
Abstract
Mutations in the genes encoding components of the renin-angiotensin system (RAS) in mice or humans cause congenital abnormalities of the kidney and urinary tract. We hypothesized that absence of angiotensin (Ang) II in angiotensinogen (AGT)-deficient mice leads to defects in ureteric bud (UB) branching and that RAS genes are critically dependent on histone deacetylase (HDAC) activity. The number of UB tips was lower in AGT−/− compared with AGT+/+ embryonic (E) day E13.5 metanephroi (24 ± 1.5 versus 36 ± 3.7, p < 0.05). Real-time RT-PCR demonstrated that pharmacological inhibition of HDAC activity with Scriptaid increases AGT, renin, angiotensin-converting enzyme (ACE), and AT1 receptor (AT1R) mRNA levels in E12.5 mouse metanephroi and early mesenchymal (MK3) cells. Furthermore, Scriptaid enhanced Ang II-induced decrease in Sprouty (Spry) 1 gene expression in cultured UB cells. Treatment of intact E12.5 mouse metanephroi grown ex vivo with Ang II (10−5 M, 24 h) increased HDAC-1 and decreased total acetylated histone H3 protein levels. These findings indicate that lack of endogenous Ang II in AGT-deficient mice inhibits UB branching. We conclude that intact RAS is critical in structural integrity of the renal collecting system and that UB morphogenetic program genes, such as AGT, renin, ACE, AT1R, or Spry1, are epigenetically controlled via HDACs.
Similar content being viewed by others


Main
Formation of the mammalian permanent (metanephric) kidney begins when the Wolffian duct (WD) gives rise to ureteric bud (UB) on embryonic (E) day E10.5 in mice and E28 in humans (1). UB outgrowth from the WD is followed by its repetitive branching and growth, a process called branching morphogenesis, to eventually form the renal collecting system (ureter, renal pelvis, calyces, and collecting ducts) (2,3). The metanephros develops via reciprocal inductive interactions between the UB and the mesenchyme (2). Each UB tip is capable of inducing the adjacent mesenchyme to form nephrons (from the glomerulus through the distal tubule). Importantly, even subtle defects in the efficiency of UB branching result in a significant decrease in nephron endowment (4). In turn, decreased nephron endowment is linked to renal hypodysplasia, CAKUT, hypertension, and eventual progression to chronic renal failure (5,6).
The renin-angiotensin system (RAS) is crucial in the regulation of blood pressure, fluid-electrolyte homeostasis, and proper kidney development (7,8). In the RAS, renin cleaves angiotensinogen (AGT) to generate angiotensin (Ang) I, which is then converted to Ang II by angiotensin converting enzyme (ACE). Ang II is the principal effector peptide growth factor of the RAS, which acts via two major G protein-coupled receptors: AT1R and AT2R (9). Mutations in the genes encoding AGT, renin, ACE, or AT1R in humans are associated with renal tubular dysgenesis (RTD) (10). Genetic inactivation of AGT, renin, ACE, or AT1R in mice causes hypoplastic renal medulla and hydronephrosis (11–16). Taken together, these findings indicate that UB and its daughter collecting ducts are principal targets for the RAS. We have recently reported that Ang II stimulates UB branching in the metanephros (17). This is accompanied by decreased expression of Sprouty (Spry) 1, an endogenous inhibitor of the GDNF/c-Ret/Wnt11 UB morphogenetic pathway (18).
Posttranslational modifications of histones such as histone acetylation and methylation affect chromatin structure and function resulting in altered gene expression (19,20). Generally, histone deacetylation of selected target genes by histone deacetylases (HDACs) mediates gene repression (21). Because aberrant gene expression is a major feature of abnormal kidney development, we investigated the role of HDACs in the transcriptional regulation of the RAS genes during organogenesis of the metanephros.
Here, we demonstrate an essential role for endogenous Ang II in UB morphogenesis. Genetic inactivation of AGT in mice results in impaired UB branching. Inhibition of HDAC activity up-regulates the expression of the RAS genes in the developing metanephros and enhances Ang II-induced decrease in Spry1 in renal epithelial cells. Finally, Ang II increases HDAC-1 expression and down-regulates histone H3 acetylation in the developing metanephros.
METHODS
Metanephric organ culture.
To determine the role of endogenous Ang II in early stages of UB growth and development in the intact kidney, we used double-transgenic Hoxb7-green fluorescent protein (GFP)/angiotensinogen (AGT) mice. AGT± C57BL6/129P2 heterozygous mice were obtained from Jackson Laboratories (Bar Harbor, ME). Hoxb7-GFP± B6xCBA transgenic mice were a kind gift of Dr. F. Costantini (Columbia University, NY) (22). GFP-positive metanephroi from Hoxb7-GFP+/AGT+/+ (n = 6) and Hoxb7-GFP+/AGT−/− (n = 4) embryos were dissected on embryonic (E) day E13.5, photographed via an Olympus IX70 phase-contrast microscope and Olympus MagnaFire FW camera, and the number of UB tips was counted. To determine the effect of exogenous Ang II on UB branching in AGT−/− mice, we compared the effect of Ang II or medium on UB morphogenesis in paired Hoxb7-GFP+/AGT−/− metanephroi isolated on E11.5. E11.5 Hoxb7-GFP+/AGT−/− metanephroi were grown on transwell filters (Corning Costar, 0.5 μm) inserted into six-well plates containing DMEM/F12 medium (GIBCO BRL) alone (control, n = 8) or combined with Ang II (10−5 M, n = 8) for 48 h. The media containing or not containing Ang II were changed every 24 h. Sequential images of branching UBs were acquired after isolation on E11.5, after 24 and 48 h of culture. Quantitative assessment of UB branching was performed by counting the number of UB tips at the time of dissection, after 24 and 48 h in culture. All experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Tulane University.
Western blot analysis.
The effect of Ang II on histone H3 acetylation and HDAC-1 expression was examined in whole CD1 mice (Charles River Laboratories, NY, NY) metanephroi grown ex vivo. Pooled E12.5 metanephroi (n = 10/sample) were grown on air-fluid interface on polycarbonate transwell filters (Corning Costar, 0.5 μm) inserted into six-well plates containing DMEM/F12 medium (GIBCO BRL) alone (control) or in the presence of Ang II (10−5 M) for 24 h at 37°C and 5% CO2. The effect of Ang II on HDAC-1 expression was also studied in cultured ureteric bud-derived UB cells (generously provided by Dr. Jonathan Barash, Columbia University). Metanephroi or UB cells were homogenized in cold lysis buffer containing a cocktail of enzyme inhibitors. The samples were centrifuged and the supernatants containing proteins (40 μg/lane) were resolved on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. After blocking nonspecific binding, the membranes were incubated with the anti-acetyl-Histone H3 (1/1000, Upstate), anti-HDAC-1 (1/1000, BioVision), anti-total histone H3 (1/1000, Upstate), or anti-β-actin (1/1500, Sigma-Aldrich Chemical Co., St. Louis, MO) antibodies at room temperature for 1 h. After washes in PBS/Tween, the nitrocellulose membrane was exposed for 1 h at room temperature to the secondary antibody. Immunoreactive bands were visualized using the enhanced chemiluminescence detection system (ECL, Amersham) as previously described (23).
Reverse-transcription RT-PCR.
Reverse-transcription PCR (RT-PCR) was used to examine the effect of HDAC inhibition with Scriptaid on the expression of angiotensinogen (AGT), renin, angiotensin-converting enzyme (ACE), and Ang II AT1 receptor (AT1R) in intact metanephroi and early mesenchymal (MK3) cells. The MK3 cell line represents early metanephric mesenchyme, before induction by the ureteric bud (24). Quiescent MK3 cells (n = 3/treatment group) or E12.5 CD1 mice metanephroi (n = 6/treatment group) were cultured in DMEM media in the presence of the HDAC inhibitor Scriptaid (2.0 μg/mL, Biomol, Plymouth Meeting, PA) (25) or its inactive analogue Nullscript (2.0 μg/mL) for 6 h. RNA was extracted using the TRIzol reagent (Invitrogen). For semiquantitative RT-PCR, MK3 cell cDNA was amplified using the Perkin Elmer Gene Amplification system 2400 (Cetus Instruments, Norwalk, CT) from 25% of RT mixture using gene-specific primers: AGT: sense-5′-TCTCTGGATTTATC-CACTGACC-3′, antisense: 5′-TCACGGAGAAGTTGTTCTGGGC-3′, renin sense: 5′-TGGGTGCCCTCCACCAAGTG-3′, antisense: 5′-CTCCCAGGGCTTGCATGATCA-3′, ACE sense: 5′-CTCCAGGCCCTGGCAGGAGGT-3′, antisense: 5′-TCTTCATATTTC-CGGGATGTGGCC-3′, AT1R sense: 5′-GCATCATCTTTGTGGTGGG-3′, antisense: 5′-GAAGAAAAGCACAATCGCC-3′.
To address the concerns of the specificity of HDAC inhibitor, we inhibited HDAC-1 expression using morpholino gene silencing in MK4 cells. The MK4 cell line represents embryonic metanephric mesenchyme undergoing epithelial conversion (24). MK4 cells were cultured in DMEM medium with 10% serum at 37°C with 5% CO2. At ∼30% confluency, cells were treated with Endo-porter (9 μM) and HDAC-1 morpholino (5′-TGCCCTGAGTCTGCGCCATCTTGCT-3′, 10 μM, n = 3) targeted at ATG codon of mouse HDAC-1 mRNA or standard control morpholino (10 μM, n = 3) (Gene Tools, Philomath, OR) for 48 h. SYBR Green quantitative RT-PCR was conducted in the Mx3000P equipment (Stratagene, LA Jolla, CA) using MxPro QPCR software (Stratagene) as described previously (17). The quantity of each target mRNA expression was normalized by that of GAPDH mRNA expression. Three RNA samples per treatment group were analyzed in triplicates in each run. PCR reaction was performed three times. To define the role of HDACs in Ang II-induced down-regulation of Spry1 gene expression in UB epithelia, we examined the effect of Ang II (10−5 M), HDAC inhibitor Scriptaid (2.0 μg/mL), Ang II combined with HDAC inhibitor Scriptaid or with inactive Scriptaid analogue, Nullscript (2.0 μg/mL) on Spry1 mRNA levels in quiescent UB cells by quantitative RT-PCR.
Data analysis.
Data are represented as mean ± SEM. Differences among the treatment groups in mRNA and protein levels and the number of UB tips were analyzed by t test. A p value < 0.05 was considered statistically significant.
RESULTS
Quantitative assessment of UB branching in Hoxb7-GFP/AGT metanephroi.
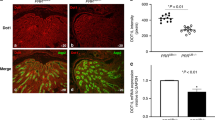
To determine whether absence of endogenous Ang II in angiotensinogen (AGT)-deficient mice leads to defects in UB branching, we examined the effect of genetic inactivation of AGT on UB branching in double transgenic Hoxb7-GFP/AGT−/− and Hoxb7-GFP/AGT+/+ mice (n = 4 per AGT genotype). The number of UB tips (24 ± 1.5 versus 36 ± 3.7, p < 0.01) and branch points (17 ± 1.6 versus 27 ± 1.2, p < 0.01) is lower in Hoxb7-GFP/AGT−/− compared with Hoxb7-GFP/AGT+/+ metanephroi (Fig. 1). These findings provide strong support for our hypothesis that lack of endogenous Ang II in AGT-deficient mice leads to defects in UB branching.

Effect of angiotensinogen (AGT) genotype on UB branching in double transgenic Hoxb7-GFP/AGT embryos on E13.5. (A–D): UB images. Numbers represent tip number. BP, branch point. Scale bar: 200 μm. Magnification ×10. (E, F): Bar graphs showing the effect of AGT genotype on the number of UB tips and branch points. *p < 0.05.
To determine whether a decrease in UB branching observed in AGT−/− mice can be rescued by the addition of exogenous Ang II, we examined the effect of Ang II or media (control) on UB branching in paired AGT−/− metanephroi. Treatment of E11.5 AGT−/− metanephroi with Ang II (10−5 M, n = 8) resulted in a variable response in UB branching. Ang II increased the number of UB tips at 24 (31 ± 1.0 versus 27 ± 0.7, p < 0.05) and 48 (36 ± 1.1 versus 30 ± 1.0, p < 0.05) hours in four of eight metanephroi and did not alter the number of UB tips in the rest four of eight metanephroi (24 h: 25 ± 2.3 versus 24 ± 2.0, p = 0.82) and (48 h: 36 ± 1.6 versus 35 ± 1.8, p = 0.61) compared with respective controls (media). Our findings differ with previous reports showing lack of the impact of AGT deficiency or exogenous Ang II administration in AGT−/− metanephroi on UB branching (11). It is conceivable that variability to Ang II response of AGT−/− kidneys may be related to the fact that by the time Ang II was added, it was already too late and some structures have already undergone degeneration and apoptosis.
HDAC inhibition up-regulates RAS gene expression.
To investigate the role of HDACs in the transcriptional regulation of the RAS gene expression during metanephric development, we used HDAC inhibitor Scriptaid. Quantitative RT-PCR demonstrated that treatment of renal mesenchymal MK3 cells with Scriptaid increased levels of AGT (2.2 ± 0.37 versus 1.0 ± 0, p < 0.05), renin (31.0 ± 2.1 versus 1.0 ± 0, p < 0.001), ACE (3.8 ± 0.48 versus 1.0 ± 0, p < 0.05), and AT1R (1.6 ± 0.05 versus 1.0 ± 0, p < 0.05) mRNAs compared with treatment with inactive Scriptaid analogue, Nullscript (Fig. 2A and B). Similar to its effects in MK3 cells, Scriptaid increased levels of AGT (11.0 ± 0.58 versus 1.0 ± 0, p < 0.001), renin (2.4 ± 0.09 versus 1.0 ± 0, p < 0.05), ACE (2.7 ± 0.06 versus 1.0 ± 0, p < 0.05), and AT1R (1.7 ± 0.06 versus 1.0 ± 0, p < 0.05) mRNAs in whole intact metanephroi compared with Nullscript (Fig. 2C). HDAC-1 knockdown in MK4 cells resulted in up-regulation of AGT (1.8 ± 0.17 versus 1.0 ± 0, p < 0.05), renin (4.6 ± 0.45 versus 1.0 ± 0, p < 0.01), ACE (10.3 ± 1.7 versus 1.0 ± 0, p < 0.01), and AT1R (1.4 ± 0.1 versus 1.0 ± 0, p < 0.05) mRNA (Fig. 2D). These findings indicate that HDAC activity is essential for the regulation of the RAS during UB morphogenesis.

Effect of HDAC inhibitor Scriptaid (S) or its inactive analogue Nullscript (N) on angiotensinogen (AGT), renin, angiotensin-convertin enzyme (ACE), and Ang II AT1 receptor (AT1R) mRNA levels in cultured MK3 cells and embryonic metanephroi. (A): Ethidium bromide-stained gel shows semi-quantitative RT-PCR analysis of the RAS mRNA in MK3 cells. (B, C): Bar graphs showing the effect of Nullscript and Scriptaid on RAS mRNA expression in MK3 cells (B) and E12.5 metanephroi (C) as determined by quantitative RT-PCR. *p < 0.05 vs. Nullscript (control = 100%), **p < 0.001 vs. Nullscript. (D): Bar graph showing the effect of HDAC-1 inhibition with morpholino in MK4 cells. *p < 0.05, **p < 0.01 vs. control (= 100%).
Angiotensin II induces HDAC-1 expression and decreases total histone H3 acetylation.
To begin testing the hypothesis that Ang II-induced stimulation of GDNF/c-Ret/Wnt11 pathway gene expression is mediated by changes in target gene acetylation status, we examined the effect of Ang II on total histone H3 acetylation and HDAC-1 expression in whole embryonic metanephroi. Treatment of E12.5 metanephroi with Ang II for 24 h resulted in an increased HDAC-1 (1.7 ± 0.1 versus 1.1 ± 0.15 densitometric units, p < 0.05) and a decreased acetylated histone H3 (2.0 ± 0.12 versus 2.5 ± 0.08 densitometric units, p < 0.05) protein levels (Fig. 3A, B, E, and F). To determine whether Ang II alters HDAC-1 expression by directly acting on the UB, we tested the effect of Ang II on HDAC-1 protein levels in cultured UB cells. Consistent with whole kidney data, Ang II increased HDAC-1 protein levels in UB cells (1.8 ± 0.15 versus 0.98 ± 0.1 densitometric units, p < 0.05) (Fig. 3C and D). These findings indicate that Ang II induces HDAC-1, which in turn acts to deacetylate histone H3. Given that HDAC inhibition impairs UB branching (Samir El-Dahr, personal communication), our findings suggest that Ang II-induced UB morphogenesis is critically dependent on HDAC activity.

Effect of media (control) or Ang II on HDAC-1 and histone H3 protein expression. (A, E): E12.5 mouse kidneys were grown ex vivo in the presence of media or Ang II (10−5 M) for 24 h. Whole kidney lysates were subjected to immunoblotting with anti-HDAC-1 (A) or anti-acetylated or total histone H3 (E) antibodies. After stripping, top membrane in “E” was reprobed with anti-total histone H3 antibody. (C): Quiescent UB cells were treated with media or Ang II (10−5 M) for 24 h and cell lysates were subjected to immunoblotting with anti-HDAC-1 antibody. (B, D, F): Bar graphs showing the effect of media or Ang II on target protein levels. Bars represent ratio of HDAC-1/β-actin (B, D) or acetylated/total histone H3 (F). *p < 0.05.
Spry1 gene and Ang II-induced inhibition of Spry1 is mediated by HDAC activity.
We have recently demonstrated that Ang II, acting via the AT1R, down-regulates Sprouty (Spry) 1 gene expression in UB epithelia (17). To determine whether Ang II-induced inhibition of Spry1 gene in the UB is mediated by HDAC activity, we examined the effect of Ang II combined with Scriptaid or its inactive analogue Nullscript on Spry1 mRNA levels in cultured UB cells. Scriptaid alone repressed Spry1 gene compared with media (Fig. 4). Ang II induced a higher decrease in Spry1 mRNA levels in the presence of Scriptaid compared with Nullscript or with Scriptaid alone (Fig. 4A). HDAC-1 knockdown in MK4 cells resulted in down-regulation of Spry1 mRNA (0.45 ± 0.1 versus 1.0 ± 0, p < 0.01) (Fig. 4B). These data indicate that Spry1 gene and Ang II-induced inhibition of Spry1 in UB cell lineage are regulated epigenetically via HDACs. Our findings suggest a model in which inhibition of HDAC activity induces RAS gene expression leading to AT1R-dependent repression of Spry1 and induction of UB branching (Fig. 5; Ref. 17). Enhanced AT1R signaling induces HDAC-1 expression leading to deacetylation of histone H3. This exerts a negative feedback on the RAS activity.

Effect of HDAC inhibition on Spry1 gene expression. (A): Bar graph showing the effect of media, HDAC inhibitor Scriptaid, Ang II combined with Scriptaid, or inactive Scriptaid analogue Nullscript on Spry1 mRNA levels in cultured UB cells. HDAC inhibition with Scriptaid decreases Spry1 mRNA levels. Scriptaid further down-regulates Ang II-induced decrease in Spry1 mRNA levels. *p < 0.05 vs. Media, **p < 0.05 vs. Scriptaid or Ang II+Nullscript. (B): Bar graph showing the effect of HDAC-1 knockdown in MK4 cells on Spry1 mRNA. *p < 0.01 vs. control (= 100%).

A proposed model for HDAC-RAS cross-talk in ureteric bud branching. Inhibition of HDAC activity induces RAS gene expression leading to AT1R-dependent repression of Spry1. This causes activation of c-Ret tyrosine kinase activity and induction of UB branching. In addition, enhanced AT1R signaling induces HDAC-1 expression leading to deacetylation of histone H3. This exerts a negative feedback on the RAS activity.
DISCUSSION
This study demonstrates that lack of endogenous Ang II in AGT-deficient mice causes a decrease in ureteric bud (UB) branching. Exogenous Ang II up-regulates HDAC-1 and down-regulates acetylated histone H3 expression in the developing metanephros. Inhibition of HDAC activity induces renin-angiotensin system (RAS) gene expression in the intact metanephros and renal mesenchymal cells, and further down-regulates Ang II-induced decrease in Spry1 mRNA levels in UB cells.
Inactivation of the RAS genes in mice causes abnormalities in the development of renal collecting system (11–16). AGT, renin, ACE, or AT1R-deficient mice exhibit pelvic dilation (hydronephrosis), small medulla, and papilla. Functionally, ACE- and AT1R-null animals have a reduced ability to concentrate urine (14,15). Mutations in the genes encoding AGT, renin, ACE, or AT1R in humans are associated with renal tubular dysgenesis (RTD) (10). In RTD, renal cortex exhibits a paucity of proximal tubules. In the medulla, collapsed collecting ducts and abundant interstitial fibrosis are observed (10). Previous studies have shown that small papilla and hydronephrosis in AT1R-deficient mice may be due to defective formation and function of the smooth muscle layer in the ureteral wall, mimicking the findings seen in urinary obstruction (26). We recently reported that exogenous Ang II can directly stimulate UB branching in the metanephric kidney cultured in vitro (17,23). Our current findings extend our previous data by showing that endogenous Ang II can directly stimulate UB morhogenesis in vivo and thus promote formation of the collecting system.
Histone acetylation plays a key role in regulating gene expression by modulating chromatin structure. Histone acetylation is controlled by antagonistic activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (27). Generally, when HDAC activity is inhibited and lysine residues on histone tails become hyperacetylated, chromatin unfolds and allows transcriptional regulators to access promoter regions of target genes and induce their expression (19). Four classes of HDACs have been identified. Class I HDACs are expressed ubiquitously and consist of HDACs 1, 2, 3, and 8 (28). Class II HDACs (HDACs 4, 5, 7, and 9) are expressed predominantly in muscle and neural systems (29). The crucial role for class I HDACs during development is demonstrated by embryonic lethality in HDAC-1- or HDAC-2-null mice due to cell proliferative defect (30). Combined conditional deletion of HDAC-1 and 2 in the cardiac lineage in mice results in neonatal lethality due to defects in heart development and is accompanied by aberrant expression of genes regulating calcium homeostasis. Although the role of HDACs in metanephric development and kidney tissue-specific gene expression have not been defined, such a role is likely. In this regard, HDACs repress Wilm's tumor (WT) 1 gene expression in human embryonic kidney epithelial cells (31). The importance of WT1 during development is demonstrated be embryonic lethality in WT1-null embryos accompanied by renal agenesis (32). In addition, inhibition of HDAC activity inhibits TGF-β1-induced epithelial-to-mesenchymal transition (EMT) in human renal epithelial cells (33).
With respect to epigenetic modifications of the RAS and its impact on kidney development, recent studies demonstrated that inhibition of HDAC activity induces the expression of renin mRNA in renal juxtaglomerular (JG) cells (34). This is accompanied by increased histone H4 acetylation in the region of cAMP (cAMP)-responsive element (CRE) of the renin promoter. In contrast, conditional deletion of HATs such as CRE-binding protein (CREB)-binding protein (CBP) and related DNA-binding protein, p300 (35), in renin-expressing JG cells results in a decreased number of renin-positive cells (36). Moreover, kidney structure is abnormal in CBP/p300-null mice: kidneys exhibit interstitial fibrosis, disorganized vasculature, medullary, and cortical cysts. In addition, inhibition of HDAC activity increases AGT mRNA levels in human hepatocellular liver carcinoma (HepG2) cells (37). Our findings of up-regulated AGT, renin, ACE, and AT1R mRNA levels in the intact metanephros and renal mesenchymal cells during HDAC inhibition are consistent with these reports, which implicate histone acetylation in RAS gene expression. Moreover, given that proper metanephric development is critically dependent on intact RAS, these data demonstrate that epigenetic modifications of the RAS play an important role in the maintenance of renal structural integrity. Our present findings of increased HDAC-1 and decreased total acetylated histone H3 by exogenous Ang II suggest that cross-talk between the RAS and target gene acetylation in the metanephros is bidirectional.
We recently reported that Ang II induces GDNF/c-Ret/Wnt11 gene expression in the metanephros (17). These changes are accompanied by down-regulation of Spry1, an endogenous inhibitor of c-Ret tyrosine kinase activity (17). Notably, GDNF/c-Ret/Wnt11 signaling pathway is a major positive regulator of UB morphogenesis (38,39). Our current findings that Ang II-induced inhibition of Spry1 is regulated via HDACs suggest that epigenetic cross-talk between the RAS and other UB morphogenetic program genes plays an important role in metanephric development. Our observations are consistent with other studies showing potential interactions of the RAS with epigenetic marks during metanephric development. For example, paired box (Pax) transactivation domain-interacting protein (PTIP), a nuclear protein that is part of a histone H3 lysine 4 (K4) (H3K4) histone methyltransferase (HMT) complex, is essential for metanephric development (40,41). In turn, methylation status of H3K4 correlates with gene expression (42) and Ang II acts to increase Pax2 gene expression in metanephros (43).
In summary, this study demonstrates that lack of endogenous Ang II in AGT-deficient mice inhibits UB branching. Exogenous Ang II up-regulates HDAC-1 and down-regulates acetylated histone H3 expression in the developing metanephros. Inhibition of HDAC activity induces RAS gene expression in the intact metanephros and further down-regulates Ang II-induced decrease in Spry1 gene in UB cells. These results indicate that intact RAS is critical in structural integrity of the renal collecting system and that UB morphogenetic program genes, such as AGT, renin, ACE, AT1R, or Spry1, are epigenetically controlled via HDACs.
Abbreviations
- ACE:
-
angiotensin-converting enzyme
- AGT:
-
angiotensinogen
- Ang:
-
angiotensin
- GFP:
-
green fluorescent protein
- HDAC:
-
histone deacetylase
- RAS:
-
renin-angiotensin system
- RTD:
-
renal tubular dysgenesis
- Spry1:
-
Sprouty 1
- UB:
-
ureteric bud
References
Saxen L 1987 Organogenesis of the Kidney. Cambridge University Press, Cambridge
Ekblom P 1989 Developmentally regulated conversion of mesenchyme to epithelium. FASEB J 3: 2141–2150
Grobstein C 1953 Inductive epithelio-mesenchymal interaction in cultured organ rudiments of the mouse metanephros. Science 118: 52–55
Sakurai H, Nigam S 1998 In vitro branching tubulogenesis: implications for developmental and cystic disorders, nephron number, renal repair, and nephron engineering. Kidney Int 54: 14–26
Brenner BM, Garcia DL, Anderson S 1988 Glomeruli and blood pressure. Less of one, more the other?. Am J Hypertens 1: 335–347
Lisle SJ, Lewis RM, Petry CJ, Ozanne SE, Hales CN, Forhead AJ 2003 Effect of maternal iron restriction during pregnancy. Br J Nutr 90: 33–39
Gomez RA, Norwood VF 1995 Developmental consequences of the renin-angiotensin system. Am J Kidney Dis 26: 409–431
Yosypiv IV, El-Dahr SS 2005 Role of the renin-angiotensin system in the development of the ureteric bud and renal collecting system. Pediatr Nephrol 20: 1219–1229
Kobori H, Nangaku M, Navar LG, Nishiyama A 2007 The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287
Gribouval O, Gonzales M, Neuhaus T 2005 Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 37: 964–968
Nagata M, Tanimoto K, Fukamizu A, Kon Y, Sugiyama F, Yagami K, Murakami K, Watanabe T 1996 Nephrogenesis and renovascular development in angiotensinogen-deficient mice. Lab Invest 75: 745–753
Niimura F, Labosky PA, Kakuchi J, Okubo S, Yoshida H, Oikawa T, Ichiki T, Naftilan AJ, Fogo A, Inagami T 1995 Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest 96: 2947–2954
Takahashi N, Lopez ML, Cowhig JE Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O 2005 Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16: 125–132
Esther CR Jr, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE 1996 Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest 74: 953–965
Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM 1998 Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA 95: 15496–15501
Tsuchida S, Matsusaka T, Chen X, Okubo S, Niimura F, Nishimura H, Fogo A, Utsunomiya H, Inagami T, Ichikawa I 1998 Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest 101: 755–760
Yosypiv IV, Boh MK, Spera M, El-Dahr SS 2008 Downregulation of Spry-1, an inhibitor of GDNF/Ret, as a mechanism for angiotensin II-induced ureteric bud branching. Kidney Int 74: 1287–1293
Basson MA, Watson-Johnson J, Shakya R, Akbulut S, Hyink D, Costantini FD, Wilson PD, Mason IJ, Licht JD 2006 Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev Biol 299: 466–477
Smith CL 2008 A shifting paradigm: histone deacetylases and transcriptional activation. Bioessays 30: 15–24
Nottke A, Colaiácovo MP, Shi Y 2009 Developmental roles of the histone lysine demethylases. Development 136: 879–889
Kurdistani SK, Grunstein M 2003 Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol 4: 276–284
Srinivas S, Goldberg MR, Watanabe T, Dagati V, Al-Awqati Q, Costantini F 1999 Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of the ureteric bud morphogenesis. Dev Genet 24: 241–251
Yosypiv IV, Schroeder M, El-Dahr SS 2006 AT1R-EGFR crosstalk regulates ureteric bud branching morphogenesis. J Am Soc Nephrol 17: 1005–1014
Valerius MT, Patterson LT, Witte DP, Potter SS 2002 Microarray analysis of novel cell lines representing two stages of metanephric mesenchyme differentiation. Mech Dev 112: 219–232
Su GH, Sohn TA, Ryu B, Kern SE 2000 A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res 60: 3137–3142
Miyazaki Y, Tsuchida S, Fogo A, Ichikawa I 1999 The renal lesions that develop in neonatal mice during angiotensin inhibition mimic obstructive nephropathy. Kidney Int 55: 1683–1695
Haberland M, Montgomery RL, Olson EN 2009 The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42
Yang XJ, Seto E 2008 The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9: 206–218
Grozinger CM, Hassig CA, Schreiber SL 1999 Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA 96: 4868–4873
Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN 2007 Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21: 1790–1802
Shao Y, Lu J, Cheng C, Cui L, Zhang G, Huang B 2007 Reversible histone acetylation involved in transcriptional regulation of WT1 gene. Acta Biochim Biophys Sin (Shanghai) 39: 931–938
Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R 1993 WT-1 is required for early kidney development. Cell 74: 679–691
Yoshikawa M, Hishikawa K, Marumo T, Fujita T 2007 Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells. J Am Soc Nephrol 18: 58–65
Pentz ES, Lopez ML, Cordaillat M, Gomez RA 2008 Identity of the renin cell is mediated by cAMP and chromatin remodeling: an in vitro model for studying cell recruitment and plasticity. Am J Physiol Heart Circ Physiol 294: H699–H707
Ogryzko VV, Schiltz RL, Russanova V, Hioward BH 1996 The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959
Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML 2009 CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol 296: H1255–H1262
Ray S, Sherman CT, Lu M, Brasier AR 2002 Angiotensinogen gene expression is dependent on signal transducer and activator of transcription 3-mediated p300/cAMP response element binding protein-binding protein coactivator recruitment and histone acetyltransferase activity. Mol Endocrinol 16: 824–836
Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V 1994 Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367: 380–383
Costantini F, Shakya R 2006 GDNF/Ret signaling and the development of the kidney. Bioessays 28: 117–127
Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K 2007 PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282: 20395–20406
Patel SR, Kim D, Levitan I, Dressler GR 2007 The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell 13: 580–592
Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E 2002 ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10: 1119–1128
Zhang SL, Moini B, Ingelfinger JR 2004 Angiotensin II increases Pax-2 expression in fetal kidney cells via the AT2 receptor. J Am Soc Nephrol 15: 1452–1465
Acknowledgements
The authors thank Colleen Garrett for the assistance with Western blot analysis and Shaowei Chen from Dr. Samir El-Dahr's laboratory for the assistance with HDAC-1 knockdown.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by NIH Grants P20 RR17659 and DK-71699.
Rights and permissions
About this article
Cite this article
Song, R., Van Buren, T. & Yosypiv, I. Histone Deacetylases Are Critical Regulators of the Renin-Angiotensin System During Ureteric Bud Branching Morphogenesis. Pediatr Res 67, 573–578 (2010). https://doi.org/10.1203/PDR.0b013e3181da477c
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181da477c
This article is cited by
-
Angiotensin II represses Npr1 expression and receptor function by recruitment of transcription factors CREB and HSF-4a and activation of HDACs
Scientific Reports (2020)
-
Foxd1 is an upstream regulator of the renin–angiotensin system during metanephric kidney development
Pediatric Research (2017)
-
Influence of gestational salt restriction in fetal growth and in development of diseases in adulthood
Journal of Biomedical Science (2016)
-
Early life obesity and chronic kidney disease in later life
Pediatric Nephrology (2015)
-
Renin–angiotensin system in ureteric bud branching morphogenesis: implications for kidney disease
Pediatric Nephrology (2014)
