
Abstract
Benzodithiophene- and quinoxaline (Qu)-based derivatives were used to obtain two kinds of D-π-A-type polymers, poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene diphenyl Qu) (PBDTAQ) and poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene dibenzophenazine (PBDTAFQ), through a Stille coupling reaction. Phenyl (Qu) and fused-phenyl (phenazine, Pz) were introduced in locations 2 and 3, respectively, of Qu derivatives. For the polymer introduced with Pz, the tilt angle (θ3) between the 2,3-carbon and the bonded phenyl side chains of Qu decreased from 44.06° to 0°. As a result, PBDTAFQ displayed a higher absorbance compared with PBDTAQ at the UV–vis absorption spectra in the film state. Also, the intra/intermolecular charge transfer (ICT) peak intensity of 500–700 nm increased relative to the solution peak. Furthermore, the results of the X-ray diffraction measurement suggest that the dπ-spacing distance for PBDTAFQ (dπ=3.89 Å) was smaller than that for PBDTAQ (dπ= 4.36Å), exhibiting a stronger intermolecular interaction. PBDTAQ showed an edge-on dominant orientation, whereas the PBDTAFQ thin films showed an increase in the face-on structure for crystallinity. As a consequence, PBDTAFQ showed an improved short current density (JSC) in organic photovoltaic cells. The PBDTAFQ:PC70BM blend-based devices that were fabricated exhibited a power conversion efficiency of 3.0% at a 1:5 ratio and reached 3.4% when treated with additive and methanol.
Similar content being viewed by others
Introduction
Over the past few decades, conjugated semiconducting polymers have been studied and applied in various fields, such as organic light-emitting diodes (OLEDs),1, 2, 3, 4 organic photovoltaics (OPVs)5, 6, 7, 8, 9 and organic thin film transistors (OTFTs).10, 11, 12, 13 In particular, OPVs have been in the spotlight owing to their low cost, light weight and flexibility.
Donor–acceptor (D–A)-type polymers have been extensively used as active material in OPVs because of their excellent light-harvesting ability (low bandgap). In addition, the electron donor unit provides a low high occupied molecular orbital (HOMO) level, and the electron acceptor unit has a unique property that can be used to control easily the polymer’s electronic bandgap and to change its electronic properties. D–A-type conjugation polymers and fullerene blends, such as [6,6′]-phenyl-C-butyricacidmethyl ester (PCBM), in bulk-heterojunction solar cells have produced good outcomes in OPVs because of the high electron affinity and ease of charge separation.14, 15
The power conversion efficiency (PCE) is generally determined by the short current density (JSC), open circuit voltage (Voc) and fill factor (FF) and a common way to increase the PCE is to reduce the bandgap and expand the absorption region into the infrared range, which accounts for ~50% of the solar spectrum. However, a low bandgap does not always guarantee a high PCE. Thus, improvements in the photon- harvesting property should be achieved through the structural engineering of the materials and devices to increase the JSC value, which can lead to an increase in the PCE. The planarity and orientation of the active layer also works as a key factor to move the electron/hole that is generated to the electrode. In addition, most of the OPVs exhibit anisotropic charge transport where the charge moves effectively from the π-stacking direction.16
Benzo-[1,2-b:4,5-b′]dithiophene (BDT) is one of the donor units of these D–A-type polymers, and it is electron rich because both sides are fused with thiopnenes, the electron-donating units that are resistant to benzene. BDT has been used in a number of OPV studies because of its long QUOTE conjugation length and high planarity.17, 18 Meanwhile, quinoxaline (Qu) and its derivatives have also become known for being acceptor units. This happens as a result of the electro-withdrawing property of the conjugation ring containing nitrogen and because of the ease of transforming the electronic properties. Another reason is that it is easy to introduce functional groups, such as side chains, providing high solubility. Of all Qu derivatives, phenazine (Pz) is one case with excellent planarity, π–π interaction and close packing properties. Pz has two fused Qu phenyl rings that expand the absorption area of the UV–vis spectrum, increasing the absorbance. It also improves the JSC for the OPV cell, which in turn enables an increase in the PCE. However, these fused-phenyl rings have a relatively low solubility and therefore are difficult to use in the solution process.19, 20, 21 However, this group previously reported an increase in solubility by introducing the octyloxy side chain of Pz.22 This is the result of introducing the alkoxy side chain, which has an effect on the inter/intramolecular interaction that results from the obtained polymer’s solution and the change in the film state’s molecular orientation.
The Takimiya group used an X-ray diffraction peak analysis to publish results on surface manipulation. The results suggested that donor polymers with face-on orientation in the film state formed efficient packing of the polymer chains, obtaining a high PCE.23 Meanwhile, our group had also presented a study on the planarity properties and π–π stacking based on the donor or acceptor’s side chains.24 In particular, D–A-type polymers where Pz derivatives were fused with various donors exhibited face-on orientation.25 Bo et al. also synthesized Pz derivatives with D–A-type polymers by using monomers with different chains introduced in BDT’s 4,7-C. They reported that the molecular weight, crystallinity and PCE were different between the two polymers. However, they did not present any results on the orientation property.26
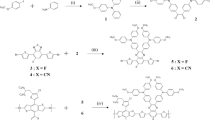
This study adopted Benzo-[1,2-b:4,5-b']dithiophene (M1, BDT) as an electron-donating unit, 6,7-bis(octyloxy)-2,3-diphenyl-5,8-di(thiophen-2-yl)quinoxaline (M2, AQ) and 11,12-bis(octyloxy)-10,13-di(thiophen-2-yl)dibenzo[a,c]phenazine (M3, AFQ) as the electron-accepting unit and thiopene as the spacer between the donor and acceptor. As a result, two D–π–A-type polymers were synthesized: poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene diphenyl Qu) (PBDTAQ) and poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene dibenzophenazine (PBDTAFQ). Both polymers exhibited a high-molecular weight (Mn=109 000–128 000 g mol−1, polydispersity index (PDI)=1.91–2.24).
In particular, this study tried to control the polymers’ planarity and the orientation in the film state according to the structure of the phenyl substitutes adopted by Qu, and the results confirmed that not only donors and accepters but also side groups had a great effect on the optical and crystal properties. According to the results of an X-ray diffraction analysis, the PBDTAFQ films formed a face-on structure, obtaining a 50% higher JSC than PBDTAQ.
Materials and methods
Instrumentation
Unless otherwise specified, all reactions were performed under a nitrogen atmosphere. The solvents were dried using the standard procedures. All column chromatography was performed with silica gel (230–400 mesh; Merck, Darmstadt, Germany) as the stationary phase. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded with a Bruker AMX400 spectrometer (Darmstadt, Germany), using the resonances of the solvent as an internal reference. Chemical shifts (d) are reported in p.p.m. downfield from tetramethylsilane. The molecular weights of the polymers were measured using the gel permeation chromatography method with polystyrene standards. Thermal gravimetric analysis measurements were performed on a TA Instruments 2050 analyzer (Netzsch, Selb, Germany). Electrochemical cyclic voltammetry was performed using a Zahner IM 6e electrochemical workstation with 0.1 m Bu4NPF6 in acetonitrile as the electrolyte. Indium tin oxide (ITO) glass coated with a thin polymer film was used as the working electrode, and a Pt wire and an Ag/Ag+ electrode were used as the counter and reference electrodes, respectively. The electrochemical potential was calibrated against Fc/Fc+. Current–voltage (I–V) curves of the PSC devices were measured using a computer-controlled Keithley 2400 source measurement unit equipped with a Peccell solar simulator under AM1.5G illumination (Yokohama, Japan, 100 mW cm−2). The illumination intensity was calibrated using a standard Si photodiode detector equipped with a KG-5 filter. The output photocurrent was adjusted to match the photocurrent of the Si reference cell to obtain a power density of 100 mW cm−2. After encapsulation, all devices were operated under ambient atmospheric conditions at 25 °C.
Synthesis of monomers
All reagents and chemicals were purchased from Aldrich (St Louis, MO, USA), TCI (Tokyo, Japan), Alfa Aesar (Ward Hill, MA, USA) and used as received unless otherwise specified. 2,6-Bis(trimethyltin)-4,8-bis(2-ethylhexyloxy)benzo[1,2-b:4,5-b′]dithiophene (M1)27 (Journal of the American Chemical Society 131: 7792–7799 (2009)) and 4,7-dibromo-5,6-bis(octyloxy)-2,1,3-benzothiadiazole (3) were prepared according to methods reported in the literature.
5,8-dibromo-6,7-bis(octyloxy)-2,3-diphenylquinoxaline (4)
Under a nitrogen atmosphere, compound 3 (4.0 g, 7.27 mmol) and zinc (5.88 g 90.0 mmol) dust were dissolved in 110 ml acetic acid. The mixture was refluxed for 3 h at 80 °C. After cooling to room temperature, the reaction mixture was washed with a NaOH solution. The solids that resulted after evaporation of the organic solvent and benzil (3.36 g, 15.92 mmol) were dissolved in 110 ml acetic acid. The mixture was refluxed for 1 day at 60 °C. After cooling to room temperature, an orange-colored mixture was observed. After filtration, the reaction mixture was purified by column chromatography on silica gel (dichloromethane as eluent) to obtain the product as a green solid (3.2 g, 63.4%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 7.37 (m, 6H; Ar), 4.20 (t, 4H; CH2), 1.92 (m, 4H; CH2), 1.55 (m, 4H; CH2), 1.33 (m, 16H; CH2), 0.90 (t, 6H; CH3), 13C-NMR (100 MHz; CDCl3; Me4Si); δ (p.p.m.): 153.81, 152.69, 138.24, 137.14, 130.22, 129.25, 128.30, 117.26, 74.86, 31.88, 30.33, 29.45, 29.31, 26.07, 22.70, 14.13. Anal. calcd for: C36H44Br2N2O2: C, 62.07; H, 6.37; N, 4.02; O, 4.59. Found: C, 61.20; H, 6.44; N, 3.92; O, 4.99.
6,7-bis(octyloxy)-5,8-di(thiophen-2-yl)-2,3-diphenylquinoxaline (5)
Compound 4 (2.0 g, 2.87 mmol) and tributyl(thiophen-2-yl)stannane (2.73 ml, 8.61 mmol) in toluene (75 ml) was added to PdCl2(PPh3)2 (0.203 g, 0.29 mmol) under a nitrogen atmosphere. After refluxing for 48 h at 80 °C, the mixture was cooled to room temperature and then poured into H2O; the organic layer was extracted by CHCl3 and dried over anhydrous Na2SO4. The crude product was purified by column chromatography on silica gel to give the product as an orange solid (1.5 g, 74.3%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 8.05 (d, 2H; Ar), 7.65 (d, 4H; Ar), 7.55 (d, 2H; Ar), 7.33 (m, 6H; Ar), 7.20 (d, 2H; Ar), 4.03 (t, 4H; CH2), 1.77 (m, 4H; CH2), 1.39 (m, 4H; CH2), 1.29 (m, 16H; CH2), 0.90 (t, 6H; CH3). 13C-NMR (100 MHz; CDCl3; Me4Si); δ (p.p.m.): 152.88, 150.24, 138.93, 136.43, 133.49, 130.88, 130.32, 128.66, 128.14, 127.85, 126.06, 124.19, 74.10, 31.87, 30.41, 29.50, 29.31, 26.07, 22.71, 14.15. Anal. calcd for: C44H50N2O2S2: C, 75.17; H, 7.17; N, 3.98; O, 4.55; S, 9.12. Found: C, 73.14; H, 7.35; N, 3.83; O, 5.13; S, 7.85.
5,8-bis(5-bromothiophen-2-yl)-6,7-bis(octyloxy)-2,3-diphenylquinoxaline (M2)
Under a nitrogen atmosphere, compound 5 (1.5 g, 2.13 mmol) was dissolved in 140 ml of tetrahydrofuran (THF), and then N-bromosuccinimide (NBS) (0.87 g, 4.9 mmol) was added in portions. The mixture was stirred for 24 h at room temperature. The mixture was then poured into water and extracted with chloroform. The combined organic layers were dried over Na2SO4, and the solvent was removed. The crude product was purified with column chromatography to give M1 as an red-orange solid (1.2 g, 65.5%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 7.97 (d, 2H; Ar), 7.63 (d, 4H; Ar), 7.36 (m, 6H; Ar), 7.14 (d, 2H; Ar), 4.07 (t, 4H; CH2), 1.83 (m, 4H; CH2), 1.42 (m, 4H; CH2), 1.30 (m, 16H; CH2), 0.90 (t, 6H; CH3). 13C-NMR (100 MHz; CDCl3; Me4Si), δ (p.p.m.): 152.68, 150.57, 138.46, 135.89, 135.19, 131.25, 130.33, 128.95, 128.89, 128.25, 123.35, 116.09, 74.22, 31.85, 30.42, 29.48, 29.30, 26.07, 22.70, 14.13. Anal. calcd for: C44H48Br2N2O2S2: C, 61.39; H, 5.62; N, 3.25; O, 3.72; S, 7.45. Found: C, 61.23; H, 5.65; N, 3.13; O, 4.28; S, 7.42.
10,13-dibromo-11,12-bis(octyloxy)dibenzo[a,c]phenazine (6)
Under a nitrogen atmosphere, compound 3 (1.75 g, 3.18 mmol) and zinc (2.58 g 39.4 mmol) dust were dissolved in 110 ml acetic acid. The mixture was refluxed for 3 h at 80 °C. After cooling to room temperature, the reaction mixture was washed with a NaOH solution. The solids that resulted after the evaporation of the organic solvent and 9,10-phenanthrenequinone (1.46 g, 7.0 mmol) were dissolved in 110 ml acetic acid. The mixture was refluxed for 1 day at 60 °C. After cooling to room temperature, an orange-colored mixture was observed. After filtration, the reaction mixture was purified by column chromatography on silica gel (dichloromethane as eluent) to obtain the product as a light-yellow solid (1.3 g, 58.9%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 9.29 (d, 2H; Ar), 8.41 (t, 2H; Ar), 7.68 (m, 4H; Ar), 4.27 (t, 4H; CH2), 1.96 (t, 4H; CH2), 1.59 (m, 4H; CH2), 1.42 (m, 16H; CH2), 0.91 (t, 6H; CH3). 13C-NMR (100 MHz; CDCl3; Me4Si), δ (p.p.m.): 153.91, 141.80, 137.92, 132.00, 130.49, 129.47, 127.96, 126.71, 122.73, 117.21, 74.94, 31.90, 30.41, 29.50, 29.35, 26.12, 22.72, 14.14. Anal. calcd for: C36H42Br2N2O2: C, 62.26; H, 6.10; N, 4.03; O, 4.61. Found: C, 60.16; H, 5.78; N, 3.79; O, 5.11.
11,12-bis(octyloxy)-10,13-di(thiophen-2-yl)dibenzo[a,c]phenazine (7)
Compound 6 (1.0 g, 1.44 mmol) and trimethyl(thiophen-2-yl)stannane (1.35 ml, 4.3 mmol) in toluene (37 ml) was added to PdCl2(PPh3)2 (0.101 g, 0.15 mmol) under a nitrogen atmosphere. After refluxing for 48 h at 80 °C, the mixture was cooled to room temperature and then poured into H2O; the organic layer was extracted by CHCl3 and dried over anhydrous Na2SO4. The crude product was purified by column chromatography on silica gel to give the product as an orange liquid (0.6 g, 59.3%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 9.11 (d, 2H; Ar), 8.30 (t, 2H; Ar), 7.98 (t, 2H; Ar), 7.58 (d, 2H; Ar), 7.53 (m, 4H; Ar), 7.20 (t, 2H; Ar), 3.98 (t, 4H; CH2), 1.72 (m, 4H; CH2), 1.34 (m, 4H; CH2), 1.21 (m, 16H; CH2), 0.82 (t, 6H; CH3). 13C-NMR (100 MHz; CDCl3; Me4Si), δ (p.p.m.): 153.10, 140.34, 138.02, 133.71, 131.89, 130.93, 130.44, 129.71, 127.85, 127.77, 127.10, 126.08, 124.26, 122.67, 74.21, 31.93, 30.51, 29.56, 29.37, 26.14, 22.76, 14.21. Anal. calcd for: C44H48N2O2S2: C, 75.39; H, 6.90; N, 4.00; O, 4.56; S, 9.15. Found: C, 74.93; H, 6.35; N, 3.77; O, 5.13; S, 8.98.
10,13-bis(5-bromothiophen-2-yl)-11,12-bis(octyloxy)dibenzo[a,c]phenazine (M3)
Under a nitrogen atmosphere, 11,12-bis(octyloxy)-10,13-di(thiophen-2-yl)dibenzo[a,c]phenazine (0.3 g, 0.427 mmol) was dissolved in 30 ml of THF, and NBS (0.174 g, 0.982 mmol) was then added in portions. The mixture was stirred for 24 h at room temperature. Then, the mixture was poured into water and extracted with chloroform. The combined organic layers were dried over Na2SO4, and then the solvent was removed. The crude product was purified with column chromatography to give M2 as a red liquid (0.24 g, 65.4%). 1H-NMR (400 MHz; CDCl3; Me4Si), δ (p.p.m.): 9.10 (d, 2H; Ar), 8.31 (t, 2H; Ar), 7.90 (t, 2H; Ar), 7.60 (d, 4H; Ar), 7.20 (m, 2H; Ar), 3.98 (t, 4H; CH2), 1.72 (m, 4H; CH2), 1.34 (m, 4H; CH2), 1.21 (m, 16H; CH2), 0.82 (t, 6H; CH3). 13C-NMR (100 MHz; CDCl3; Me4Si), δ (p.p.m.): 152.75, 140.39, 137.18, 135.21, 131.88, 131.26, 130.03, 029.87, 128.94, 127.83, 127.14, 123.29, 122.62, 115.74, 74.23, 31.91, 30.54, 29.73, 29.55, 29.37, 26.15, 22.75, 14.18. Anal. calcd for: C44H46Br2N2O2S2: C, 61.54; H, 5.40; Br, 18.61; N, 3.26; O, 3.73; S, 7.47. Found: C, 61.26; H, 5.31; N, 3.09; O, 3.53; S, 7.34.
Polymerization
P-type monomer (M1) (0.3 mmol), n-type monomer (M2 and M3) (0.3 mmol), Pd2dba3(0) (0.01 g, 0.012 mmol) and P(o-tolyl)3 (0.0146 g, 0.048 mmol) were dissolved in anhydrous toluene (10 ml). The flask was degassed and refilled with nitrogen gas twice. The polymerization mixture was stirred at 90 °C for 48 h, and a small quantity 2-bromothiophene were added. After 6 h, a small quantity 2-tributylstannyl thiophene were also added for the end-capping reaction. The reaction mixture was cooled to room temperature and poured into methanol. The precipitate was filtered and purified with methanol, acetone, hexane and chloroform in a Soxhlet apparatus. The polymer was precipitated in methanol. Finally, the polymer was collected as a material.
Poly(4,8-Bis(2-ethylhexyloxy)benzo[1,2-b:4,5-b′]dithiophene-alt-6,7-bis(octyloxy)-2,3-diphenyl-5,8-di(thiophen-2-yl)quinoxaline) (PBDTAQ): yield=78%. 1H-NMR (400 MHz, CDCl3), δ (p.p.m.): 8.70–8.40 (m), 7.7–7.20 (m), 7.10–7.00 (m), 4.40–4.00 (m), 2.70–2.50 (m), 2.20–1.80 (m), 1.80–1.10 (m), 1.00–0.80 (m). Anal. calcd for: C70H86N2O4S4: C, 73.25; H, 7.55; N, 2.44; O, 5.58; S, 11.18. Found: C, 73.16; H, 7.37; N, 2.34; O, 5.66; S, 11.02.
Poly(4,8-Bis(2-ethylhexyloxy)benzo[1,2-b:4,5-b′]dithiophene-alt-11,12-bis(octyloxy)-10,13-di(thiophen-2-yl)dibenzo[a,c]phenazine) (PBDTAFQ): yield=65%. 1H-NMR (400 MHz, CDCl3), δ (p.p.m.): 9.70–9.40 (m), 8.70–8.40 (m), 8.35–8.15 (m), 8.10–8.00 (m), 7.80–7.20 (m), 4.40–4.00 (m), 2.20–1.80 (m), 1.80–1.20 (m), 1.20–0.80 (m). Anal. calcd for: C70H84N2O4S4: C, 73.38; H, 7.39; N, 2.45; O, 5.59; S, 11.20. Found: C, 73.19; H, 7.24; N, 2.35; O, 5.72; S, 11.11.
OPVs fabrication and characterization
All of the bulk-heterojunction PV cells were prepared using the following device fabrication procedure. The glass/ITO substrates (Sanyo, Osaka, Japan (10 X/c)) were lithographically patterned, cleaned with detergent and sonicated in deionized water, acetone and isopropyl alcohol. The substrates were then dried on a hot plate at 120 °C for 10 min and treated with oxygen plasma for 30 min to improve the contact angle immediately before the film coating process. Poly(3,4-ethylene-dioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS; Baytron P 4083; Bayer AG, St Louis, MO, USA) was passed through a 0.45-lm filter before being deposited onto the ITO substrates at a thickness of ca. 32 nm by spin coating at 4000 r.p.m. in air and then dried at 120 °C for 20 min inside a glove box. The solutions were then passed through a 0.45-lm PTFE filter and spin coated (500–4000 r.p.m., 30 s) on top of the PEDOT:PSS layer device fabrication was completed by depositing layers of Al (200 nm) at pressures of <10−6 torr. The active area of the devices was 4.0 mm2. Finally, the cell was encapsulated using a UV-curing glue (Nagase, Tokyo, Japan). The conventional devices were fabricated with the following structure: ITO glass/PEDOT:PSS/polymer:PC70BM/BaF2/Ba/Al encapsulation glass.
Results and discussion
Synthesis and characterization of polymers
Scheme 1 shows the chemical structure and synthetic routes for the monomers and polymers. The monomers were synthesized based on methods described in the literature.28, 29 PBDTAQ and PBDTAFQ were polymerized through a Stille coupling reaction by using 2,7-Bis(trimethyltin)-4,5-di(2-ethylhexyloxy)benzo-[1,2-b:4,5-b′]dithiophene (M1) as donor and 5,8-bis(4-bromocyclopenta-1,3-dienyl)-6,7-bis(octyloxy)-2,3-diphenylquinoxaline (Qu, M2) and 10,13-bis(4-bromocyclopenta-1,3-dienyl)-11,12-bis(octyloxy)dibenzo[a,c]phenazine(Pz, M3) as acceptor.
The polymerization process took place for 48 h at 90 °C using Pd2(dba)3 and P(o-tol)3 as catalysts and toluene as solvent. Once the process was completed, bromothiophene and tributhyl stannyl thiophene were each used for a 3-h long end-capping process. The obtained powders were retrieved through reprecipitation in methanol. Then, methanol, acetone and chloroform were purified, in that order, through the Soxhlet apparatus. Finally, by the chloroform-soluble portion was extracted to obtain dark purple (PBDTAQu) and dark bluish purple (PBDTAFQ) powders with transference numbers at 78% and 65%, respectively.
The polymers dissolved well in general organic solvents, such as THF, chloroform, chlorobenzene and orthodichlorobenzene. The synthesized monomer and polymer structure were confirmed through 1H-NMR and 13C-NMR, as illustrated in Supplementary Figures S1–S6. The hydrogen peak of phenyl 2-C above Qu in PBDTAQ was at 8.0–7.0 p.p.m. Meanwhile, the proton peak of Pz was at 9.1 p.p.m. because of the fused ring. In addition, the aromatic ring peaks were at 8.4–7.2 p.p.m., an up-shift compared with Qu. Similar to the NMR peak in the monomers, the aromatic peaks in PBDTAQ and PBDTAFQ were at 8.7–7.0 and 9.7–7.2 p.p.m., respectively.
Table 1 displays an estimate of the polymer’s molecular weights and the PDI. The average molecular weight (Mn) for PBDTAQ and PBDTAFQ was at 128 000 and 109000 g mol−1 each, and the PDI was narrow at 2.24 and 1.91. This is higher than the molecular weight of the polymer that adopted Qu and Pz as acceptor without the alkoxy chain (Mn=ca. 17 000–30 000), as presented by Jo and co-workers.30, 31 The D–A-type polymers presented by our group that used Qu and Pz with octyloxy side chains as acceptor also had low molecular weights (Mn=ca. 20 000–40 000).24 The polymer in Bo et al.’s study that polymerized BDT derivatives and the acceptor with F adopted by the Pz core had higher molecular weights (Mn=ca. 28 000–64 000). This is similar to the results presented in Bumjoon, Alan McLean and Qionghua Jin’s studies, which exhibited BDT’s linear curvature and planarity properties providing higher molecular weights than any other donors.32, 33, 34
This paper thus confirmed that polymers using Qu and Pz with BDT derivatives and octyloxy side chains have a high Mn, and this is the result of an effective increase in solubility due to the synergy between the donor’s linear property and the octyloxy side chains adopted by the acceptor. In other words, monomers with a higher solubility prevent the extraction of polymers in the polymerization process, resulting in a higher degree of polymerization.
Thermal properties
As can be seen in Supplementary Figure S7 and Table 1, the polymers that were obtained showed 5wt% loss in temperature exceeding 311 °C because of the high-molecular-weight structure, exhibiting a high thermal stability. Both of the polymers displayed their applicability in OPVs and optoelectronic devices that require a high thermal stability.21
Computational study of the polymers
The electrical properties, molecular geometries and electron density of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the polymers were measured through a simulation using density functional theory (DFT). The DFT calculation was performed using the Gaussian 09 program as a hybrid B3LYP correlation functional and split valence 6–31G(d) basis set. Oligomers with one repeat unit were used as the calculation model, and the calculated HOMO and LUMO orbitals are shown in Figure 1 and Table 2.

Density functional theory (DFT) simulation for (a) tilt angle and (b) electron cloud of polymers. A full color version of this figure is available at Polymer Journal online.
The results of the estimation for the dihedral angles of the donor (BDT), thiophene spacer and acceptor (Qu and Pz) are displayed in Figure 1a and Table 2. The tilt angle between the donor–spacer (θ1) and spacer–acceptor (θ2) for the model compound showed similar values: BDT-thiophene-Qu (θ1=4.69°, θ2=23.03°) and BDT-thiophene-Pz (θ1=7.21°, θ2=28.56). As a consequence, the intramolecular electron transfer seemed to be favorable to PBDTAQ with its smaller main chain tilt. The tilt angle (θ3) between the 2,3-carbon and bonded phenyl side chains of Qu was 44.06°, but was 0° for the fused Pz. Compared with Pz, Qu increases the steric hindrance and reduces the π–π stacking between the polymers. That is, the intermolecular charge transport decreases accordingly.19 Therefore, introducing Pz to the D–π–A polymers’ acceptor is anticipated to reduce the steric hindrance between the main polymer chains and increase the intermolecular interaction between the polymer chains.
Electrons in HOMO orbitals are delocalized in the overall polymer main chain, as illustrated in Figure 1b. Meanwhile, electrons in LUMO orbitals are delocalized in the acceptor. This is due to the structural property of the quinoid formed between N’s non-bonding electron pair and C.20 In addition, the electron cloud in PBDTAFQ extended from the LUMO orbital to the acceptor’s phenyl ring because of fusing in the Qu core. Furthermore, PBDTAFQ’s HOMO level was calculated to be −5.74 eV, which is relatively lower compared with PBDTAQ’s HOMO level (−5.66 eV). A high oxidation stability and Voc can be expected based on these results.
Optical–electrochemical properties
Figure 2 illustrates the UV–visible absorption spectra of the two polymers’ solution state (orthodichlorobenzene, a) and the film state (thickness=60 nm, b) obtained through spin coating. The results are listed in Table 3. In addition, the absorption coefficient (ɛ) was calculated by estimating the solution with five different concentrations (Supplementary Figure S8).

Ultraviolet (UV–visible) absorption of the copolymers in (a) orthodichlorobenzene (ODCB) solution (10 μg ml−1) and (b) UV–vis absorption of thin film (60 nm) of the polymers. A full color version of this figure is available at Polymer Journal online.
As can be seen in Figures 2a and b, the PBDTAQ and PBDTAFQ polymers are both composed largely of two absorption bands, with a wide absorption range of 300–700 nm. The absorption peak in the high-energy region between 300 and 400 nm is based on localized π–π* transitions,35 and the peak in the long wavelength region between 450 and 700 nm is based on the ICT between the polymer’s acceptor and donor units. 36 PBDTAQ’s λmax in the solution state was at 393 and 518 nm each, whereas PBDTAFQ’s λmax was at 421 and 562 nm. PBDTAQ displayed a higher intensity in the ICT transition peak (ɛ518 nm=3.85 × 104 M−1cm−1) than for the π–π* transition peak (ɛ393nm=3.08 × 104 M−1cm−1). In contrast, PBDTAFQ exhibited a lower intensity in the ICT transition peak (ɛ421nm=3.43 × 104 M−1cm−1) compared with the π–π* transition peak (ɛ421nm=4.71 × 104 M−1cm−1) because the intramolecular electron transfer is not easy in PBDTAFQ because of the relatively bigger tilting angle of the polymer’s main chain, as can be seen in the DFT calculation.
The UV–vis spectra of PBDTAQ and PBDTAFQ in the film state showed a red-shift of 27 and 34 nm relative to the solution state, respectively, as illustrated in Figure 2b. This suggests that the interaction between the polymer chains in the solid state is strengthened through two-dimensional aggregation, which is consistent with this group’s previous study and Yong Cao et al.’s study that indicated the result of an effective intermolecular interaction between polymeric backbones based on the regular arrangement formed by molecules in the process of developing films.30, 35, 37, 38
In addition to the red-shift in the UV–vis spectra of the film state compared with the solution state for PBDTAFQ, the π–π* transition intensity also decreased significantly (ca. 30%) when compared with the ICT absorption intensity. This study thus demonstrated a low ICT intramolecular effect in PBDTAFQ because of the steric hindrance (θ1 and θ2) of the main chain. However, the ICT effect increased in the film state because the intermolecular stacking became easier because of the presence of the fused-phenyl ring. The relative improvement in the ICT peak resulted in PBDTAFQ with a generally high ɛ within the UV–visible region in the film state. The reason for which the ICT absorption peak increases is because PBDTAFQ is relatively well stacked, which improves the intermolecular charge transfer. This is consistent study, which shows that under different circumstances, the π–π* transition and the ICT peak of the same substance are reinforced depending on the changes in the electron attracting strength.39 This is also consistent with Bo et al.’s study, which exhibited a red-shift and a relative increase in the ICT peak, which is similar to the outcome of this study. In addition, the change in the absorbance was the greatest when the molecular weight increased for the same polymer. In this instance, the polymer with a the larger molecular weight exhibited a relatively higher mobility and PCE because intramolecular electron transfer is faster than intermolecular electron transfer. Thus, an improvement in the solubility of polymers was confirmed to improve JSC and electron transfer in the OPV devices.
PBDTAQ and PBDTAFQ were estimated to have an absorption edge at 642 and 687 nm and an optical bandgap at 1.93 and 1.80 eV, respectively. In other words, PBDTAFQ developed a better planar conformation than that of PBDTAQ by introducing a fused Pz. This led to an extended electronic delocalization, increasing the conjugation length. These results are consistent with those reported by Lidzey and co-workers and Jo and co-workers.31, 40
The electrochemical properties of the PBDTAQ and PBDTAFQ polymer thin films were measured using cyclic voltammogram spectroscopy. The polymers were dissolved in CHCl3 and drop-casted on ITO glass electrode to form polymer films that were used as the working electrode. The measurements were made in an N2 atmosphere under 0.1 m tetrabutylammonium-hexafluorophosphate in acetonitrile solution. The results are displayed in Supplementary Figure S8 and Table 3.
PBDTAQ and PBDTAFQ had an oxidation onset potential (Eox onset) of +1.53 V and +1.47 V and a HOMO level of −5.47 and −5.61 eV, respectively (Supplementary Figure S9). The HOMO level for PBDTAQ is lower compared with that for PBDTAFQ because of an influence of the fused Pz and the stabilized polymer structure. As the oxidation threshold of air is −5.27 eV, the HOMO level needs to be lower for the substance to have oxidation stability,41 and so both polymers are expected to have excellent oxidation stability. In addition, PBDTAFQ is anticipated to have a higher Voc compared with PBDTAQ, which is determined by the difference between the HOMO energy level of the donor polymer and the LUMO level of the fullerene acceptor. It was thus confirmed that this had the same disposition as the value calculated through the DFT.
X-ray analysis
X-ray diffraction was used to determine the ordering structure of both polymers, as seen in Figure 3. The PBDTAQ and PBDTAFQ thin films that were measured in the out-of-plane mode both showed (100) diffraction peak and broad (0 1 0) diffraction peak, as shown in Figure 3a, which means that both polymers form a bimodal lamellar structure. PBDTAQ exhibited a relatively stronger (1 0 0) peak, and PBDTAFQ had a stronger (0 1 0) peak, which indicates that PBDTAFQ is face-on dominant while PBDTAQ is edge-on dominant.42 The polymers’ (1 0 0) diffraction peak (2θ) was at 5.17° and 4.33°, respectively. Based on Bragg’s law (λ=2d sin θ), the lamellar d-spacing (dl) was calculated at 17.08 and 20.04 Å for each, and the (0 1 0) diffraction peak related to π–π stacking was at 20.36° and 22.68° for PBDTAQ and PBDTAFQ, respectively. This result thus indicates that the dπ-spacing was 4.36 and 3.91 Å for each, suggesting that the distance between the side chains within the molecule was closer in PBDTAQ and the distance between the main chains was closer in PBDTAFQ. These findings are consistent with results of the DFT, which indicated that there is little steric hindrance within the main chain molecules of PBDTAQ and that the intermolecular interaction is easier for PBDTAFQ due to the fused acceptor core. This is also consistent with the optical property where PBDTAQ has a larger ICT effect than PBDTAFQ in the absorption region of the solution state while PBDTAFQ has a bigger ICT effect in the film state.

X-ray diffraction patterns of polymers (a) out-of-plane (b) in-plane. A full color version of this figure is available at Polymer Journal online.
Figure 3b shows the X-ray diffraction measurements in the in-plane mode, and both polymers showed peaks in a low angle of around 5°. However, a peak in the high angle close to 15–25° was only seen in PBDTAQ, which means that PBDTAFQ has a strong tendency of having a face-on structure and PBDTAQ dominantly has an edge-on crystal structure.43 Bo et al. reported that two polymers with the same Pz acceptor had different types of crystal structure depending on the BDT donor’s side chain. While P1 polymers had the same main monomer as the PBDTAFQ polymers in this study, a low (010) diffraction peak and a dominant edge-on orientation was observed owing to the impact of the spacer’s alkyl side chain that was introduced to enhance solubility and also increased the tilt angle (θ1). When the 4,5-didecylthienyl group was adopted as a side chain in the BDT, steric hindrance occurred between the side chains, resulting in a decrease in the crystal peak intensity. On the other hand, this paper used thiophene without any side chains and Pz adopting octyloxy side chains in 11 and 12-C as the spacer, which effectively improved the solubility and resulted in a high crystallinity and planarity that was face-on dominant. In other words, the powerful π–π stacking between the polymer chain backbones was significantly affected by the molecular planarity based on not the donor/acceptor type but on the location and type of the side chain.
Photovoltaic properties
Figures 4a and b show the J–V (current–voltage) curves for the PBDTAQ and PBDTAFQ photovoltaic devices. Figures 4c and d illustrate the corresponding IPCE spectra, and Table 4 displays the results of the estimation. The photovoltaic properties of both polymers were assessed by producing devices with a tITO/PEDOT:PSS/polymer:PC70BM/BaF2/Ba/Al structure. The ratio of the polymer to PC70BM was adjusted from 1:3 to 1:6 (by weight), and the oprimum condition was found to have a ratio of 1:5 (Supplementary Figure S10 and Supplementary Table S1). The devices were measured under illumination with AM1.5G, 100 mW cm−2. PBDTAQ had JSC of 5.0 mA cm−2, Voc of 0.84 V and FF of 61.7%, exhibiting a PCE of 2.59%. At 1:5 (w w%−1), PBDTAFQ had a JSC of 7.5 mA cm−2, Voc of 0.84V and FF of 47.6%, exhibiting a PCE of 3.00%. Compared with PBDTAQ, PBDTAFQ had 1.5 times the JSC because the conjugation length and the planarity of the fused-phenyl ring increased when compared with that of the separated phenyl ring. This is consistent with Marks and Xie co-workers42 results indicating an increase in intermolecular charge transfer because of the effective π–π stacking of PBDTAFQ with its relatively high planarity.44

(a, b) J–V characteristics and (c, d) external quantum efficiency of photovoltaic devices fabricated with PBDTAQ:PCBM, PBDTAFQ:PCBM, PBDTAFQ:PCBM DIO and methanol (MeOH) treatment. DIO, dioxane; J–V, current–voltage; MeOH, methanol; PBDTAQ, poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene diphenyl Qu); PBDTAFQ, poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene dibenzophenazine; PCBM, [6,6′]-phenyl-C-butyricacidmethyl ester.
The addition of 3 vol% DIO increased the efficiency of the PBDTAFQ:PC70BM blend-based devices (at a 1:5 ratio) to 3.2%. FF then rose to 52.2% after MeOH treatment, increasing the PCE to 3.40%. This was achieved by improving the nanomorphology of the blending surface for the PC70BM polymer and therefore reducing the charge recombination, as confirmed by measuring the morphology through atomic force microscopy (AFM).
These devices translated into a sufficiently extensive EQE graph in the visible range (300–700 nm). The EQEmax value exceeded 55% for PBDTAFQ but went below 30% for PBDTAQ, which is alignment with PBDTAFQ polymers with high JSC values. When the theoretical JSC value was calculated using the EQE area, PBDTAQ:PC70BM (1:5 (w w−1)) was estimated at 4.19 mA cm−2 and PBDTAFQ:PC70BM (1:5 (w w−1)) at 7.27 mA cm−2, which were similar to the values calculated with the J–V curve.
Morphology
Figure 5 illustrates the morphologies of the polymer:PC70BM blend film obtained with AFM.

Atomic force microscopy (AFM) images of films spin coated from (a) PBDTAQ:PCBM 1:5 (w w%−1), (b) PBDTAFQ:PCBM 1:5 (w w%−1), (c) PBDTAFQ:PCBM 1:5 (w w%−1) DIO and (d) PBDTAFQ:PCBM 1:5 w w%−1 DIO MeOH. DIO, dioxane; MeOH, methanol; PBDTAQ, poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene diphenyl Qu); PBDTAFQ, poly(diethylhexyloxy benzo dithiophene-dioctyloxy dithiophene dibenzophenazine; PCBM, [6,6′]-phenyl-C-butyricacidmethyl ester. A full color version of this figure is available at Polymer Journal online.
The overall root mean square of the devices was below 1nm, and the film using PBDTAQ had a low value of 0.518 nm, whereas the PBDTAFQ film had a high value of 0.720 nm. Owing to the increase in the root mean square in the film state, the device using PBDTAQ showed a higher FF value than the device using PBDTAFQ.32
The nanonetwork channel for PBDTAFQ polymers and PC70BM was a larger domain, as illustrated in Figure 5b. On the other hand, the nanonetwork channel for devices treated with DIO additive and films postprocessed with MeOH was a smaller domain (Figures 5c and d). The decrease in the nanonetwork channel improves the charge separation in the active layer while reducing the exciton diffusion length and the charge recombination, leading to a high photocurrent.45
Conclusion
This study synthesized two kinds of Qu-type electron-accepting units (Qu and Pz), adopting octyloxy and polymerized D–π–A-type polymers—PBDTAQ and PBDTAFQ—using Stille cross-coupling with BDT derivatives as the electron-donating unit. Both polymers had a significantly higher molecular weight when compared with existing D–π–A polymers adopting Qu and Pz. PBDTAFQ displayed a higher absorbance and absorption region than PBDTAQ because of the expanded conjugation based on the fused-phenyl ring. Moreover, θ3 fell to 0°, increasing the intermolecular interaction and forming a face-on ordered structure. Therefore, PBDTAFQ had a superior JSC value when applied in an OPV, showing a high PCE, and it exhibited a PCE performance of 3.0% under the PBDTAFQ:PC70BM structure (1:5 (w w−1)). The morphology improved following the addition of DIO and MeOH treatment, improving the PCE to 3.40%.

Synthesis and characterization of monomers and polymers.
References
Bouzakraoui, S., Bouzzine, S. M., Bouachrine, M. & Hamidi, M. Theoretical investigation of electroluminescent alkoxy substituted 4,4′-bis(2-phenylethenyl)biphenyls as guest in blue OLEDs. Sol. Energy Mater. Sol. Cells 90, 1393–1402 (2006).
Armstrong, N. R., Wang, W., Alloway, D. M., Placencia, D., Ratcliff, E. & Brumbach, M. Organic/organic' heterojunctions: organic light emitting diodes and organic photovoltaic devices. Macromol. Rapid Commun. 30, 717–731 (2009).
Friend, R. H., Gymer, R. W., Holmes, A. B., Burroughes, J. H., Marks, R. N., Taliani, C., Bradley, D. D. C., Santos, D. A. D., Das, J. L. B., Gdlund, M. L. & Salaneck, W. R. Electroluminescence in conjugated polymers. Nature 397, 121–128 (1999).
Liu, B., Zou, J., Zhou, Z., Wang, L., Xu, M., Tao, H., Gao, D., Lan, L., Ning, H. & Peng, J. Efficient single-emitting layer hybrid white organic light-emitting diodes with low efficiency roll-off, stable color and extremely high luminance. J. Ind. Eng. Chem. 30, 85–91 (2015).
Kanal, I. Y., Owens, S. G., Bechtel, J. S. & Hutchison, G. R. Efficient computational screening of organic polymer photovoltaics. J.Phys. Chem. Lett. 4, 1613–1623 (2013).
Kim, J. R., Jung, J. H., Shin, W. S., So, W. W. & Moon, S. J. Efficient TCO-free organic solar cells with modified poly(3,4-ethylenedioxythiophene): poly(styrenesulfonate) anodes. J. Nanosci. Nanotechnol. 11, 326–330 (2011).
Jørgensen, M. & Krebs, F. C. Stepwise unidirectional synthesis of oligo phenylene vinylenes with a series of monomers. use in plastic solar cells. J. Org. Chem. 70, 6004–6017 (2005).
Wan, X., Long, G., Huang, L. & Chen, Y. Graphene—a promising material for organic photovoltaic cells. Adv. Mater. 23, 5342–5358 (2011).
Dadkhah, M., Salavati-Niasari, M. & Mir, N. Synthesis and characterization of TiO2 nanoparticles by using new shape controllers and its application in dye sensitized solar cells. J. Ind. Eng. Chem. 20, 4039–4044 (2014).
Kumar, P., Sharma, A., Yadav, S. & Ghosh, S. Morphology optimization for achieving air stable and high performance organic field effect transistors. Org. Electron. 14, 1663–1672 (2013).
Troshin, P. A., Ponomarenko, S. A., Luponosov, Y. N., Khakina, E. A., Egginger, M., Meyer-Friedrichsen, T., Elschner, A., Peregudova, S. M., Buzin, M. I. & Razumov, V. F. Quaterthiophene-based multipods as promising materials for solution-processible organic solar cells and field effect transistors. Sol. Energy Mater. Sol. Cells 94, 2064–2072 (2010).
Praveen, V. K., George, S. J., Varghese, R., Vijayakumar, C. & Ajayaghosh, A. Self-assembled ð-nanotapes as donor scaffolds for selective and thermally gated fluorescence resonance energy transfer (FRET). J. Am. Soc 128, 7542–7550 (2006).
Davar, F., Loghman-Estarki, M. R. & Ashiri, R. From inorganic/organic nanocomposite based on chemically hybridized CdS–TGA to pure CdS nanoparticles. J. Ind. Eng. Chem. 21, 965–970 (2015).
Lee, J. Y., Choi, M. H., Heo, S. W. & Moon, D. K. Synthesis of random copolymers based on 3-hexylthiophene and quinoxaline derivative: influence between the intramolecular charge transfer (ICT) effect and π-conjugation length for their photovoltaic properties. Synth. Met 161, 1–6 (2011).
Lee, J. -Y., Choi, M. -H., Song, H. -J. & Moon, D. -K. Random copolymers based on 3-hexylthiophene and benzothiadiazole with induced p-conjugation length and enhanced open-circuit voltage property for organic photovoltaics. J. POLYM. SCI. A 48, 4875–4883 (2010).
Zhang, J., Zhang, Y., Fang, J., Lu, K., Wang, Z., Ma, W. & Wei, Z. Conjugated polymer-small molecule alloy leads to high efficient ternary organic solar cells. J. Am. Chem. Soc. 137, 8176–8183 (2015).
Zhang, Z. G., Min, J., Zhang, S., Zhang, J., Zhang, M. & Li, Y. Alkyl chain engineering on a dithieno[3,2-b:2',3'-d]silole-alt-dithienylthiazolo[5,4-d]thiazole copolymer toward high performance bulk heterojunction solar cells. Chem. Commun. 47, 9474–9476 (2011).
Zhang, G., Fu, Y., Zhang, Q. & Xie, Z. Benzo[1,2-b:4,5-b']dithiophene-dioxopyrrolothiophen copolymers for high performance solar cells. Chem. Commun. 46, 4997–4999 (2010).
Lee, J. -Y., Kim, S. -H., Song, I. -S. & Moon, D. -K. Efficient donor–acceptor type polymer semiconductors with well-balanced energy levels and enhanced open circuit voltage properties for use in organic photovoltaics. J. Mater. Chem. 21, 16480 (2011).
Du, C., Li, C., Li, W., Chen, X., Bo, Z., Veit, C., Ma, Z., Wuerfel, U., Zhu, H., Hu, W. & Zhang, F. 9-Alkylidene-9H-fluorene-containing polymer for high-efficiency polymer solar cells. Macromolecules 44, 7617–7624 (2011).
Biniek, L., Chochos, C. L., Hadziioannou, G., Leclerc, N., Leveque, P. & Heiser, T. Electronic properties and photovoltaic performances of a series of oligothiophene copolymers incorporating both thieno[3,2-b]thiophene and 2,1,3-benzothiadiazole moieties. Macromol. Rapid Commun. 31, 651–656 (2010).
Song, H. -J., Kim, D. -H., Lee, E. -J. & Moon, D. -K. Conjugated polymers consisting of quinacridone and quinoxaline as donor materials for organic photovoltaics: orientation and charge transfer properties of polymers formed by phenyl structures with a quinoxaline derivative. J. Mater. Chem. A 1, 6010 (2013).
Osaka, I., Saito, M., Koganezawa, T. & Takimiya, K. Thiophene–thiazolothiazole copolymers: significant impact of side chain composition on backbone orientation and solar cell performances. Adv. Mater. 26, 331–338 (2014).
Song, H. J., Lee, T. H., Han, M. H., Lee, J. Y. & Moon, D. K. Synthesis of donor–acceptor polymers through control of the chemical structure: improvement of PCE by planar structure of polymer backbones. Polymer 54, 1072–1079 (2013).
Song, H. J., Lee, J. Y., Lee, E. J. & Moon, D. K. Conjugated polymer consisting of dibenzosilole and quinoxaline as donor materials for organic photovoltaics. Eur. Polym. J. 49, 3261–3270 (2013).
Li, G., Lu, Z., Li, C. & Bo, Z. The side chain effect on difluoro-substituted dibenzo[a,c]phenazine based conjugated polymers as donor materials for high efficiency polymer solar cells. Polym. Chem 6, 1613–1618 (2015).
Liang, Y., Feng, D., Wu, Y., Tsai, S. -T., Li, G., Ray, C. & Yu, L. Highly efficient solar cell polymers developed via fine-tuning of structural and electronic properties. J. Am. Chem. Soc. 131, 7792–7799 (2009).
Liang, Y., Feng, D., Wu, Y., Tsai, S. -T., Li, G., Ray, C. & Yu, L. Highly efficient solar cell polymers developed via fine-tuning of structural and electronic properties. J. Am. Chem. Soc. 131, 7792–7799 (2009).
Hou, J., Park, M. -H., Zhang, S., Yao, Y., Chen, L. -M., Li, J. -H. & Yang, Y. Bandgap and molecular energy level control of conjugated polymer photovoltaic materials based on benzo[1,2-b:4,5-b′]dithiophene. Macromolecules 41, 6012–6018 (2008).
Zhang, J., Cai, W., Huang, F., Wang, E., Zhong, C., Liu, S., Wang, M., Duan, C., Yang, T. & Cao, Y. Synthesis of quinoxaline-based donor−acceptor narrow-band-gap polymers and their cyclized derivatives for bulk-heterojunction polymer solar cell applications. Macromolecules 44, 894–901 (2011).
Lee, Y., Nam, Y. M. & Jo, W. H. Enhanced device performance of polymer solar cells by planarization of quinoxaline derivative in a low-bandgap polymer. J. Mater. Chem. 21, 8583 (2011).
Kang, T. E., Cho, H. -H., Kim, H. j., Lee, W., Kang, H. & Kim, B. J. Importance of optimal composition in random terpolymer-based polymer solar cells. Macromolecules 46, 6806–6813 (2013).
Keller, B., McLean, A., Kim, B. -G., Chung, K., Kim, J. & Goodson, T. Ultrafast spectroscopic study of donor–acceptor benzodithiophene light harvesting organic conjugated polymers. J. Phys. Chem. C 120, 9088–9096 (2016).
Xue, X., Fan, B., Liu, T., Sun, X., Huo, L., Ha, S. R., Choi, H., Kim, T., Kim, J. Y., Wei, D., Yu, M., Jin, Q. & Sun, Y. Influence of aromatic heterocycle of conjugated side chains on photovoltaic performance of benzodithiophene-based wide-bandgap polymers. Polym. Chem. 4036–4045 (2016).
Wang, E., Wang, L., Lan, L., Luo, C., Zhuang, W., Peng, J. & Cao, Y. High-performance polymer heterojunction solar cells of a polysilafluorene derivative. Appl. Phys. Lett. 92, 033307 (2008).
Zhou, E., Yamakawa, S., Tajima, K., Yang, C. & Hashimoto, K. Synthesis and photovoltaic properties of diketopyrrolopyrrole-based donor−acceptor copolymers. Chem. Mater. 21, 4055–4061 (2009).
Lee, J. Y., Song, K. W., Ku, J. R., Sung, T. H. & Moon, D. K. Development of DA-type polymers with phthalimide derivatives as electron withdrawing units and a promising strategy for the enhancement of photovoltaic properties. Sol. Energy Mater. Sol. Cells 95, 3377–3384 (2011).
Lee, J. Y., Song, H. J., Lee, S. M., Lee, J. H. & Moon, D. K. Synthesis and investigation of photovoltaic properties for polymer semiconductors based on porphyrin compounds as light-harvesting units. Eur. Polym. J. 47, 1686–1693 (2011).
Gowrisankar, M., Venkateswarlu, P., Siva Kumar, K. & Sivarambabu, S. Thermodynamics of amine+ketone mixtures 3. Volumetric, speed of sound data and viscosity at (303.15 and 308.15K) for the binary mixtures of N,N-dimethylaniline+propiophenone, +p-methylacetophenone, +p-chloroacetophenone. J. Mol. Liquids 173, 172–179 (2012).
Krebs, F. C., Tromholt, T. & Jorgensen, M. Upscaling of polymer solar cell fabrication using full roll-to-roll processing. Nanoscale 2, 873–886 (2010).
Gadisa, A., Mammo, W., Andersson, L. M., Admassie, S., Zhang, F., Andersson, M. R. & Inganäs, O. A new donor–acceptor–donor polyfluorene copolymer with balanced electron and hole mobility. Adv. Funct. Mater. 17, 3836–3842 (2007).
Lu, G., Usta, H., Risko, C., Wang, L., Facchetti, A., Ratner, M. A. & Marks, T. J. Synthesis, characterization, and transistor response of semiconducting silole polymers with substantial hole mobility and air stability. Experiment and theory. J. Am. Chem. Soc. 130, 7670–7685 (2008).
Lu, G., Usta, H., Risko, C., Wang, L., Facchetti, A., Ratner, M. A. & Marks, T. J. Synthesis, characterization, and transistor response of semiconducting silole polymers with substantial hole mobility and air stability. Experiment and theory. J. Am. Chem. Soc. 130, 7670–7685 (2008).
Zhang, G., Fu, Y., Zhang, Q. & Xie, Z. Synthesis and photovoltaic properties of conjugated copolymers with benzo[1,2-b:4,5-b′]dithiophene and bis(thiophene)phthalimide units. Macromol. Chem. Phys. 211, 2596–2601 (2010).
Sanjaykumar, S. R., Badgujar, S., Song, C. E., Shin, W. S., Moon, S. -J., Kang, I. -N., Lee, J., Cho, S., Lee, S. K. & Lee, J. -C. Synthesis and characterization of a novel naphthodithiophene-based copolymer for use in polymer solar cells. Macromolecules 45, 6938–6945 (2012).
Acknowledgements
This work was supported by the Konkuk University’s research support program in 2014.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Lee, T., Choi, M., Jeon, S. et al. Improvement of short circuit current density by intermolecular interaction between polymer backbones for polymer solar cells. Polym J 49, 177–187 (2017). https://doi.org/10.1038/pj.2016.104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2016.104
This article is cited by
-
Subtle Effect of Alkyl Substituted π-Bridges on Dibenzo[a,c]phenazine Based Polymer Donors towards Enhanced Photovoltaic Performance
Chinese Journal of Polymer Science (2022)
