
Abstract
Necrotizing enterocolitis (NEC) is the most common life-threatening gastrointestinal emergency encountered in the neonatal intensive care unit. Despite advances in neonatal care, NEC remains a leading cause of morbidity and mortality among premature infants. Epidemiologic studies have identified multiple factors that increase an infant's risk for the development of NEC, although premature birth, bacterial colonization, and enteral feeding are thought to play central roles in disease pathogenesis. Appreciating factors that underlie the susceptibility of prematurely born infants to NEC is important for the development of new strategies aimed at the prevention and treatment of disease. In this review, we discuss defense mechanisms in the intestine and discuss how these systems may be insufficient in the prematurely born infant and thereby further contribute to initiation of NEC. In addition, based on a review of the literature, we suggest that, although numerous bacterial and viral pathogens have been associated with NEC, no individual organism is known to be responsible for disease. Finally, we comment on the possible role for probiotics in promoting maturation of intestinal defense mechanisms thereby attenuating or preventing the sequence of events that lead to NEC.
Similar content being viewed by others

Main
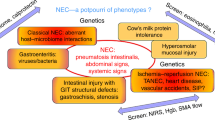
Necrotizing enterocolitis (NEC) is the most common surgical emergency affecting the gastrointestinal tract of infants in the neonatal intensive care unit (NICU). The incidence of NEC varies from 0.3 to 2.4 infants per 1000 live births, with nearly 70% of cases occurring in infants born at less than 36 wk of gestation. Although the national incidence of NEC varies, NEC affects 2–5% of all premature infants and accounts for up to 8% of all NICU admissions (1). The overall mortality for NEC ranges from 10% to 50% but approaches 100% for infants with the most severe form of the disease, characterized by full-thickness destruction of the intestine leading to intestinal perforation, peritonitis, bacterial invasion, and sepsis. The majority of these infants are extremely low birth weight infants whose disease requires surgical intervention (2). Despite optimal medical and surgical management of NEC, infants that recover from disease may still require prolonged hospitalization for related complications, such as intestinal obstruction from scarring, liver failure due to a prolonged requirement for total parenteral nutrition, short bowel syndrome with intestinal failure and associated nutritional deficiencies, and associated defects in growth and development. Hence, NEC is a significant and growing heath concern for prematurely born infants.
The events that lead to NEC in premature infants are multifactorial and complex and include a history of a complicated early neonatal course and poor intrauterine environment and perinatal transition. The only consistent epidemiologic risk factors for NEC are prematurity and a history of enteral feeding, which may include a rapid advancement in feeding or high osmotic strength formula feeding (2,3). Despite its predilection for premature infants, NEC has also been described in infants born at term. The onset of disease in these infants occurs within days of birth and is often associated with a history of hypoxia such as in cyanotic congenital heart disease or ischemia as single risk factors, suggesting that the pathogenic sequence leading to NEC in term infants may be distinct from that in the premature infant (3). Although recent evidence supports that susceptibility to NEC, in premature infants, is linked to gastrointestinal tract immaturity, the mechanisms whereby these factors incite or promote disease are poorly defined. Understanding the defense mechanisms in the premature intestine and their contribution to NEC susceptibility and pathogenesis is therefore of great importance. Elucidation of these mechanisms may allow for the development of strategies to target components that normally maintain integrity of the epithelial barrier, and to prevent the characteristic inflammatory cascade of NEC. This review will focus on defense mechanisms in the gastrointestinal tract of premature infants and highlight how the state of immaturity in the intestine may permit the initiation and propagation of inflammation and pathogenesis in NEC.
INTESTINAL EPITHELIUM AND NONIMMUNOLOGIC DEFENSE MECHANISMS IN NEC
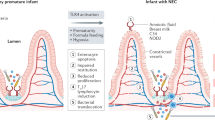
A primary function of the intestinal epithelium is to provide a physical barrier between the inside of the body and the luminal environment of the intestinal tract. Although epithelial cells lining the intestine provide a physical barrier, barrier function is incomplete in the absence of both nonimmunologic and immunologic components of the intestinal defense system. The nonimmunologic components include the mucus layer overlying epithelial cells, which is composed of glycoproteins called mucins, as well as several peptides such as trefoil factor, which are produced by goblet interspersed within epithelial layer (4). The degree of mucus production and its composition changes with postnatal age, in response to bacterial challenge, following colonization by commensal microbial flora, and during intestinal epithelial restitution following injury (5). Therefore, deficits in production or in the composition of intestinal mucus or in its associated peptides are hypothesized to contribute to the ability of bacteria to invade and participate in the induction and propagation of NEC (6). Tight junctions between epithelial cells maintain the semipermeable properties of epithelial cells, thereby limiting the passage of bacteria as well its permeability to a myriad of other luminal macromolecules, while promoting the transcytosis and absorption of macromolecules generated during the normal processes of digestion. Immaturity in the composition and function of tight junctions, through the interactions of occludins and claudins (7) most likely accounts for the increased permeability of the epithelial barrier in the newborn, a finding that is further exaggerated in the premature infant. Moreover, cytokines produced by epithelial cells and cells of the innate and adaptive immune system in response to bacteria and their products, such as lipopolysaccharide (LPS), may further disrupt the function of epithelial tight junctions, thereby promoting the translocation of bacteria and their products and other macromolecules (8). LPS, a known ligand of TLR4, has been shown to stimulate transcription and translation of several pro-inflammatory cytokines by epithelial cells, including tumor necrosis factor (TNF), IL-6, and IL-8, all of which independently promote the inflammatory cascade characteristic of NEC (9). Indeed, immature human epithelial cells secrete more IL-8 than did a mature human epithelial cell line (10). Additionally, inflammatory cytokines such as TNF-α, interferon (IFN)-γ, and IL-1β, produced by epithelial cells and cells of the innate and adaptive immune systems (see below), further contribute to epithelial barrier dysfunction through the up-regulation of inducible nitric oxide synthase (iNOS), local overproduction of nitric oxide (NO), and the generation of reactive nitrogen intermediate, peroxynitrite (ONOO–) (11). ONOO– has been shown to be directly responsible for increases in epithelial cell apoptosis and death and results in the decreases in epithelial cell proliferation and migration characteristic of NEC.
Proper barrier function in the intestine also includes the action of nonimmunologic defense mechanisms, such as physical and chemical factors at the epithelial-luminal interface, including acid secretion by gastric enterochromaffin cells, bile salt production by the liver, and the release of proteolytic enzymes by the pancreas in concert with the regular peristaltic contractions along the length of the intestine. Gastric acidity provides a first-line defense against bacterial passage into the proximal intestine. The premature human infant's gastric pH is initially high and then lowers toward mature levels with increasing age, ultimately reaching pH of <4 (12). In full-term infants and adults, migrating motor complexes propagate as waves along the intestine; however, these complexes are not present until approximately 34 wk of gestation (13). Disordered peristalsis in premature infants may therefore contribute to stasis of intestinal luminal contents and bacterial overgrowth, particularly in concert with formula feeding. Thus, pro-inflammatory cytokines and mediators produced by epithelial cells in conjunction with cells of the innate and adaptive immune system compound baseline defects and immaturity in barrier function associated with the premature infant at birth, thereby promoting the inflammatory cascade characteristic of NEC.
INNATE AND ADAPTIVE IMMUNOLOGIC DEFENSE MECHANISMS AND NEC
The gastrointestinal tract is home to the largest lymphoid tissue in the body and is responsible for coordinating immunologic defense mechanisms between the adaptive and innate immune systems (14). The gut-associated lymphoid tissue (GALT) consists of cells of the adaptive immune system called lymphocytes, as well as cells of the innate immune system, including resident tissue macrophages, dendritic cells, specialized epithelial cells called M cells overlying the Peyer's patches, and Paneth cells located in the crypt region of the intestine.
Macrophages are well-recognized phagocytic cells of the gut. However, both macrophages and dendritic cells act as professional antigen-presenting cells (APC), directly sampling antigens from within the lumen of the bowel, as well as functioning in the uptake of antigens processed by the M cell epithelium overlying Peyer's patches. In addition, professional APC, M cells, and epithelial cells lining the gut function as nonprofessional APC, presenting antigens to resident lymphocytes (15). The majority of antigens sampled directly from the lumen must be processed before their presentation to the cells of the adaptive immune system (16). Antigen degradation, processing, and presentation are vital steps in the initiation of an immune response. The processing and presentation of antigens is less efficient in the newborn, thereby reducing the ability of the adaptive and innate immune system to detect and respond to pathogenic organisms. Paneth cells produce a variety of antibacterial substances, including defensins, lysozymes, secretory phospholipase A2, and lectins. Defensins are small cationic peptides with broad-spectrum antimicrobial actions (17,18). Paneth cell secretion is stimulated by bacteria or by components of bacterial cell walls, such as LPS and lipoteichoic acid (LTA) (19). In a study by Salzman et al. (20), expression of α-defensin was found at significantly lower levels during fetal life when compared with the term newborn and adult. Therefore, the decreased production of antibacterial products by Paneth cells may predispose premature infants to bacterial overgrowth, thus allowing NEC to develop. Although polymorphonuclear leukocytes (PMN) are not regular inhabitants of the healthy intestine, an increased number of PMN may be detected in the intestinal epithelium early in NEC. PMN production by the bone marrow and function is impaired in the newborn, potentially contributing to intestinal bacterial overgrowth and invasion (21). Likewise, the inflammatory cascade induced by bacteria may include a reduced oxidative burst in the premature infant due to low levels of NADPH, thereby reducing the function of the innate immune system (22).
Lymphocytes, including B and T cells, are found dispersed throughout the intestinal wall and lymphoid aggregates. Intestinal intraepithelial lymphocytes are T cells that reside between enterocytes. Both subsets are found in the lamina propria, as well as in defined lymphoid structures, including Peyer's patches, small lymphoid aggregates, and mesenteric lymph nodes. Despite considerable development of human T and B cells during fetal life, complete maturation of the systemic and intestinal immune system occurs after birth and in response to colonization by commensal microbial flora (23). The adult human small intestine contains 200–300 Peyer's patches composed of germinal centers with distinct B and T cell zones. Although Peyer's patches are present in the newborn, they are reduced in number and size and lack germinal centers. Furthermore, the newborn lamina propria is largely devoid of immunoglobulin A (IgA)-secreting plasma cells. IgA, which is normally secreted into the mucus layer, is a potent inhibitor of bacterial and viral epithelial attachment. As human intestinal IgA production does not peak until 4 y of age, a relative deficiency of IgA contributes to the susceptibility of the newborn to infections at the mucosal surface (24). Additionally, T cells in the newborn are less responsive to antigenic stimulation and have a decreased proliferative response to a variety of mitogenic stimuli (25). Therefore, it is plausible that intestinal immune system of the premature infant (similar to the systemic immune system has a reduced capacity to control or respond to the overgrowth and invasion by pathogenic and commensal bacteria that is characteristic of infants with NEC.
BACTERIA AND THEIR PRODUCTS IN THE PATHOGENESIS OF NEC
Whether bacteria are primary in the initiation of NEC, or whether bacterial invasion occurs secondarily following the breakdown of the epithelial barrier is not known. The presence of pneumatosis intestinalis, air within the intestinal wall, however, indicates that bacterial fermentation at least accompanies disease. Thus far, however, a single bacterial species or virus has not been consistently isolated in cases of NEC (26–29). Enterobacteriaceae sp. are the most commonly described bacteria to be found in association with NEC (30–45). Clostridia sp. and Staphylococcus sp. have also been isolated from infants with NEC (37,46–57). Although bacteria are clearly the most commonly associated microbe with disease, isolation of viruses and fungus have been described (58–64) (Table 1). The data in Table 1 were derived from a PUBMED-based search of the English literature for NEC-associated pathogens using a modified Cochrane Collaboration data-quality scoring system. Due to the overall low quality of the data, however, a formal meta-analysis was not possible. The data are, however, consistent with the notion that NEC is unlikely to be a primary infectious or toxin-mediated disease. Although outbreaks of NEC have been described in association with feeding formula contaminated by Enterobacter sakazakii and breast milk containing Staphylococcus sp., it appears more likely that a dominant bacterial species gains access to the submucosa and circulation secondary to breakdown of the gut epithelial barrier, thereby playing a secondary but requisite role in disease.
Bacteria and their products have also been implicated in inflammatory bowel disease in humans (65) and in rodent models of inflammatory colitis and NEC (66–70). Detection and responses to microbial flora in the intestine occur through interactions with various pattern recognition receptors known as toll-like receptors (TLR) (71,72). These receptors recognize specific conserved pathogen-associated molecular patterns (PAMP), including glycoproteins, lipoproteins, glycolipids, peptidoglycans, fatty acids, and nucleic acids (73,74). Different TLR family members have been identified and are expressed in a variety of cell types including macrophages, dendritic cells, and enterocytes (73,75,76) (Table 2). TLR expression varies with postnatal maturation and cell type, indeed TLR deficiency has been demonstrated in macrophages and in the lung of neonatal mice (77). Differences in the expression of TLR may therefore alter a host's response to a commensal or pathogenic microorganism. For example, LPS is an outer membrane virulence factor associated with Gram-negative bacteria and the ligand for TLR-4. In vitro studies have demonstrated that LPS may facilitate bacterial transcytosis in Caco-2 cells (78) and increase bacterial translocation (79). Although TLR ligands are expressed by several pathogenic species, commensal microbes also express these ligands. In fact, commensal signaling through TLR appear important for normal intestinal maturation and epithelial homeostasis (80). Hence, intestinal epithelial expression patterns of TLR-4 in the presence of altered intestinal flora may result in a variety of local responses, including altered bacterial translocation, gut homeostasis, and induction of cytokine pathways (71,81). Indeed, the administration of LPS to mice replicates the findings of human NEC (82,83). In mouse models of colitis and NEC, TLR-4 knockout mice have reduced inflammatory infiltrates when compared with wild-type mice (84). Thus, signaling via TLR by both enterocytes and cells of the innate and adaptive immune system may play a crucial role in triggering the inflammatory sequence in NEC.
BREAST MILK FEEDING AND PROBIOTICS IN PROTECTION FROM NEC
The microbial ligands recognized by TLR are not specific to pathogens, and signaling through TLR by commensal microbial flora in the gut may in fact enable normal assembly and function of the intestinal immune system (85,86). The intestine is devoid of bacterial flora at birth but is rapidly colonized by the recto-vaginal flora of the mother (55). Colonization by commensal bacteria is required for the normal development and maturation of the newborn intestine. Bacterial-host cross-talk modulates gut vascular development and promotes epithelial barrier function (87). For example, the intestinal flora stimulates the production of IgA in animal models (88). Indeed, the presence of IgA correlates with a decrease in bacterial translocation in the gut. In fact, the presence of Bacteroides fragilis in vaginally born infants, when compared with infants born by cesarean section, correlates with higher levels of IgA and IgM secreting plasma cells in peripheral blood (89).
The sequence of intestinal bacterial colonization after birth may provide protection from overgrowth by potential pathogens. The earliest bacterial species to colonize the intestine are facultative anaerobes including Enterobacteriaceae sp. and Lactobacilli sp., followed by anaerobic bacteria such as Bifidobacterium sp., Bacteroides sp., Clostridium sp., and Eubacterium sp. (90). Lactobacillus and Bifidobacteria sp. predominate by approximately 10-d post partum in the full-term breast milk–fed infant, whereas the microbial flora in the formula-fed infant is more heterogeneous, with only 50% of the level of Bifidobacteria sp. found in breast milk–fed infants. Significant differences in the microbial flora are seen in prematurely born infants hospitalized in the NICU, when compared with term infants in a non-NICU setting. Several factors contribute to the abnormal flora found in NICU infants, including the microbes endemic to a particular NICU and the frequent use of antibiotics. The intestine of hospitalized premature infants is likely to be colonized by pathogenic bacteria such as Klebsiella sp., Enterobacter sp., and Clostridium sp., with a marked paucity of Bifidobacteria sp. (91). Even in breast milk–fed very low birth weight infants it takes several weeks to approach the levels of Bifidobacteria sp. found in a full-term breast-fed infant (92,93). Premature infants fed human breast milk do, however, have a reduced incidence of NEC, when compared with formula-fed infants (94). A prospective multicenter study of preterm infants found an almost 10-fold increase in the incidence of NEC in formula-fed infants as compared with those who were fed breast milk (95). The positive effects of breast milk feeding may be due to a variety of antimicrobial products present in breast milk, including immunoglobulins, cytokines, oligosaccharides, lactoferrin, and glycoproteins with antiadhesive capacity for bacteria (96). In addition, the high levels of prebiotics, particularly in colostrums, such as fructo-oligosaccharides, may promote colonization of breast milk–fed infants with Bifidobacteria sp. thereby abrogating colonization by other pathogenic organisms (97). Moreover, lactoferrin is a milk protein found in high concentration in colostrum, which has been shown to decrease bacterial translocation from the gut and improve survival in LPS-fed animal models (98). Breast milk also contains cellular factors that likely facilitate the development and maturation of the immune system. For example, colostrum contains concentrated leukocytes, including macrophages (55–60%), neutrophils (30–40%), and lymphocytes (5–10%) (99). These cells are believed to be active and viable, and have been isolated from the feces of infants fed human breast milk.
The identification of probiotic bacterial species involved in gut homeostasis has stimulated interest in their use in the prevention and treatment of a variety of intestinal diseases. Although their mechanisms of action are poorly understood, it is believed that probiotic bacteria may impair growth of pathogenic species by stimulating production of nonfunctional receptor “decoys” in the mucosal lining, by promoting pathogen binding without internalization by epithelial cells, or the activation of an anti-inflammatory cascade (Fig. 1). Nonpathogenic enteric microbes may exact an immunosuppressive effect on epithelial cells through TLR interactions (100). The potential therapeutic benefit of probiotics has led to interest in their role in the prevention of NEC. Lactobacillus and Bifidobacteria sp. produce acidic end products during cellular metabolism, thereby lowering the pH of the intestinal microenvironment creating a locally unfavorable environment for pathogens. In mice, Lactobacillus sp. associate with Peyer's patches and exert effects on T-helper cells (101). Furthermore, it has been shown that Lactobacillus sp. binds to mucins and intestinal epithelial cells, and may be able to reverse the permeability of the immature gut (102). Additionally, it is hypothesized that Lactobacillus sp. may stimulate cytokine secretion and pathogen aggregation. In adults the administration of probiotic bacteria reduced the mucosal expression of several key pro-inflammatory cytokines including IL-1β, IFN-γ, and IL-8, suggesting that probiotics inhibit the production of pro-inflammatory cytokines (103) and in turn reduce epithelial cell apoptosis and the loss of epithelial barrier function. In addition, other studies demonstrate that Lactobacillus sp. increase the expression of intestinal mucins, which may directly enhance mucosal barrier function (104). Several randomized prospective studies in human infants suggest that oral probiotics decreased the incidence of NEC, without the development of other infectious complications such as sepsis (105–107). However, isolated case reports of sepsis following Lactobacillus sp. feeding have been described in both pediatric and adult populations following probiotic therapy (108,109). Hence, until further studies of efficacy and safety are available, routine use of probiotic therapy in premature newborns cannot be recommended (Fig. 1).

Immaturity of the intestinal epithelial barrier and neonatal mucosal immune system predispose the premature infant to bacterial infection/invasion triggering the pathogenic sequence in NEC. The immature intestinal barrier may lack several key protective mechanisms that normally prevents invasion by luminal bacterial flora. The compromised gut barrier together with an altered bacterial flora in premature infants may stimulate the production of pro-inflammatory cytokines further compromising intestinal defense mechanisms. The resulting imbalance between epithelial cell injury and repair leads to a vicious cycle of bacterial invasion, immune activation, uncontrolled inflammation, and gut barrier failure.
SUMMARY
Necrotizing enterocolitis accounts for significant morbidity and mortality in the premature newborn. The sequence of events leading to the development of NEC is complex and still incompletely defined. Well-developed models of disease and clinical studies will be required to understand why the premature newborn is susceptible to NEC. Our consensus supports that proposed by others: damage to the immature intestine after birth results in invasion of the intestine by bacteria. Bacteria, in turn, initiate a cascade of inflammation that leads to further destruction, or perforation of the intestine and to systemic infection. This may lead to the infant's death or to long-term morbidity in survivors that includes significant alterations in growth and development. Despite published reports over the last 30 years implicating many different microbial pathogens in the pathogenesis of NEC, no individual bacteria, fungus, or virus has been definitively shown to be causative of NEC. Although no single organism fulfills Koch's postulate, the role of microbes in the pathogenesis of NEC, similar to other inflammatory bowel diseases, is crucial. Probiotic therapy is an area of great potential, providing several strategies to improve epithelial barrier function perhaps through TLR signaling, precocious maturation of the mucosal immune system, or simple exclusion of pathogens from critical niches in the intestine. As our understanding of the microbial-immunologic underpinnings of NEC continue to progress, we believe that successful modalities in the prevention and treatment of NEC will emerge.
Abbreviations
- APC:
-
antigen-presenting cells
- LPS:
-
lipopolysaccharide
- NEC:
-
necrotizing enterocolitis
- PMN:
-
polymorphonuclear leukocytes
- TLR:
-
Toll-like receptor
References
Kafetzis DA, Skevaki C, Costalos C 2003 Neonatal necrotizing enterocolitis: an overview. Curr Opin Infect Dis 16: 349–355
Blakely ML, Lally KP, McDonald S, Brown RL, Barnhart DC, Ricketts RR, Thompson WR, Scherer LR, Klein MD, Letton RW, Chwals WJ, Touloukian RJ, Kurkchubasche AG, Skinner MA, Moss RL, Hilfiker ML 2005 Postoperative outcomes of extremely low birth-weight infants with necrotizing enterocolitis or isolated intestinal perforation: a prospective cohort study by the NICHD Neonatal Research Network. Ann Surg 241: 984–989
Bolisetty S, Lui K, Oei J, Wojtulewicz J 2000 A regional study of underlying congenital diseases in term neonates with necrotizing enterocolitis. Acta Paediatr 89: 1226–1230
Plaut AG 1997 Trefoil peptides in the defense of the gastrointestinal tract. N Engl J Med 336: 506–507
Deplancke B, Gaskins HR 2001 Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr 73: 1131S–1141S
Vieten D, Corfield A, Carroll D, Ramani P, Spicer R 2005 Impaired mucosal regeneration in neonatal necrotising enterocolitis. Pediatr Surg Int 21: 153–160
Muresan Z, Paul DL, Goodenough DA 2000 Occludin 1B, a variant of the tight junction protein occludin. Mol Biol Cell 11: 627–634
Shen L, Turner JR 2006 Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am J Physiol Gastrointest Liver Physiol 290: G577–G582
Nanthakumar NN, Fusunyan RD, Sanderson I, Walker WA 2000 Inflammation in the developing human intestine: a possible pathophysiologic contribution to necrotizing enterocolitis. Proc Natl Acad Sci U S A 97: 6043–6048
Claud EC, Lu L, Anton PM, Savidge T, Walker WA, Cherayil BJ 2004 Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc Natl Acad Sci U S A 101: 7404–7408
Ford H, Watkins S, Reblock K, Rowe M 1997 The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg 32: 275–282
Kelly EJ, Newell SJ, Brownlee KG, Primrose JN, Dear PR 1993 Gastric acid secretion in preterm infants. Early Hum Dev 35: 215–220
Berseth CL 1989 Gestational evolution of small intestine motility in preterm and term infants. J Pediatr 115: 646–651
Mowat AM, Viney JL 1997 The anatomical basis of intestinal immunity. Immunol Rev 156: 145–166
Neutra MR, Mantis NJ, Kraehenbuhl JP 2001 Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat Immunol 2: 1004–1009
Shao L, Serrano D, Mayer L 2001 The role of epithelial cells in immune regulation in the gut. Semin Immunol 13: 163–176
Ganz T 2005 Defensins and other antimicrobial peptides: a historical perspective and an update. Comb Chem High Throughput Screen 8: 209–217
Ouellette AJ 2004 Defensin-mediated innate immunity in the small intestine. Best Pract Res Clin Gastroenterol 18: 405–419
Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ 2000 Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 1: 113–118
Salzman NH, Polin RA, Harris MC, Ruchelli E, Hebra A, Zirin-Butler S, Jawad A, Martin Porter E, Bevins CL 1998 Enteric defensin expression in necrotizing enterocolitis. Pediatr Res 44: 20–26
Carr R 2000 Neutrophil production and function in newborn infants. Br J Haematol 110: 18–28
Akella SS, Harris C 2001 Developmental ontogeny of NAD+ kinase in the rat conceptus. Toxicol Appl Pharmacol 170: 124–129
Hannet I, Erkeller-Yuksel F, Lydyard P, Deneys V, DeBruyere M 1992 Developmental and maturational changes in human blood lymphocyte subpopulations. Immunol Today 13: 215–218
Mayer L 2003 Mucosal immunity. Pediatrics 111: 1595–1600
Adkins B 1999 T-cell function in newborn mice and humans. Immunol Today 20: 330–335
Peter CS, Feuerhahn M, Bohnhorst B, Schlaud M, Ziesing S, von der Hardt H, Poets CF 1999 Necrotising enterocolitis: is there a relationship to specific pathogens?. Eur J Pediatr 158: 67–70
Guinan M, Schaberg D, Bruhn FW, Richardson CJ, Fox WW 1979 Epidemic occurrence of neonatal necrotizing enterocolitis. Am J Dis Child 133: 594–597
Anderson CL, Collin MF, O'Keefe JP, Challapalli M, Myers TF, Caldwell CC, Ahmed GS 1984 A widespread epidemic of mild necrotizing enterocolitis of unknown cause. Am J Dis Child 138: 979–983
Gaynes RP, Palmer S, Martone WJ, Holt CL, Buchter DS, Frawley LW, Perlino C, Kanto WP Jr 1984 The role of host factors in an outbreak of necrotizing enterocolitis. Am J Dis Child 138: 1118–1120
Hill HR, Hunt CE, Matsen JM 1974 Nosocomial colonization with Klebsiella, type 26, in a neonatal intensive-care unit associated with an outbreak of sepsis, meningitis, and necrotizing enterocolitis. J Pediatr 85: 415–419
Speer ME, Taber LH, Yow MD, Rudolph AJ, Urteaga J, Waller S 1976 Fulminant neonatal sepsis and necrotizing enterocolitis associated with a “nonenteropathogenic” strain of Escherichia coli. J Pediatr 89: 91–95
Bell MJ, Feigin RD, Ternberg JL 1979 Changes in the incidence of necrotizing enterocolitis associated with variation of the gastrointestinal microflora in neonates. Am J Surg 138: 629–631
Powell J, Bureau MA, Pare C, Gaildry ML, Cabana D, Patriquin H 1980 Necrotizing enterocolitis. Epidemic following an outbreak of Enterobacter cloacae type 3305573 in a neonatal intensive care unit. Am J Dis Child 134: 1152–1154
Broadhead RL, Sehgal KC 1981 Necrotising enterocolitis associated with Salmonella paratyphi B type 4, 5. Ann Trop Paediatr 1: 65–68
Cushing AH 1983 Necrotizing enterocolitis with Escherichia coli heat-labile enterotoxin. Pediatrics 71: 626–630
Mollitt DL, Tepas JJ 3rd, Talbert JL 1988 The microbiology of neonatal peritonitis. Arch Surg 123: 176–179
Uauy RD, Fanaroff AA, Korones SB, Phillips EA, Phillips JB, Wright LL 1991 Necrotizing enterocolitis in very low birth weight infants: biodemographic and clinical correlates. National Institute of Child Health and Human Development Neonatal Research Network. J Pediatr 119: 630–638
Millar MR, MacKay P, Levene M, Langdale V, Martin C 1992 Enterobacteriaceae and neonatal necrotising enterocolitis. Arch Dis Child 67: 53–56
Gregersen N, Van Nierop W, Von Gottberg A, Duse A, Davies V, Cooper P 1999 Klebsiella pneumoniae with extended spectrum beta-lactamase activity associated with a necrotizing enterocolitis outbreak. Pediatr Infect Dis J 18: 963–967
Hoy CM, Wood CM, Hawkey PM, Puntis JW 2000 Duodenal microflora in very-low-birth-weight neonates and relation to necrotizing enterocolitis. J Clin Microbiol 38: 4539–4547
van Acker J, de Smet F, Muyldermans G, Bougatef A, Naessens A, Lauwers S 2001 Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol 39: 293–297
Llanos AR, Moss ME, Pinzon MC, Dye T, Sinkin RA, Kendig JW 2002 Epidemiology of neonatal necrotising enterocolitis: a population-based study. Paediatr Perinat Epidemiol 16: 342–349
Mehall JR, Kite CA, Saltzman DA, Wallett T, Jackson RJ, Smith SD 2002 Prospective study of the incidence and complications of bacterial contamination of enteral feeding in neonates. J Pediatr Surg 37: 1177–1182
Krediet TG, van Lelyveld N, Vijlbrief DC, Brouwers HA, Kramer WL, Fleer A, Gerards LJ 2003 Microbiological factors associated with neonatal necrotizing enterocolitis: protective effect of early antibiotic treatment. Acta Paediatr 92: 1180–1182
Coates EW, Karlowicz MG, Croitoru DP, Buescher ES 2005 Distinctive distribution of pathogens associated with peritonitis in neonates with focal intestinal perforation compared with necrotizing enterocolitis. Pediatrics 116: e241–e246
Duffy LC, Zielezny MA, Carrion V, Griffiths E, Dryja D, Hilty M, Rook C, Morin F 3rd 1997 Concordance of bacterial cultures with endotoxin and interleukin-6 in necrotizing enterocolitis. Dig Dis Sci 42: 359–365
Blakey JL, Lubitz L, Campbell NT, Gillam GL, Bishop RF, Barnes GL 1985 Enteric colonization in sporadic neonatal necrotizing enterocolitis. J Pediatr Gastroenterol Nutr 4: 591–595
Kosloske AM, Ball WS Jr, Umland E, Skipper B 1985 Clostridial necrotizing enterocolitis. J Pediatr Surg 20: 155–159
Howard FM, Flynn DM, Bradley JM, Noone P, Szawatkowski M 1977 Outbreak of necrotising enterocolitis caused by Clostridium butyricum. Lancet 2: 1099–1102
Cashore WJ, Peter G, Lauermann M, Stonestreet BS, Oh W 1981 Clostridia colonization and clostridial toxin in neonatal necrotizing enterocolitis. J Pediatr 98: 308–311
Han VK, Sayed H, Chance GW, Brabyn DG, Shaheed WA 1983 An outbreak of Clostridium difficile necrotizing enterocolitis: a case for oral vancomycin therapy?. Pediatrics 71: 935–941
Laverdiere M, Robert A, Chicoine R, Salet D, Rosenfeld R 1978 Clostridia in necrotising enterocolitis. Lancet 2: 377
Alfa MJ, Robson D, Davi M, Bernard K, Van Caeseele P, Harding GK 2002 An outbreak of necrotizing enterocolitis associated with a novel clostridium species in a neonatal intensive care unit. Clin Infect Dis 35: S101–S105
Isaacs D 2003 A ten year, multicentre study of coagulase negative staphylococcal infections in Australasian neonatal units. Arch Dis Child Fetal Neonatal Ed 88: F89–F93
Hallstrom M, Eerola E, Vuento R, Janas M, Tammela O 2004 Effects of mode of delivery and necrotising enterocolitis on the intestinal microflora in preterm infants. Eur J Clin Microbiol Infect Dis 23: 463–470
Liu RX, Johnson A 1984 Necrotizing enterocolitis with epidemic Staphylococcus in a neonatal intensive care unit. Chin Med J (Engl) 97: 278–282
Ng KY, Desmond PV, Collier N 1993 Relapsing pancreatitis due to juxta-pancreatic duodenal duplication cyst with pancreatic ductal communication. Aust N Z J Surg 63: 224–229
Mogilner BM, Bar-Yochai A, Miskin A, Shif I, Aboudi Y 1983 Necrotizing enterocolitis associated with rotavirus infection. Isr J Med Sci 19: 894–896
Rotbart HA, Nelson WL, Glode MP, Triffon TC, Kogut SJ, Yolken RH, Hernandez JA, Levin MJ 1988 Neonatal rotavirus-associated necrotizing enterocolitis: case control study and prospective surveillance during an outbreak. J Pediatr 112: 87–93
Rotbart HA, Levin MJ, Yolken RH, Manchester DK, Jantzen J 1983 An outbreak of rotavirus-associated neonatal necrotizing enterocolitis. J Pediatr 103: 454–459
Birenbaum E, Handsher R, Kuint J, Dagan R, Raichman B, Mendelson E, Linder N 1997 Echovirus type 22 outbreak associated with gastro-intestinal disease in a neonatal intensive care unit. Am J Perinatol 14: 469–473
Chany C, Moscovici O, Lebon P, Rousset S 1982 Association of coronavirus infection with neonatal necrotizing enterocolitis. Pediatrics 69: 209–214
Lodha A, de Silva N, Petric M, Moore AM 2005 Human torovirus: a new virus associated with neonatal necrotizing enterocolitis. Acta Paediatr 94: 1085–1088
Karlowicz MG 1993 Risk factors associated with fungal peritonitis in very low birth weight neonates with severe necrotizing enterocolitis: a case-control study. Pediatr Infect Dis J 12: 574–577
Sartor RB 2003 Targeting enteric bacteria in treatment of inflammatory bowel diseases: why, how, and when. Curr Opin Gastroenterol 19: 358–365
Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC, Owen MJ 1997 T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 150: 91–97
Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE Jr, Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB 1996 Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest 98: 945–953
Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernandez-Sueiro JL, Balish E, Hammer RE 1994 The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med 180: 2359–2364
Videla S, Vilaseca J, Guarner F, Salas A, Treserra F, Crespo E, Antolin M, Malagelada JR 1994 Role of intestinal microflora in chronic inflammation and ulceration of the rat colon. Gut 35: 1090–1097
Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I 1993 Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75: 253–261
Beutler B, Rietschel ET 2003 Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol 3: 169–176
Takeda K, Akira S 2003 Toll receptors and pathogen resistance. Cell Microbiol 5: 143–153
Chabot S, Wagner JS, Farrant S, Neutra MR 2006 TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol 176: 4275–4283
Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS 2006 The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol 177: 3273–3282
Hornef MW, Normark BH, Vandewalle A, Normark S 2003 Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med 198: 1225–1235
Takeda K 2005 Evolution and integration of innate immune recognition systems: the Toll-like receptors. J Endotoxin Res 11: 51–55
Harju K, Glumoff V, Hallman M 2001 Ontogeny of Toll-like receptors Tlr2 and Tlr4 in mice. Pediatr Res 49: 81–83
Panigrahi P, Bamford P, Horvath K, Morris JG Jr, Gewolb IH 1996 Escherichia coli transcytosis in a Caco-2 cell model: implications in neonatal necrotizing enterocolitis. Pediatr Res 40: 415–421
Deitch EA, Berg R, Specian R 1987 Endotoxin promotes the translocation of bacteria from the gut. Arch Surg 122: 185–190
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R 2004 Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241
Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, Hackam DJ 2006 Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol 176: 3070–3079
Caplan MS, Kelly A, Hsueh W 1992 Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet activating factor. Pediatr Res 31: 428–434
Musemeche C, Caplan M, Hsueh W, Sun X, Kelly A 1991 Experimental necrotizing enterocolitis: the role of polymorphonuclear neutrophils. J Pediatr Surg 26: 1047–1049 discussion 1049–1050.
Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT 2005 Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 288: G1055–G1065
Janeway CA Jr, Medzhitov R 2002 Innate immune recognition. Annu Rev Immunol 20: 197–216
Medzhitov R, Janeway CA Jr 2002 Decoding the patterns of self and nonself by the innate immune system. Science 296: 298–300
Stappenbeck TS, Hooper LV, Gordon JI 2002 Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A 99: 15451–15455
Shroff KE, Meslin K, Cebra JJ 1995 Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun 63: 3904–3913
Gronlund MM, Arvilommi H, Kero P, Lehtonen OP, Isolauri E 2000 Importance of intestinal colonisation in the maturation of humoral immunity in early infancy: a prospective follow up study of healthy infants aged 0-6 months. Arch Dis Child Fetal Neonatal Ed 83: F186–F192
Fanaro S, Chierici R, Guerrini P, Vigi V 2003 Intestinal microflora in early infancy: composition and development. Acta Paediatr Suppl 91: 48–55
Sakata H, Yoshioka H, Fujita K 1985 Development of the intestinal flora in very low birth weight infants compared to normal full-term newborns. Eur J Pediatr 144: 186–190
Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW 2000 Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr 30: 61–67
Blakey JL, Lubitz L, Barnes GL, Bishop RF, Campbell NT, Gillam GL 1982 Development of gut colonisation in pre-term neonates. J Med Microbiol 15: 519–529
Schanler RJ, Shulman RJ, Lau C 1999 Feeding strategies for premature infants: beneficial outcomes of feeding fortified human milk versus preterm formula. Pediatrics 103: 1150–1157
Lucas A, Cole TJ 1990 Breast milk and neonatal necrotising enterocolitis. Lancet 336: 1519–1523
Goldman AS 1993 The immune system of human milk: antimicrobial, antiinflammatory and immunomodulating properties. Pediatr Infect Dis J 12: 664–671
Gibson GR 1998 Dietary modulation of the human gut microflora using prebiotics. Br J Nutr 80: S209–S212
Lee WJ, Farmer JL, Hilty M, Kim YB 1998 The protective effects of lactoferrin feeding against endotoxin lethal shock in germfree piglets. Infect Immun 66: 1421–1426
Michie CA 1998 The long term effects of breastfeeding: a role for the cells in breast milk?. J Trop Pediatr 44: 2–3
Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, Rao AS, Madara JL 2000 Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 289: 1560–1563
Plant L, Conway P 2001 Association of Lactobacillus spp. with Peyer's patches in mice. Clin Diagn Lab Immunol 8: 320–324
Baken KA, Ezendam J, Gremmer ER, de Klerk A, Pennings JL, Matthee B, Peijnenburg AA, van Loveren H 2006 Evaluation of immunomodulation by Lactobacillus casei Shirota: immune function, autoimmunity and gene expression. Int J Food Microbiol 112: 8–18
Bergonzelli GE, Granato D, Pridmore RD, Marvin-Guy LF, Donnicola D, Corthesy-Theulaz IE 2006 GroEL of Lactobacillus johnsonii La1 (NCC 533) is cell surface associated: potential role in interactions with the host and the gastric pathogen Helicobacter pylori. Infect Immun 74: 425–434
Mack DR, Michail S, Wei S, McDougall L, Hollingsworth MA 1999 Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am J Physiol 276: G941–G950
Dani C, Biadaioli R, Bertini G, Martelli E, Rubaltelli FF 2002 Probiotics feeding in prevention of urinary tract infection, bacterial sepsis and necrotizing enterocolitis in preterm infants. A prospective double-blind study. Biol Neonate 82: 103–108
Lin HC, Su BH, Chen AC, Lin TW, Tsai CH, Yeh TF, Oh W 2005 Oral probiotics reduce the incidence and severity of necrotizing enterocolitis in very low birth weight infants. Pediatrics 115: 1–4
Bin-Nun A, Bromiker R, Wilschanski M, Kaplan M, Rudensky B, Caplan M, Hammerman C 2005 Oral probiotics prevent necrotizing enterocolitis in very low birth weight neonates. J Pediatr 147: 192–196
Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK 2005 Lactobacillus sepsis associated with probiotic therapy. Pediatrics 115: 178–181
Ledoux D, Labombardi VJ, Karter D 2006 Lactobacillus acidophilus bacteraemia after use of a probiotic in a patient with AIDS and Hodgkin's disease. Int J STD AIDS 17: 280–282
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases grant AI-49473 (HRF, JSU, and VC).
Rights and permissions
About this article
Cite this article
Hunter, C., Upperman, J., Ford, H. et al. Understanding the Susceptibility of the Premature Infant to Necrotizing Enterocolitis (NEC). Pediatr Res 63, 117–123 (2008). https://doi.org/10.1203/PDR.0b013e31815ed64c
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31815ed64c
This article is cited by
-
Effects of prophylactic probiotics supplementation on infants born very preterm or very low birth weight
Journal of Perinatology (2023)
-
CCL3 aggravates intestinal damage in NEC by promoting macrophage chemotaxis and M1 macrophage polarization
Pediatric Research (2023)
-
Dedifferentiated fat cells administration ameliorates abnormal expressions of fatty acids metabolism-related protein expressions and intestinal tissue damage in experimental necrotizing enterocolitis
Scientific Reports (2023)
-
Intestine and brain TLR-4 modulation following N-acetyl-cysteine treatment in NEC rodent model
Scientific Reports (2023)
-
Advances in our understanding of the molecular pathogenesis of necrotizing enterocolitis
BMC Pediatrics (2022)
