
Abstract
Genomic imprinting refers to parent-of-origin–specific gene expression. Human chromosome band 11p15.5 houses a large cluster of genes that are imprinted. Dysregulation of this gene cluster is associated with the overgrowth and tumor predisposition syndrome, Beckwith-Wiedemann syndrome. Several genes in this imprinted cluster encode proteins involved in growth regulation, e.g. the paternally expressed IGF2 and the maternally expressed cell-cycle regulator cyclin dependent kinase inhibitor, CDKN1C. Disruption of imprinted gene expression can result from genetic or epigenetic alterations. Genetic alterations such as duplication, deletion, translocation, inversion, and mutation in imprinted regions have been shown to cause disease. In addition, epimutations that are extrinsic to the primary DNA sequence have also been shown to cause disease. These epimutations usually involve gain or loss of methylation at regulatory differentially methylated regions. Recently, several human diseases in addition to Beckwith-Wiedemann syndrome have been reported to have molecular alterations at chromosome 11p15.5. These include isolated hemihyperplasia, Russell-Silver syndrome, and transient neonatal diabetes mellitus. These molecular alterations and their phenotypic effects on growth are discussed.
Similar content being viewed by others

Main
Most mammalian autosomal genes are expressed from both the maternally inherited and paternally inherited copies of the chromosomes. However, some genes are expressed in a parent-of-origin–specific manner. This phenomenon, known as genomic imprinting, is regulated by epigenetic mechanisms (1). Epigenetics refers to transmissible changes in gene expression that are not accompanied by a change in primary nucleotide sequence (2). Epigenetic marks include alterations in DNA methylation, chromatin conformation (specified in part by the covalent modifications of histone proteins), and noncoding RNA transcripts. Different combinations of these epigenetic marks have been demonstrated to be critical for normal imprinted gene regulation among various genomic regions. There is considerable evidence of dysregulation of imprinted gene expression in human disorders of growth and development. This can occur via primary epigenetic alterations or genetic alterations that change the contribution of parental alleles, such as duplications, deletions, and UPD. UPD refers to the presence of two chromosomal regions from one parent and none from the other.
To date, approximately 50 imprinted genes have been identified in the human genome (3–5). These genes tend to be clustered together in imprinted chromosomal domains. Such domains have been mapped to chromosomes 7q32, 11p15, 15q11, and 20q13. These clusters of genes are characterized by cis regulation via an IC. IC are regions of DNA that regulate the expression of imprinted genes in cis over large distances up to one megabase. IC are also often associated with noncoding RNA transcripts and differential methylation of parental alleles known as DMR (6). One of the best studied clusters of imprinted genes is located on human chromosome 11p15. This cluster contains several imprinted genes that control growth and development (7). Dysregulation of imprinted genes in the 11p15 cluster causes BWS (8–10). Imprinted genes in this cluster can also be dysregulated in IH (11–15). Recently, some patients with RSS, a disorder clinically diagnosed in children with severe pre- and postnatal growth restriction and few dysmorphic features, have also been shown to have methylation alterations in this domain (16–18). Finally, some patients with TNDM with loss of methylation at a locus on chromosome 6q24 have also been shown to have loss of methylation at 11p15 (19). Thus, the 11p15 imprinted region plays an important role in development and growth control. The modulation of epigenetic marks and imprinted gene expression appears now to be important in several conditions other than BWS.
11p15.5 IMPRINTED CHROMOSOMAL REGION
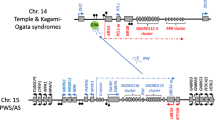
The regulation of imprinted genes on chromosome 11p15.5 is functionally divided into two domains that control the expression of genes over one megabase (Fig. 1). In Domain 1, the imprinted genes IGF2 and H19 are found. IGF2 is a paternally expressed fetal growth factor that has been implicated in the pathomechanism of BWS (20). H19 is a maternally expressed noncoding RNA for which the function is not well understood. Imprinted expression of these genes is regulated by the action of H19 DMR or IC. Differential methylation of the H19 DMR is important in the regulation of IGF2 and H19 expression. The H19 DMR is usually methylated on the paternal allele and this prohibits the binding of CTCF, a zinc finger protein with insulating activity (21). The maternal allele is unmethylated and thus CTCF binding is permitted. H19 and IGF2 share a set of downstream mesodermal and endodermal enhancers. Thus, they must compete for access to them for their expression. When CTCF is bound to the maternal allele, it blocks access to the IGF2 promoters and IGF2 is expressed solely from the paternal allele where access is not obstructed by CTCF.

Schematic representation of imprinted gene clusters on human chromosome 11p15.5 and 6q24.2. Maternally expressed genes are indicated as red lines with arrows showing transcription and direction, repressed genes are a light red without arrows. Paternally expressed genes are indicated as blue lines with arrows, and light blue lines are repressed. Hollow squares show the location on normally unmethylated DMR and filled squares indicated the DMR is normally methylated. Methylation alterations, such as loss of methylation (red box) and gain of methylation (green box), show the locations of these changes in each of the four syndromes: BWS, IH, RSS spectrum, and TNDM. This is a simplified representation of these very complex regions, showing the imprinted regions relevant to the discussion in the text. Full details on this region can be seen at UCSC Genome Browser http://genome.ucsc.edu, chr11:1,950,000-2,950,000. For detailed information on imprinted genes, see the Catalogue of Imprinted Genes and Parent-of-Origin Effects at http://igc.otago.ac.nz/home.html.
Domain 2 contains the imprinted genes KCNQ1, KCNQ1OT1, CDKN1C, SLC22A18, and PHLDA2. The IC for domain 2 is contained within intron 10 of the gene KCNQ1OT1 (22,23). This differentially methylated region (KCNQ1OT1 DMR, formerly KvDMR) contains the promoter for KCNQ1OT1, a noncoding transcript, that is thought to regulate in cis the expression of imprinted genes in the domain 2 cluster (24–26)
BWS
BWS (OMIM#130650) is an overgrowth syndrome originally characterized by the presence of macrosomia, macroglossia, and abdominal wall defects. However, many other features are often observed in BWS such as neonatal hypoglycemia, visceromegaly, helical ear pits and creases, hemihyperplasia, and an elevated risk for pediatric embryonal tumors such as Wilms tumor and hepatoblastoma (9,10).
There are several molecular alterations associated with BWS. Genetic alterations that involve both domain 1 and 2 include paternal uniparental disomy for a segment of chromosome 11 that always encompasses 11p15.5. This alteration, which occurs in 20% of BWS patients (9,10), results in a bipaternal pattern of expression, that is, there is a double dose of all paternally expressed genes and no expression of maternally expressed genes. Rarely (1%), BWS patients have duplications of chromosome 11p15.5. Maternally transmitted translocations/inversions also occur rarely (1%), almost always disrupting the gene KCNQ1 (9).
In domain 1, gain of methylation for the maternal allele of the H19 DMR occurs sporadically in 5–10% of BWS patients (Fig. 1). Recently, Sparago et al. (27,28) reported five heritable partial microdeletions of the H19 DMR, resulting in gain of methylation presumably due to disruption of CTCF binding to the DMR. Prawitt et al. (29) also reported a microdeletion of the H19 DMR. However, methylation was not altered and was accompanied by duplication of the IGF2 gene. In this case, the molecular basis for BWS is unusually complex.
In domain 2, sporadic loss of methylation of the KCNQ1OT1 DMR occurs in 50% of patients (9) (Fig. 1). There has only been one reported microdeletion, deleting 70 kb and likely the entire KCNQ1OT1 transcript (30). Mutations of the gene CDKN1C (p57KIP2) itself are seen both sporadically (5%) and in autosomal dominant pedigrees (25%). Loss of methylation at the KCNQ1OT1 DMR has been shown to be associated with a decreased amount of CDKN1C expression indicating a pivotal role for CDKN1C in the pathophysiology of BWS (25).
IH
IH (OMIM# 235000) is an abnormality of cell proliferation leading to asymmetric overgrowth of one or more regions of the body. IH occurs with an estimated incidence of 1 in 86,000 (31), with some authors reporting a higher frequency in females (32,33). Hemihyperplasia can occur as an isolated clinical finding or as a feature of other syndromes such as BWS. In IH, a broad range of tumors has been reported (34) which in some cases are distinct from the profile seen in BWS, supporting the concept that IH is a distinct clinical entity associated with heterogeneous molecular etiologies (15).
A subset of IH cases has been shown to be associated with alterations to the chromosome 11p15.5 region. Mosaic paternal uniparental disomy for chromosome 11, gain of methylation of H19 DMR, loss of methylation of the KCNQ1OT1 DMR have all been demonstrated (Fig. 1) (11–15). In all of the above reports, some cases of IH did not demonstrate a detectable alteration on chromosome 11p15. Because uniparental disomy and epigenetic alterations can often be mosaic in somatic cells (15), it is possible that this molecular change was present in those cases of IH but not in the sampled tissue. This is an important consideration given that tissue is often not available directly from the overgrown area and most diagnostic samples are derived from blood. It is also possible that 11p15 is only one of several loci involved in the growth dysregulation observed in patients with IH.
RSS
In striking contrast with BWS, RSS (OMIM 180860) is characteristically diagnosed in individuals with severe prenatal and postnatal growth compromise, usually with normal head size, and a variable combination of facial dysmorphic features (triangular face, down-turned angles of the mouth, prominent forehead/nasal bridge, and small jaw), often with limb or facial asymmetry, with hypotrophy of a limb or side, and developmental delay (35–37). Indeed, several aspects of the phenotype are the reverse of BWS prompting clinicians to hypothesize that the same gene could be involved in both conditions (38).
Most cases of RSS are sporadic; occasionally there are familial cases. There is, indeed, familial and chromosomal evidence for genetic heterogeneity (37). Most cases of RSS have a normal karyotype, however, some reports describe structural chromosomal anomalies of chromosomes 7, 8, 11, 15, 17, and 18 (39,40). Using the information from chromosomal aberrations has been helpful in the identification of genomic regions where genes involved in the etiology of RSS are located. To date, however, no DNA sequence mutations have been found in any of the candidate genes tested (40).
There is considerable controversy regarding the clinical criteria required for a clinical diagnosis of RSS. Prenatal growth compromise, with or without postnatal short stature, has been the most consistent feature described in patients categorized as RSS or RSS-like. Significant variation occurs in the remaining phenotypic features. Molecular results should therefore be examined in the context of the clinical criteria used for ascertainment of the subjects. Maternal uniparental disomy of chromosome 7, a chromosome with known imprinted loci, has been found in 7–10% of RSS cases ascertained using stringent diagnostic criteria. These data demonstrate that imprint dysregulation on human chromosome 7 can cause RSS (40). Further, duplication of maternal 11p15 has also been reported in individuals with reasonably stringent RSS diagnostic criteria (39). Because growth dysregulation in the two imprinted domains on 11p15 has been observed with BWS, it was therefore postulated that hypomethylation of H19 promoter and/or IC1 may be a mechanism leading to RSS or RSS-like clinical features. Gicquel et al. (18) published a study of nine patients diagnosed using strict RSS criteria, who tested negative for maternal UPD7. In this study, five of the nine showed loss of methylation for several sites in domain 1 of 11p15.5 including the H19 promoter, IC1, and IGF2 DMR2 (Fig. 1). For domain 2, no methylation alterations were found at the KCNQ1OT1 DMR. They concluded that loss of methylation of the telomeric imprinted domain of region 11p15 could be an important cause of RSS.
In RSS, the molecular signature, loss of methylation of the H19 DMR, is the opposite of that found in BWS, which is gain of methylation of the H19 DMR. However, the small number of cases studied by Gicquel et al. (18) made it difficult to determine the prevalence of this alteration in RSS. Eggermann et al. (17) screened 51 patients with prenatal and postnatal growth restriction with three or more phenotypic features of RSS. They found loss of methylation at H19 DMR in 16/51 (31%) of their patients. Bliek et al. (16) reported nine further cases with loss of methylation of the H19 promoter, only two of which were diagnosed with RSS using strict criteria. The remaining cases had prenatal and/or postnatal growth compromise plus asymmetry, and lacked sufficient criteria to be classified as RSS. Bliek et al. (16) looked for mutations in CTCF binding sites but none were found in their limited series. They concluded that, although it seems that there is a correlation between the methylation index and the severity of the phenotype, hypomethylation at the H19 DMR can occur in individuals who are just small. In summary, the main phenotypic feature consistently seen among patients with H19 DMR hypomethylation is the severe prenatal growth restriction with the postnatal maintenance of short stature (more then 2 SD below the mean).
AN IMPRINTED REGION ON 6q24 AFFECTS IMPRINTING ON CHROMOSOME 11p15.5
Recent evidence suggests that imprint regulation of chromosome 11p15.5 may be impacted by expression of imprinted genes on chromosome 6q24. This hypothesis has been developed to explain recent molecular data reported for two imprinting disorders, BWS and TNDM (19,41). TNDM (OMIM# 601410) has an incidence of 1 in 400,000 live births. In this condition, growth-retarded neonates present within the first 6 wk of life with persistent hyperglycemia and low or undetectable insulin levels (42). Another feature noted in the neonatal period by many authors is macroglossia (43) and umbilical hernia (44). The observation of partial pat UPD6 in patients with TNDM suggests that somatic mosaicism may affect the clinical presentation of TNDM. That is, the presence of different genetic mechanisms could contribute to the variability in phenotype seen in patients, as is the case with BWS.
TNDM has been associated with abnormalities involving chromosome 6. To date, three types of molecular abnormalities are identified in patients with TNDM, including paternal uniparental disomy of chromosome 6 (UPD6), paternally inherited duplication of 6q24, and loss of methylation at the PLAGL1 (ZAC) DMR (Fig. 1) (42).
The PLAGL1 gene, maps to 6q24–25, a region implicated in the origin of several cancers (45). This gene is a widely expressed zinc finger protein that shows transactivation and DNA-binding activity. PLAGL1 exhibits tumor suppressor function through the induction of both apoptosis and cell cycle arrest (46,47). PLAGL1 encodes a zinc finger protein and is preferentially expressed from the paternal allele with the maternal allele silent and methylated (48).
A comparison of phenotypes between BWS and TNDM demonstrates concordance for the tongue and umbilical findings but discordance for the growth findings. The overlap in the target tissues affected in these two disorders may reflect a regulatory association between imprinted regions at 6q24 and 11p15. In fact, several studies have reported, in TNDM patients, aberrant methylation at imprinted loci on both chromosomes 6q24 and 11p15.5, respectively. Mackay et al. (19) identified, among 43 TNDM patients tested, two with maternal loss of methylation (LOM) at the KCNQ1OT1 DMR, both of whom also exhibited maternal LOM at the PLAGL1 DMR. These two TNDM patients presented with clinical manifestations of severe growth restriction at birth, hyperglycemia, umbilical hernia, and macroglossia. None of the 16 BWS patients tested in the same study showed aberrant methylation at the PLAGL1 DMR. Another report by Arima et al. (41) showed similar findings. They examined the methylation status of the PLAGL1 DMR and KCNQ1OT1 DMR in 8 BWS and 17 TNDM patients. All BWS patients showed normal methylation at the PLAGL1 DMR, but two TNDM patients showed loss of methylation at KCNQ1OT1 DMR. One of these patients was characterized with TNDM complicated with umbilical hernia and macroglossia, features commonly seen in BWS patients. These results support the hypothesis that there is a biologic interaction between these two loci. Further credence for this position can be derived from a comparison of the expression pattern in mouse of the Zac 1 and p57Kip2 genes, which show a strikingly similar expression pattern in many tissues including the tongue (41). The presence of a zinc finger motif within the PLAGL1 protein suggests a DNA-binding capability that directed the authors to look for PLAGL1 binding sites in the CpG island of the human CDKN1C (p57KIP2) gene. Their work showed that the PLAGL1 protein binds to, and activates expression of, KCNQ1OT1 located on chromosome 11p15.5, which is known to negatively regulate the p57KIP2 gene in cis (41). These data suggest that PLAGL1 may affect p57KIP2 gene expression through the KCNQ1OT1 DMR, forming part of a novel signaling pathway that regulates cell growth.
The interaction of these two regions will require further investigation in terms of both basic molecular biology and clinical phenotype. Mackay et al. (49) suggested that methylation defects at more than one locus can modify the clinical presentation of the TNDM phenotype. Most of the TNDM patients presenting with loss of methylation at KCNQ1OT1 DMR have macroglossia and abdominal wall defects, clinical features commonly seen in BWS.
SUMMARY
Epigenetic alterations of differentially methylated imprinting centers on chromosome 11p15.5 have been linked to several growth-related disorders. These data highlight the fact that interactions among imprinted genes are likely important factors in the expression of clinical disease. Recent observations have demonstrated that many imprinted growth regulatory genes are co-regulated in a network of imprinted genes (50).
Abbreviations
- BWS:
-
Beckwith-Wiedemann syndrome
- DMR:
-
differentially methylated region
- IC:
-
imprinting center
- IH:
-
isolated hemihyperplasia
- RSS:
-
Russell-Silver syndrome
- TNDM:
-
transient neonatal diabetes mellitus
- UPD:
-
uniparental disomy
References
Bartolomei MS, Tilghman SM 1997 Genomic imprinting in mammals. Annu Rev Genet 31: 493–525
Wolffe AP, Matzke MA 1999 Epigenetics: regulation through repression. Science 286: 481–486
Glaser RL, Ramsay JP, Morison IM 2006 The imprinted gene and parent-of-origin effect database now includes parental origin of de novo mutations. Nucleic Acids Res 34: D29–D31
Morison IM, Ramsay JP, Spencer HG 2005 A census of mammalian imprinting. Trends Genet 21: 457–465
Morison IM, Reeve AE 1998 A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet 7: 1599–1609
Ferguson-Smith AC, Surani MA 2001 Imprinting and the epigenetic asymmetry between parental genomes. Science 293: 1086–1089
Weksberg R, Smith AC, Squire J, Sadowski P 2003 Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet 1: R61–R68
Enklaar T, Zabel BU, Prawitt D 2006 Beckwith-Wiedemann syndrome: multiple molecular mechanisms. Expert Rev Mol Med 8: 1–19
Weksberg R, Shuman C, Smith AC 2005 Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 137: 12–23
Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, Bowdin SC, Riccio A, Sebastio G, Bliek J, Schofield PN, Reik W, Macdonald F, Maher ER 2005 Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet 13: 1025–1032
Grundy P, Telzerow P, Paterson MC, Haber D, Berman B, Li F, Garber J 1991 Chromosome 11 uniparental isodisomy predisposing to embryonal neoplasms. Lancet 338: 1079–1080
Hertel NT, Carlsen N, Kerndrup G, Pedersen IL, Clausen N, Hahnemann JM, Jacobsen BB 2003 Late relapse of adrenocortical carcinoma in Beckwith-Wiedemann syndrome. Clinical, endocrinological and genetic aspects. Acta Paediatr 92: 439–443
West PM, Love DR, Stapleton PM, Winship IM 2003 Paternal uniparental disomy in monozygotic twins discordant for hemihypertrophy. J Med Genet 40: 223–226
Niemitz EL, Feinberg AP, Brandenburg SA, Grundy PE, DeBaun MR 2005 Children with idiopathic hemihypertrophy and Beckwith-Wiedemann syndrome have different constitutional epigenotypes associated with Wilms tumor. Am J Hum Genet 77: 887–891
Shuman C, Smith AC, Steele L, Ray PN, Clericuzio C, Zackai E, Parisi MA, Meadows AT, Kelly T, Tichauer D, Squire JA, Sadowski P, Weksberg R 2006 Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet A 140: 1497–1503
Bliek J, Terhal P, van den Bogaard MJ, Maas S, Hamel B, Salieb-Beugelaar G, Simon M, Letteboer T, van der Smagt J, Kroes H, Mannens M 2006 Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet 78: 604–614
Eggermann T, Schonherr N, Meyer E, Obermann C, Mavany M, Eggermann K, Ranke MB, Wollmann HA 2006 Epigenetic mutations in 11p15 in Silver-Russell syndrome are restricted to the telomeric imprinting domain. J Med Genet 43: 615–616
Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, Bertrand AM, Netchine I, Le Bouc Y 2005 Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet 37: 1003–1007
Mackay DJ, Hahnemann JM, Boonen SE, Poerksen S, Bunyan DJ, White HE, Durston VJ, Thomas NS, Robinson DO, Shield JP, Clayton-Smith J, Temple IK 2006 Epimutation of the TNDM locus and the Beckwith-Wiedemann syndrome centromeric locus in individuals with transient neonatal diabetes mellitus. Hum Genet 119: 179–184
Weksberg R, Shen DR, Fei YL, Song QL, Squire J 1993 Disruption of insulin-like growth factor 2 imprinting in Beckwith-Wiedemann syndrome. Nat Genet 5: 143–150
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM 2000 CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405: 486–489
Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, Feinberg AP 1999 Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci U S A 96: 5203–5208
Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB, Higgins MJ 1999 A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A 96: 8064–8069
Fitzpatrick GV, Soloway PD, Higgins MJ 2002 Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 32: 426–431
Diaz-Meyer N, Day CD, Khatod K, Maher ER, Cooper W, Reik W, Junien C, Graham G, Algar E, Der Kaloustian VM, Higgins MJ 2003 Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J Med Genet 40: 797–801
Du M, Zhou W, Beatty LG, Weksberg R, Sadowski PD 2004 The KCNQ1OT1 promoter, a key regulator of genomic imprinting in human chromosome 11p15.5. Genomics 84: 288–300
Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A 2004 Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet 36: 958–960
Sparago A, Russo S, Cerrato F, Ferraiuolo S, Castorina P, Selicorni A, Schwienbacher C, Negrini M, Battista Ferrero G, Cirillo Silengo M, Anichini C, Larizza L, Riccio A 2006 Mechanisms causing imprinting defects in familial Beckwith-Wiedemann syndrome with Wilms' tumour. Hum Mol Genet 16: 254–264
Prawitt D, Enklaar T, Gartner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, Schmidtke P, Reutzel D, Fees S, Lucito R, Korzon M, Brozek I, Limon J, Housman DE, Pelletier J, Zabel B 2005 Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc Natl Acad Sci U S A 102: 4085–4090
Niemitz EL, DeBaun MR, Fallon J, Murakami K, Kugoh H, Oshimura M, Feinberg AP 2004 Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am J Hum Genet 75: 844–849
Parker DA, Skalko RG 1969 Congenital asymmetry: report of 10 cases with associated developmental abnormalities. Pediatrics 44: 584–589
Hoyme HE, Seaver LH, Jones KL, Procopio F, Crooks W, Feingold M 1998 Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79: 274–278
Ringrose RE, Jabbour JT, Keele DK 1965 Hemihypertrophy. Pediatrics 36: 434–448
Lapunzina P 2005 Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 137: 53–71
Russell A 1954 A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med 47: 1040–1044
Silver HK, Kiyasu W, George J, Deamer WC 1953 Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics 12: 368–376
Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC 1999 The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 36: 837–842
Chitayat D, Friedman JM, Anderson L, Dimmick JE 1988 Hepatocellular carcinoma in a child with familial Russell-Silver syndrome. Am J Med Genet 31: 909–914
Eggermann T, Meyer E, Obermann C, Heil I, Schuler H, Ranke MB, Eggermann K, Wollmann HA 2005 Is maternal duplication of 11p15 associated with Silver-Russell syndrome?. J Med Genet 42: e26
Hitchins MP, Stanier P, Preece MA, Moore GE 2001 Silver-Russell syndrome: a dissection of the genetic aetiology and candidate chromosomal regions. J Med Genet 38: 810–819
Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, Oshimura M, Soejima H, Mukai T, Wake N 2005 ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res 33: 2650–2660
Shield JP, Temple IK 2002 Neonatal diabetes mellitus. Pediatr Diabetes 3: 109–112
Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP 2000 Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes 49: 1359–1366
Christian SL, Rich BH, Loebl C, Israel J, Vasa R, Kittikamron K, Spiro R, Rosenfield R, Ledbetter DH 1999 Significance of genetic testing for paternal uniparental disomy of chromosome 6 in neonatal diabetes mellitus. J Pediatr 134: 42–46
Fujii H, Zhou W, Gabrielson E 1996 Detection of frequent allelic loss of 6q23-q25.2 in microdissected human breast cancer tissues. Genes Chromosomes Cancer 16: 35–39
Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, Journot L 1997 Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J 16: 2814–2825
Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, Bockaert J, Spengler D, Journot L 1998 hZAC encodes a zinc finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Proc Natl Acad Sci U S A 95: 8835–8840
Kamiya M, Judson H, Okazaki Y, Kusakabe M, Muramatsu M, Takada S, Takagi N, Arima T, Wake N, Kamimura K, Satomura K, Hermann R, Bonthron DT, Hayashizaki Y 2000 The cell cycle control gene ZAC/PLAGL1 is imprinted–a strong candidate gene for transient neonatal diabetes. Hum Mol Genet 9: 453–460
Mackay DJ, Boonen SE, Clayton-Smith J, Goodship J, Hahnemann JM, Kant SG, Njolstad PR, Robin NH, Robinson DO, Siebert R, Shield JP, White HE, Temple IK 2006 A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet 120: 262–269
Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L 2006 Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11: 711–722
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smith, A., Choufani, S., Ferreira, J. et al. Growth Regulation, Imprinted Genes, and Chromosome 11p15.5. Pediatr Res 61, 43–47 (2007). https://doi.org/10.1203/pdr.0b013e3180457660
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e3180457660
This article is cited by
-
RhoGDI1 interacts with PHLDA2, suppresses the proliferation, migration, and invasion of trophoblast cells, and participates in the pathogenesis of preeclampsia
Human Cell (2022)
-
Genetic basis and identification of candidate genes for wooden breast and white striping in commercial broiler chickens
Scientific Reports (2021)
-
Large parental differences in chromatin organization in pancreatic beta cell line explaining diabetes susceptibility effects
Nature Communications (2021)
-
Associations between the maternal circulating lipid profile in pregnancy and fetal imprinted gene alleles: a cohort study
Reproductive Biology and Endocrinology (2018)
-
IGF2 stimulates fetal growth in a sex- and organ-dependent manner
Pediatric Research (2018)
