPF by 32% (P < 0.01) in normal PAECs, restoring tube formation to normal ( Figure 7c ). In PPHN PAECs, Y-27632 increased tube length by 75% (P < 0.01) ( Figure 7c ) and branch points per HPF by 95% (P < 0.01) ( Figure 7c ), achieving similar values as measured in normal PAECs ( Figure 7c ). Y-27632 alone increased tube length by 13 (P < 0.05) and 36% (P < 0.01) and branch points per HPF by 18 (P < 0.05) and 44% (P < 0.01) in normal and PPHN PAECs, respectively (data not shown).
Effect of Bosentan on ROCK Activity in Normal and PPHN PAECs
The effect of bosentan on ROCK activity was measured by MYPT-1 and phosphorylated-MYPT-1 protein by western blot analysis in normal and PPHN PAEC whole-cell lysates (n = 4 clones) with and without ET-1 (100 nmol/l) treatment. In normal PAECs, bosentan decreased the phosphorylated-MYPT-1:MYPT-1 protein ratio by 39% and 41% with and without ET-1 treatment, respectively (P < 0.05 and P < 0.01, respectively), whereas in PPHN PAECs, the ratio was decreased by 36% and 32% with and without ET-1 treatment, respectively (P < 0.01 for both comparisons) ( Figure 8 ). Both in the presence and absence of ET-1 treatment, bosentan decreased ROCK activity to levels similar to those seen in normal untreated controls.

Effect of bosentan on Rho-kinase activity in normal and PPHN PAECs. The effect of bosentan on Rho-kinase activity was measured by MYPT-1 and phosphorylated-MYPT-1 (p-MYPT-1) protein by western blot analysis in normal and PPHN PAEC whole-cell lysates (n = 4 clones) with and without ET-1 (100 nmol/l) treatment. Bosentan decreased p-MYPT-1:MYPT-1 protein ratio by 39% (*P < 0.01 vs. control) with ET-1 treatment and by 41% (**P < 0.05 vs. control) without ET-1 treatment in normal PAECs and by 36% (*P < 0.01 vs. control) with ET-1 treatment and by 32% (*P < 0.01 vs. control) without ET-1 treatment in PPHN PAECs. ET-1, endothelin-1; PAEC, pulmonary artery endothelial cell; PPHN, persistent pulmonary hypertension of the newborn.
Discussion
We report that ET-1 had no effect on PAEC growth, but ET-1 decreased tube formation in normal and PPHN fetal PAECs. In comparison with control PAECs, tube formation by PPHN PAECs was decreased. ET-1 protein and gene expression were increased and ETB receptor protein decreased in PPHN PAECs. Treatment of PPHN PAECs with ET-1 siRNA, ET-1mAbs, and bosentan, but not BQ-123, increased tube formation. As compared with controls, ROCK activity was increased in PPHN PAECs and was decreased with ET-1 siRNA and bosentan treatments. Although these findings are from in vitro studies, they suggest that ET-1 directly alters PAEC function and impairs angiogenesis of fetal PAECs through ROCK activation and that disruption of ET-1–ROCK interactions may ameliorate endothelial dysfunction and increase vascular growth in severe PPHN.
In addition to increased vascular tone and marked hypertensive remodeling of the vascular wall, decreased arterial density also contributes to high PVR in severe PPHN, especially in the setting of lung hypoplasia (2,4,6,7,26). Earlier studies have shown that pulmonary hypertension during late gestation impairs fetal lung vascular growth in vivo (7) and causes abnormalities in PAECs that persist in vitro (6). The PAEC PPHN phenotype is characterized by decreased growth and tube formation, altered vascular endothelial growth factor–nitric oxide signaling, and increased ROCK activity (6,26). Although these findings provide some insights into mechanisms responsible for impaired angiogenesis in severe PPHN, the mechanism by which sustained elevations in pulmonary arterial pressure inhibit lung angiogenesis during development is poorly understood. ET-1 contributes to the development of pulmonary hypertension (8,9,10,13,14), but whether ET-1 regulates angiogenesis in the developing lung and contributes to impaired angiogenesis in PPHN has not been previously studied.
ET-1 has been implicated in the pathogenesis of diverse neonatal diseases. Newborn infants with meconium aspiration, respiratory distress syndrome, birth asphyxia, or sepsis all have increased serum ET-1 levels, which correlate with disease severity (27,28,29). ET-1 also contributes to the development of bronchopulmonary dysplasia, a chronic lung disease of infancy characterized by decreased alveolar and vascular growth. In newborn infants with respiratory distress syndrome, the degree of elevation in plasma ET-1 concentrations predicted higher risk for the subsequent development of bronchopulmonary dysplasia (28,29). Earlier work further suggests that lung fibrosis in bronchopulmonary dysplasia may also be ET-1 dependent (30). In newborn lambs with respiratory distress syndrome and PPHN, serum ET-1 levels correlated with the degree of elevation in pulmonary artery pressure (31), and human newborns with severe PPHN have high circulating levels of ET-1 (15). In infants with congenital diaphragmatic hernia, plasma ET-1 levels predict disease severity and degree of pulmonary hypertension (3), and in infants who died from congenital diaphragmatic hernia, lung ET-1 levels are also markedly increased (16). Overall, these studies suggest the importance of ET-1 in the pathogenesis of severe neonatal pulmonary hypertension, especially in the setting of lung hypoplasia. Our findings of increased ET-1 mRNA and protein expression in PPHN PAECs and striking improvement in PAEC tube formation with ET-1 inhibition further demonstrate the importance of ET-1 signaling pathways in the pathobiology of PPHN.
This is the first study to suggest that ET-1 may be responsible for decreased fetal lung vascular growth in experimental PPHN. The findings that exogenous ET-1 treatment decreased PAEC tube formation and ET-1 siRNA increased tube formation in normal and PPHN PAECs suggest that ET-1, through disrupting PAEC function, may be responsible for impaired angiogenesis in PPHN. Although these are in vitro findings, they do provide some insights into how PAEC dysfunction contributes to impaired angiogenesis in PPHN. In contrast to our findings in the fetal lung, studies of tumor angiogenesis have demonstrated a proangiogenic role for ET-1 and that ET-1 increases tumor vascularity in lung cancer through ETA receptor activation (17). In addition, others have shown that ET-1 increases vascular network formation through ETB receptor activation in human umbilical vein endothelial cells (18). These findings differ from the current results and may be explained in part by differences in cell type and developmental timing, or perhaps differential receptor activation.
A previous report has suggested that ETA receptor protein may be expressed in PAECs from adults with idiopathic pulmonary hypertension (32). However, although we could readily detect ETA receptor protein expression in fetal pulmonary artery smooth muscle cells, we could not detect the ETA receptor in PAECs in our study. In addition, BQ-123, a selective ETA receptor blocker, had no effect on tube formation in PAECs and did not prevent the decrease in tube formation during ET-1 treatment. By contrast, the effects of ET-1 on both ROCK activation and tube formation were prevented by bosentan. These interesting findings support our speculation that ETB receptors may mediate ET-1 activation of ROCK and contribute to impaired tube formation in PPHN. Although ET-1 activation of ETB receptors increases nitric oxide production, other published studies have shown that ET-1 increases reactive oxygen species (33) and thromboxane A2 release (34) through ETB receptor activation. Both reactive oxygen species and thromboxane A2 are potent activators of ROCK (35,36), suggesting a putative mechanism linking ETB receptor stimulation with ROCK activation. We speculate that in the setting of neonatal pulmonary hypertension with impaired vascular growth, combined ETA/ETB receptor blockade may be more beneficial than selective ETA receptor blockade alone.
High ET-1 and ROCK activities contribute to high pulmonary vascular tone and myogenic reactivity in the normal fetus (11,12,21,22). With chronic intrauterine pulmonary hypertension, lung ET-1 gene and protein expression and ROCK activity are increased (13,26). However, links between these two potent cell-signaling pathways in neonatal pulmonary hypertension have not been previously studied. The current study reports that ET-1 treatment increases ROCK activity in fetal PAECs and that the adverse effects of ET-1 on tube formation are reversed by ROCK inhibition, suggesting that ET-1 is responsible for increased ROCK activity in PPHN PAECs and that ROCK mediates ET-1–induced reduction of tube formation in vitro. siRNA knockdown of ET-1 increases tube formation in normal and PPHN PAECs and decreases ROCK activity, suggesting that ET-1–ROCK interactions may be responsible for decreased PAEC function and impaired angiogenesis in PPHN. In contrast to our findings in the fetal lung, the role of ROCK in the regulation of angiogenesis in chronic pulmonary hypertension is controversial. In adult rats with pulmonary hypertension due to chronic hypoxia, ROCK inhibition inhibits angiogenesis (37). These findings differ from the current results and may be explained in part by differences between species or development timing.
Potential limitations of this study include the use of fetal PAECs harvested from relatively large vessels; differences may exist in the behavior of these cells as compared with microvascular PAECs. Although microvascular PAECs may better represent where lung angiogenesis occurs during development, studying organ-specific cells from a developmentally relevant model of PPHN and the use of multiple clones of cells from different animals is a strength of our study. Future studies are needed to compare and contrast ET-1–ROCK interactions in microvascular PAECs to determine if similar cellular mechanisms are responsible for impaired microvascular endothelial cell function. Another potential limitation is the fact that angiogenesis was only measured in vitro. Whether increased ET-1 production activates ROCK to impair angiogenesis and if prolonged ET-1 or ROCK inhibition enhances angiogenesis in vivo remain unknown.
In conclusion, we found that ET-1 impairs fetal PAEC function through ROCK activation and may contribute to decreased vascular growth in severe PPHN. We speculate that treatment strategies that inhibit ET-1 production, block ETB receptor activity, or disrupt ET-1–ROCK interactions in PPHN may improve PAEC function and enhance lung angiogenesis, which may be especially important in treating pulmonary hypertension in the presence of endothelial dysfunction and lung hypoplasia.
Methods
Isolation and Culture of Fetal Ovine Pulmonary Arterial Endothelial Cells
All procedures and protocols were reviewed and approved by the Animal Care and Use Committee at the University of Colorado Health Sciences Center, Aurora, CO. The left and right pulmonary arteries were isolated at 135–140 d of gestation (term = 147 d) from late-gestation normal fetal sheep (n = 4) (mixed-breed Columbia-Rambouillet pregnant ewes) and from fetal sheep that had undergone partial ligation of the DA in utero at 125–130 d of gestation, 7–10 d before euthanasia (PPHN) (n = 4) as previously described (5). Proximal PAECs were isolated and PAEC phenotype was confirmed by cobblestone appearance and positive immunostaining for von Willebrand factor, endothelial nitric oxide synthase, and vascular endothelial cadherin. Vascular endothelial growth factor-receptor 2 (kinase insert domain receptor), positive uptake of acetylated low-density lipoprotein, and negative staining for desmin as previously described (6). Cells from passages 4 and 5 were used for the experiments, and all experiments were performed in 3% oxygen to mimic the in utero fetal environment. Cells from each animal were kept separate throughout all passages and experiments.
Cell Growth
Fetal PAECs from normal and PPHN lambs were plated at 2 × 105 cells per well and allowed to adhere overnight. Cells were grown in Dulbecco’s modified essential medium (Sigma, St. Louis, MO) supplemented with 5% fetal bovine serum (Sigma) with and without ET-1 treatment. Daily cell counts were performed for 4 d using a hemocytometer.
Three-Dimensional Tube Formation Assay
PAECs were grown to 90% confluence in T150 flasks and trypsinized. A volume of 2.5 × 105 cells per well in a 24-well plate was resuspended in a collagen mix, which is prepared by diluting bovine type 1 collagen (5 mg/ml) (R&D, Minneapolis, MN) in sterile water for a 1.3 mg/ml final concentration and 10× phosphate-buffered saline with phenol red for quantification of pH. After mixing the collagen–PAEC suspension, 0.5 ml was added per well to a 24-well plate, and the plate was placed in the incubator for 1 h to allow for polymerization. Various combinations of ET-1 (100 nmol/l) (Sigma), Y-27632 (1 µmol/l) (Cayman Chemical, Ann Arbor, MI), BQ-123 (1 µmol/l) (Sigma), and bosentan (1 µmol/l) (Actelion Pharmaceuticals, San Francisco, CA) were added, and media were changed daily to achieve maximal tube formation. Tube formation was also assessed after exposure to ET-1 siRNA and control siRNA. Tube formation was quantified using Fovea 3 software analysis (Microsoft, Redmond, WA) by counting the number of branch points per HPF and measuring tube length.
ET-1 Assay
Enzyme-linked immunosorbent assay (EIA) was performed using the EIA for Endothelin-1 kit (Peninsula Laboratories, San Carlos, CA). PAECs from control and PPHN fetal sheep were grown to 90% confluence in T75 flasks, and cell supernatant collected for solid-phase extraction by Sep-Pak C18 columns (Peninsula Laboratories). Before loading onto the column, 0.25 ml 2 mol/l HCL was added to each sample to acidify the solution. Solid-phase extraction was performed and samples lyophilized overnight using a centrifugal concentrator. Purified lyophilized samples were resuspended in EIA buffer, and EIA was performed in triplicate per manufacturer’s recommendations. Differences in ET-1 protein between supernatant from normal and PPHN PAECs were measured and quantified.
ET-1 siRNA
ET-1 siRNA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and siRNA knockdown was performed per manufacturer’s recommendations using control siRNA as a negative control.
RNA Isolation and Real-Time PCR
RNA was isolated from 95% confluent PAECs using RLT buffer (Qiagen, Valencia, CA) for lysis of cells and the RNAeasy minikit (Qiagen) per manufacturer’s protocol. Reverse-transcriptase reactions were prepared using 2 μg of RNA and Superscript III RT with random hexamers (Invitrogen, Carlsbad, CA). Real-time PCR primers and assays were optimized for ET-1 (Integrated DNA Technologies, Coralville, IA) (forward sequence: AGGTTGGAGACCATCAGCAA; reverse sequence: AGCACGGCTGTAGATCACTT) and S15 (Integrated DNA Technologies). Real-time PCR was performed using 2 ng of diluted cDNA and LightCycler 480 SYBR Green I Master (Roche, Indianapolis, IN) on the Roche-Light Cycle 480 II real-time PCR instrument (Roche). A relative standard curve of pooled PAEC lysate was generated (six standards prepared at fourfold serial dilutions) and used for quantification of unknown sample expression. Results were adjusted to S15 expression and expressed relative to the average of the control for each gene.
Western Blot Analysis
PAECs from normal and PPHN fetal sheep were grown on 150 mm cloning dishes in Dulbecco’s modified essential medium supplemented with 5% serum. At 70% confluence, PAECs were treated with ET-1 (100 nmol/l) with or without bosentan (1 µmol/l) for 24 h. Seventy percent confluence was chosen because previous studies have demonstrated a downregulation of important cell-signaling pathways with confluence (38). At the time of harvest, PAECs were 95% confluent. For ETA receptor antibody western blot analysis, fetal pulmonary artery smooth muscle cells were used as a positive control. Cell lysates were also collected after exposure to ET-1 siRNA and control siRNA. Cell lysates from PAECs for all experiments were collected as previously described (6,26). Protein content in the supernatant was determined by the bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL) using bovine serum albumin as the standard. Twenty micrograms of protein sample per lane was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and proteins from the gel were transferred to a nitrocellulose membrane. ETB receptor antibodies (USA Biologicals, Boston, MA), ETA receptor antibodies (USA Biologicals), MYPT-1 (Cell Signaling, Danvers, MA), and phospho-MYPT1 (Thr853) antibodies (Cell Signaling) were detected per manufacturer’s protocol using appropriate controls and molecular weight as identified by the manufacturer for the protein of interest. Densitometry was performed using NIH Image J (v1.61; NIH, Bethesda, MD), and changes in protein expression were analyzed after normalizing for β-actin expression.
Statistical Analysis
Data are presented as means ± SEM. Statistical analysis was performed with the Prism 4 software package (GraphPad Software, San Diego, CA). Statistical comparisons were carried out using ANOVA for tube formation assays and western blot analysis using Bonferroni posttest analysis. Unpaired t-test was used for EIA and real-time PCR analysis. A value of P < 0.05 was considered significant.
Statement of Financial Support
This study was supported by an Entelligence Young Investigator Award and National Institutes of Health (Bethesda, MD) grants 5K08HL102261 and R01 HL068702-05A2.
References
Levin DL, Heymann MA, Kitterman JA, Gregory GA, Phibbs RH, Rudolph AM . Persistent pulmonary hypertension of the newborn infant. J Pediatr 1976;89:626–30.
Hopkins N, McLoughlin P . The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat 2002;201:335–48.
Keller RL, Tacy TA, Hendricks-Munoz K, et al. Congenital diaphragmatic hernia: endothelin-1, pulmonary hypertension, and disease severity. Am J Respir Crit Care Med 2010;182:555–61.
Goldman AP, Tasker RC, Haworth SG, Sigston PE, Macrae DJ . Four patterns of response to inhaled nitric oxide for persistent pulmonary hypertension of the newborn. Pediatrics 1996;98(4 Pt 1):706–13.
Abman SH, Shanley PF, Accurso FJ . Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 1989;83:1849–58.
Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH . Chronic intrauterine pulmonary hypertension impairs angiogenesis and growth of ovine fetal pulmonary artery endothelial cells in vitro. Am J Respir Crit Care Med 2007;176:1146–53.
Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH . Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2005;288:L648–54.
Chen SJ, Chen YF, Meng QC, Durand J, Dicarlo VS, Oparil S . Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol 1995;79:2122–31.
Davie N, Haleen SJ, Upton PD et al. ETA and ETB receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2002;165:398–405.
Miyauchi T, Yorikane R, Sakai S, et al. Contribution of endogenous endothelin-1 to the progression of cardiopulmonary alterations in rats with monocrotaline-induced pulmonary hypertension. Circ Res 1993;73:887–97.
Ivy DD, le Cras TD, Parker TA, et al. Developmental changes in endothelin expression and activity in the ovine fetal lung. Am J Physiol Lung Cell Mol Physiol 2000;278:L785–93.
Wong J, Fineman JR, Heymann MA . The role of endothelin and endothelin receptor subtypes in regulation of fetal pulmonary vascular tone. Pediatr Res 1994;35:664–70.
Ivy DD, Ziegler JW, Dubus MF, Fox JJ, Kinsella JP, Abman SH . Chronic intrauterine pulmonary hypertension alters endothelin receptor activity in the ovine fetal lung. Pediatr Res 1996;39:435–42.
Ivy DD, Parker TA, Ziegler JW, et al. Prolonged endothelin A receptor blockade attenuates chronic pulmonary hypertension in the ovine fetus. J Clin Invest 1997;99:1179–86.
Rosenberg AA, Kennaugh J, Koppenhafer SL, Loomis M, Chatfield BA, Abman SH . Elevated immunoreactive endothelin-1 levels in newborn infants with persistent pulmonary hypertension. J Pediatr 1993;123:109–14.
Kobayashi H, Puri P . Plasma endothelin levels in congenital diaphragmatic hernia. J Pediatr Surg 1994;29:1258–61.
Zhao YD, Springall DR, Hamid Q, Levene M, Polak JM . Localization and characterization of endothelin-1 receptor binding in the blood vessels of human pulmonary tumors. J Cardiovasc Pharmacol 1995;26:Suppl 3: S336–40.
Salani D, Taraboletti G, Rosanò L, et al. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 2000;157:1703–11.
Arbajal JM . RhoA inactivation enhances endothelial cell barrier function. Am J Physiol 1999;277:C955–64.
McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M . Hypoxia and Rho/Rho-kinase signaling. Lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol 2003;543:127–37.
Parker TA, Roe G, Grover TR, Abman SH . Rho kinase activation maintains high pulmonary vascular resistance in the ovine fetal lung. Am J Physiol Lung Cell Mol Physiol 2006;291:L976–82.
Tourneux P, Chester M, Grover T, Abman SH . Fasudil inhibits the myogenic response in the fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 2008;295:H1505–13.
Nagaoka T, Morio Y, Casanova N, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2004;287:L665–72.
Oka M, Homma N, Taraseviciene-Stewart L, et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 2007;100:923–9.
Homma N, Nagaoka T, Morio Y, et al. Endothelin-1 and serotonin are involved in activation of RhoA/Rho kinase signaling in the chronically hypoxic hypertensive rat pulmonary circulation. J Cardiovasc Pharmacol 2007;50:697–702.
Gien J, Seedorf GJ, Balasubramaniam V, Tseng N, Markham N, Abman SH . Chronic intrauterine pulmonary hypertension increases endothelial cell Rho kinase activity and impairs angiogenesis in vitro. Am J Physiol Lung Cell Mol Physiol 2008;295:L680–7.
Figueras-Aloy J, Gómez-Lopez L, Rodríguéz-Miguélez JM, et al. Plasma endothelin-1 and clinical manifestations of neonatal sepsis. J Perinat Med 2004;32:522–6.
El Sayed M, Sherif L, Said RN, El-Wakkad AS, El-Refay A, Aly H . Endothelin-1 and L-arginine in preterm infants with respiratory distress. Am J Perinatol 2011;28:129–36.
Niu JO, Munshi UK, Siddiq MM, Parton LA . Early increase in endothelin-1 in tracheal aspirates of preterm infants: correlation with bronchopulmonary dysplasia. J Pediatr 1998;132:965–70.
Kambas K, Chrysanthopoulou A, Kourtzelis I, et al. Endothelin-1 signaling promotes fibrosis in vitro in a bronchopulmonary dysplasia model by activating the extrinsic coagulation cascade. J Immunol 2011;186:6568–75.
de Vroomen M, Lopes Cardozo RH, Steendijk P, Frölich M, Baan J, van Bel F . Endothelin-1 plasma concentration increases in the early phase of pulmonary hypertension development during respiratory distress syndrome: a study in newborn lambs. Early Hum Dev 2001;63:9–21.
Hall SM, Davie N, Klein N, Haworth SG . Endothelin receptor expression in idiopathic pulmonary arterial hypertension: effect of bosentan and epoprostenol treatment. Eur Respir J 2011;38:851–60.
Dong F, Zhang X, Wold LE, Ren Q, Zhang Z, Ren J . Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br J Pharmacol 2005;145:323–33.
D’Orléans-Juste P, Claing A, Télémaque S, Maurice MC, Yano M, Gratton JP . Block of endothelin-1-induced release of thromboxane A2 from the guinea pig lung and nitric oxide from the rabbit kidney by a selective ETB receptor antagonist, BQ-788. Br J Pharmacol 1994;113:1257–62.
Jernigan NL, Walker BR, Resta TC . Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2008;295:L515–29.
Martin C, Göggel R, Ressmeyer AR, Uhlig S . Pressor responses to platelet-activating factor and thromboxane are mediated by Rho-kinase. Am J Physiol Lung Cell Mol Physiol 2004;287:L250–7.
Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P . Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 2005;97:185–91.
Zöllner S, Aberle S, Harvey SE, Polokoff MA, Rubanyi GM . Changes of endothelial nitric oxide synthase level and activity during endothelial cell proliferation. Endothelium 2000;7:169–84.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gien, J., Tseng, N., Seedorf, G. et al. Endothelin-1 impairs angiogenesis in vitro through Rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr Res 73, 252–262 (2013). https://doi.org/10.1038/pr.2012.177
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2012.177
This article is cited by
-
Challenges, priorities and novel therapies for hypoxemic respiratory failure and pulmonary hypertension in the neonate
Journal of Perinatology (2016)
