
Abstract
Prenatal diagnosis of esophageal atresia (EA) may improve the outcome of affected neonates by allowing optimization of both prenatal and postnatal care. Prenatal sonographic detection is based on polyhydramnios and/or nonvisualization of the fetal stomach bubble, two signs with a large number of etiologies. We evaluated a biochemical approach to improving diagnostic efficiency. We compared amniotic fluid biochemical markers in 44 EA cases with 88 polyhydramnios and 88 nonpolyhydramnios controls. Both matched for GA with cases. Total proteins, alpha-fetoprotein (AFP), and digestive enzyme activities were assayed, including gamma-glutamyl transpeptidase (GGTP). We defined an EA index (AFP multiplied by GGTP). A significant difference (p < 0.0001) was observed for total protein, AFP, GGTP, and EA index between the EA group and each of the two control groups. No statistical difference was observed for any marker between the two most frequent EA subgroups (type I and type III) or between the two control groups. Using a cutoff of 3 for the EA index, 98% sensitivity and 100% specificity were observed for amniotic fluid prenatal diagnosis of EA, whatever the anatomical type. A large prospective series is required to confirm these results.
Similar content being viewed by others


Main
Esophageal atresia (EA) and tracheoesophageal fistula are major congenital malformations affecting 1/3500 live births, characterized by disruption of the continuity of the esophagus with or without a persistent communication with the trachea (1). Five subtypes (type I to type V) have been defined based on the location of the atresia and on the type of connection between trachea and esophagus (2). Type III (EA with distal tracheoesophageal fistula) is the most frequent variety, representing 86% of cases (3). Associated anomalies occur in 50% of patients and include vertebral, anal, cardiovascular, renal, and limb anomalies [occurring together in the Vertebral, Anal atresia, Cardiac defect, TracheoEsophageal fistula, Renal, Limb association (VACTERL)] (4–6) or chromosomal anomalies (7). The etiology of EA is unknown and is thought to be multifactorial with involvement of genetic and environmental factors (3,8–10).
Prenatal diagnosis of EA may improve the outcome of affected neonates by allowing optimization of both prenatal and postnatal care (11). The prenatal sonographic detection of EA relies on the finding of a small or nonvisualizable fetal stomach bubble associated with polyhydramnios (12). However, because of the large number of etiologies for these two signs (13,14) and to the subjective nature of a small fetal stomach (12,15,16), EA prenatal diagnosis remains difficult and is characterized both by a high false-positive rate and a poor detection rate. Several studies have documented that the sensitivity of sonography ranges from 8.9 to 42% (6,17–22) with Sparey et al. (19) reporting a 44% positive predictive value and Stringer et al. (22) 56%. Direct visualization of the fluid-filled, blind-ending esophagus during fetal swallowing or upper neck “pouch sign” was described as early as at 23 wk of gestation. However, visualization of the pouch may be challenging owing to technical difficulties or time limitation (20,23,24).
Polyhydramnios can be observed in large number of etiologies, such as maternal diabetes, digestive tract atresia, chloride diarrhea, Pierre Robin syndrome, and Bartter syndrome. In addition to fetal karyotyping, amniotic fluid digestive enzyme assay could be of interest for polyhydramnios etiology (25–27). The aim of this study was to evaluate the potential value of amniotic fluid digestive enzyme assay for the EA prenatal diagnosis, when suggested at routine ultrasound scan by indirect signs.
METHODS
Amniocentesis was performed for fetal karyotyping in all pregnancies. Amniotic fluid (kept frozen −20°C) sent by different institutions to our laboratory for biochemistry assay during the period 2003–2009 was retrospectively studied in 44 cases of esophageal atresia (EA) suspected at routine ultrasound scan (polyhydramnios as defined by Nwosu et al. (28) and/or nonvisualization of the stomach bubble). The pouch sign was not taken into account because it was not recorded in the medical chart on a regular basis. Ultrasound scan screening for associated malformation was performed in all pregnancies. Median GA at amniocentesis was 32 wk (23.3–38.5).
EA (with type classification) was clinically diagnosed using postnatal finding at birth or at pathological examination. In utero fetal death occurred in EA cases, termination of pregnancy due to severe associated malformations was performed at parent's request in 5, and of the 35 live birth infants, 2 died at neonatal period. Ultrasound signs at amniocentesis, outcome of pregnancy, GA at delivery, newborn weight, type of EA, presence or not or associated anomalies were recorded. IUGR was defined as all fetal measurement below the third percentile.
The EA group was divided into two subgroups depending on the presence or not of a tracheoesophageal fistula (type I versus other types). Two control groups, both matched with EA cases for GA, were defined: an idiopathic polyhydramnios control group (n = 88) and a nonpolyhydramnios control group of 88 fetuses in which karyotyping was performed because of maternal age ≥38 y or because of Down syndrome risk >1/250 at maternal serum marker screening. Twin pregnancies, fetal aneuploidy, oligohydramnios, fetal morphological abnormalities, and pregnancies with a clear etiology of polyhydramnios (e.g. duodenal atresia) were excluded from controls.
Biochemical markers were assayed in amniotic fluid (Olympus, Hamburg, Germany): total protein (urinary/cerebrospinal fluid protein) expressed in g/L and digestive enzyme activities (26,27), including gamma-glutamyl transpeptidase (GGTP), l-leucine-aminopeptidase (AMP; EC 3.4.11.1), and alkaline phosphatase isoenzymes including the intestinal form (iALP). Amniotic fluid alpha-fetoprotein (AFP; DualKit, PerkinElmer, Turku, Finland) was also assayed. We previously established normal values of all markers for each gestational week based on 10,000 amniotic fluid samples. Results are expressed in multiple of median (MoM) corresponding to the ratio between the observed raw value and the median raw value defined for the same GA. An EA index was defined corresponding to the multiplication of GGTP and of AFP (MoM values).
The nonparametric Mann-Whitney U test was used for group comparisons. p < 0.01 was considered as significant. The χ2 test was used for comparison of percentages. The study was exempt from institutional review board approval, because at the time of prenatal diagnosis amniotic fluid sampling was part of the routine diagnostic work-up. In accordance with French law, consent for amniocentesis and for laboratory testing was obtained from each patient.
RESULTS
Table 1 shows the prenatal ultrasound findings. EA was isolated in 28 cases (64%) and syndromic in 16 (36%). Polyhydramnios was associated with nonvisualization of the stomach bubble in 29 cases and was isolated in 12. Nonvisualization of stomach bubble without polyhydramnios was observed in three cases. A single umbilical artery was present in 15/44 EA cases (34%), with significant discrepancies (χ2 = 13.7) between syndromic EA cases (11/16; 69%) and isolated EA (4/28; 14%).
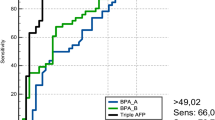
Table 2 shows the amniotic fluid biochemical results in the three groups we defined. A significant difference (p < 0.0001) was observed for total protein, AFP, GGTP, and the EA index between the EA group and each of the two control groups. No statistical difference was observed for AMP or iALP. No statistical difference was observed for any marker between the two EA subgroups (type I versus other types) or between the two control groups (with and without polyhydramnios). No difference in biochemical patterns was observed between fetal deaths and live births. The Figure 1 shows the results of this EA index in the three groups. Using a cutoff of 3 for the EA index, 98% sensitivity and 100% specificity were observed.

Amniotic Fluid Esophageal Atresia Index = (AFP MoM) × (GGTP MoM), in the three groups: control 1 = polyhydramnios control group (n = 88); control 2 = nonpolyhydramnios control group (n = 88); EA group = (n = 44). The solid dots correspond to the different individual values situated out of boxes representing mean and SD.
DISCUSSION
In this series of 44 EA-affected fetuses presenting at least one indirect ultrasound sign, a 98% sensitivity for a 100% specificity was observed with amniotic fluid biochemical EA index for EA prenatal diagnosis. The percentage of type III EA in our study (59%) and the 36% of associated malformations is the same as in published studies (4–6). VACTERL syndrome represents 20% of our cases, which is similar to the 23.1% observed by De Jong et al. (9).
Five amniotic fluid biochemical markers were assayed: two proteins (total protein and AFP) and three digestive enzymes (GGTP, AMP, and iALP). All of them (but AMP and iALP) were significantly different (p < 0.001) between the EA group and the control groups. We have previously demonstrated that amniotic fluid biochemistry assay is helpful for the diagnosis of various gastrointestinal diseases (25,29,30). Four abnormal patterns were defined (Table 3): 1) 16–22 wk, “obstruction” pattern characterized by all enzyme activities below the first percentile, normal total protein, and normal AFP, a pattern useful for the prenatal diagnosis of cystic fibrosis or anal atresia; 2) after 22 wk, “bilious vomiting” pattern characterized by GGTP and AMP activities over the 99th percentile, with normal values for iALP, total proteins, and AFP, a pattern useful for diagnosing upper digestive tract stenosis or atresia; 3) after 22 wk, “anal leakage” characterized by all enzymes over the 99th percentile, normal total protein, and normal AFP a pattern observed in urodigestive fistula, infectious diseases, and fetal distress; 4) after 22 wk, “Bartter syndrome pattern” characterized by low digestive enzymes, low total protein, and low AFP, specific to this renal tubular disease. All these profiles have pathophysiological explanations. Amniotic fluid digestive enzymes come from microvillar secretions. Fetal tissues studies have specified the location of each enzyme: biliary epithelia for GGTP, proximal small bowel enterocytes for AMP, and distal small bowel for iALP (29). In whatever type of EA, we observed a new fifth pattern characterized by high GGTP, normal AMP, high total protein, and high AFP. The high GGTP values (over 3 MoM) observed in both EA types cannot be explained by bile vomiting because the anatomical defect is located upstream of the Oddi sphincter, thus creating an obstacle. In type I, because of the esophageal sac, amniotic fluid is regurgitated but this does not correspond to bile vomiting. In types with tracheoesophageal fistula, a short circuit exists for the digestive tract, therefore making bile evacuation impossible. Local GGTP synthesis at the cul-de-sac level by metaplastic tissue could be hypothesized and the parallel increase in total proteins and AFP (over 2.5 MoM) could be due to the existence of cutaneous lesions caused by amniotic fluid movements in the cul-de-sac or around the fistula. Another hypothesis is that the global and similar elevation of GGTP, total proteins, and AFP is because of the absence of amniotic fluid swallowing. However, this hypothesis would not explain the normal AMP value (1 MoM), unless the half-life of AMP in amniotic fluid is shorter than the half-lives of other markers, or if a specific AMP activity inhibitor due to EA is present. We therefore have no firm explanation for this amniotic fluid biochemical pattern typical of fetal EA.
EA prenatal diagnosis is perceived to be advantageous for several reasons. First, it provides an opportunity to counsel and prepare parents for the birth of an affected newborn. Second, it allows better planning of the delivery and neonatal transfer to a pediatric surgical center. Third, it avoids the institution of oral feeding for an affected newborn and therefore may reduce the incidence of aspiration pneumonia. There is an obvious need for improvement in the prenatal diagnosis of this abnormality.
In conclusion, when EA is suspected at routine ultrasound scan (polyhydramnios and/or nonvisualization of stomach bubble), we suggest a biochemical approach to the prenatal diagnosis of EA based on amniotic fluid EA index. This approach must be confirmed in larger studies. Such a policy would not carry additional risk for the patient because amniocentesis is routinely performed to rule out karyotype abnormalities.
Abbreviations
- AFP:
-
alpha-fetoprotein
- AMP:
-
leucine-amino-peptidase (EC 3.4.11.1)
- EA:
-
esophageal atresia
- GGTP:
-
gamma-glutamyl transpeptidase (EC 2.3.2.2)
- iALP:
-
intestinal alkaline phosphatase (EC 3.1.3.1)
- MoM:
-
multiple of median
References
Depaepe A, Dolk H, Lechat MF 1993 The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Child 68: 743–748
Vogt EC 1929 Congenital esophageal atresia. AJR Am J Roentgenol 22: 463–465
Spitz L 2007 Oesophageal atresia. Orphanet J Rare Dis 2: 24
Torfs CP, Curry CJ, Bateson TF 1995 Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology 52: 220–232
Chittmittrapap S, Spitz L, Kiely EM, Brereton RJ 1989 Oesophageal atresia and associated anomalies. Arch Dis Child 64: 364–368
Geneviève D, de Pontual L, Amiel J, Sarnacki S, Lyonnet S 2007 An overview of isolated and syndromic oesophageal atresia. Clin Genet 71: 392–399
Nicolaides KH, Snijders RJ, Cheng HH, Gosden C 1992 Fetal gastro-intestinal and abdominal wall defects: associated malformations and chromosomal abnormalities. Fetal Diagn Ther 7: 102–115
Felix JF, de Jong EM, Torfs CP, de Klein A, Rottier RJ, Tibboel D 2009 Genetic and environmental factors in the etiology of esophageal atresia and/or tracheoesophageal fistula: an overview of the current concepts. Birth Defects Res A Clin Mol Teratol 85: 747–754
de Jong EM, Felix JF, Deurloo JA, van Dooren MF, Aronson DC, Torfs CP, Heij HA, Tibboel D 2008 Non-VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association. Birth Defects Res A Clin Mol Teratol 82: 92–97
Morini F, Cozzi DA, Ilari N, Casati A, Cozzi F 2001 Pattern of cardiovascular anomalies associated with esophageal atresia: support for a caudal pharyngeal arch neurochristopathy. Pediatr Res 50: 565–568
De Vigan C, Goujard J, Vodovar V, Uzan S 1997 Management of the fetus with a correctable malformation in Paris maternity units: evolution 1985–1994. Fetal Diagn Ther 12: 216–220
Farrant P 1980 The antenatal diagnosis of oesophageal atresia by ultrasound. Br J Radiol 53: 1202–1203
Dashe JS, McIntire DD, Ramus RM, Santos-Ramos R, Twickler DM 2002 Hydramnios: anomaly prevalence and sonographic detection. Obstet Gynecol 100: 134–139
Houben CH, Curry JI 2008 Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula. Prenat Diagn 28: 667–675
Millener PB, Anderson NG, Chisholm RJ 1993 Prognostic significance of nonvisualization of the fetal stomach by sonography. AJR Am J Roentgenol 160: 827–830
McKenna KM, Goldstein RB, Stringer MD 1995 Small or absent fetal stomach: prognostic significance. Radiology 197: 729–733
Pretorius DH, Drose JA, Dennis MA, Manchester DK, Manco-Johnson ML 1987 Tracheoesophageal fistula in utero. Twenty-two cases. J Ultrasound Med 6: 509–513
Stoll C, Alembik Y, Dott B, Roth MP 1996 Evaluation of prenatal diagnosis of congenital gastro-intestinal atresias. Eur J Epidemiol 12: 611–616
Sparey C, Jawaheer G, Barrett AM, Robson SC 2000 Esophageal atresia in the Northern Region Congenital Anomaly Survey 1985–1997: prenatal diagnosis and outcome. Am J Obstet Gynecol 182: 427–431
Choudhry M, Boyd PA, Chamberlain PF, Lakhoo K 2007 Prenatal diagnosis of tracheo-oesophageal fistula and oesophageal atresia. Prenat Diagn 27: 608–610
Shulman A, Mazkereth R, Zalel Y, Kuint J, Lipitz S, Avigad I, Achiron R 2002 Prenatal identification of esophageal atresia: the role of ultrasonography for evaluation of functional anatomy. Prenat Diagn 22: 669–674
Stringer MD, McKenna KM, Goldstein RB, Filly RA, Adzick NS, Harrison MR 1995 Prenatal diagnosis of esophageal atresia. J Pediatr Surg 30: 1258–1263
Kalache KD, Chaoui R, Mau H, Bollmann R 1998 The upper neck pouch sign: a prenatal sonographic marker for esophageal atresia. Ultrasound Obstet Gynecol 11: 138–140
Salomon LJ, Sonigo P, Ou P, Ville Y, Brunelle F 2009 Real-time fetal magnetic resonance imaging for the dynamic visualization of the pouch in esophageal atresia. Ultrasound Obstet Gynecol 34: 471–474
Muller F, Dommergues M, Ville Y, Lewin F, Delvalez-Morichon N, Nihoul-Fekete C, Bargy F, Dumez Y, Boué A 1994 Amniotic fluid digestive enzymes: diagnostic value in fetal gastrointestinal obstructions. Prenat Diagn 14: 973–979
Garnier A, Dreux S, Vargas-Poussou R, Oury JF, Benachi A, Deschênes G, Muller F 2010 Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res 67: 300–303
Magann EF, Chauhan SP, Doherty DA, Lutgendorf MA, Magann MI, Morrison JC 2007 A review of idiopathic hydramnios and pregnancy outcomes. Obstet Gynecol Surv 62: 795–802
Nwosu EC, Welch CR, Manasse PR, Walkinshaw SA 1993 Longitudinal assessment of amniotic fluid index. Br J Obstet Gynaecol 100: 816–819
Muller F, Oury JF, Dumez Y, Boué J, Boué A 1988 Microvillar enzyme assays in amniotic fluid and fetal tissues at different stages of development. Prenat Diagn 8: 189–198
Oca F, Dreux S, Gérard B, Simon-Bouy B, de Becdelièvre A, Ferec C, Girodon E, Muller F 2009 Amniotic fluid digestive enzyme analysis is useful for identifying CFTR gene mutations of unclear significance. Clin Chem 55: 2214–2217
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Czerkiewicz, I., Dreux, S., Beckmezian, A. et al. Biochemical Amniotic Fluid Pattern for Prenatal Diagnosis of Esophageal Atresia. Pediatr Res 70, 199–202 (2011). https://doi.org/10.1203/PDR.0b013e318220c08a
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e318220c08a
This article is cited by
-
Oesophageal atresia
Nature Reviews Disease Primers (2019)
-
Hyperechogenic kidneys and polyhydramnios associated with HNF1B gene mutation
Pediatric Nephrology (2016)
-
The diagnosis of fetal esophageal atresia and its implications on perinatal outcome
Pediatric Surgery International (2014)
-
Specific biochemical amniotic fluid pattern of fetal isolated esophageal atresia
Pediatric Research (2013)
-
Peut-on diagnostiquer l’atrésie de l’œsophage en prénatal ?
Revue de médecine périnatale (2012)
