
Abstract
Extensive cancer research in the past few decades has identified the existence of a rare subpopulation of stem cells in the grove of cancer cells. These cells are known as the cancer stem cells marked by the presence of surface biomarkers, multi-drug resistance pumps and deregulated self-renewal pathways (SRPs). They have a crucial role in provoking cancer cells leading to tumorigenesis and its progressive metastasis. Cancer stem cells (CSCs) are much alike to normal stem cells in their self-renewal mechanisms. However, deregulations in the SRPs are seen in CSCs, making them resistant to conventional chemotherapeutic agents resulting in the tumor recurrence. Current treatment strategies in cancer fail to detect and differentiate the CSCs from their non-tumorigenic progenies owing to absence of specific biomarkers. Now, it has become imperative to understand complex functional biology of CSCs, especially the signaling pathways to design improved treatment strategies to target them. It is hopeful that the SRPs in CSCs offer a promising target to alter their survival strategies and impede their tumorigenic potential. However, there are many perils associated with the direct targeting method by conventional therapeutic agents such as off targets, poor bioavailability and poor cellular distribution. Recent evidences have shown an increased use of small molecule antagonists directly to target these SRPs may lead to severe side-effects. An alternative to solve these issues could be an appropriate nanoformulation. Nanoformulations of these molecules could provide an added advantage for the selective targeting of the pathways especially Hedgehog, Wnt, Notch and B-cell-specific moloney murine leukemia virus integration site 1 in the CSCs while sparing the normal stem cells. Hence, to achieve this goal a complete understanding of the molecular pathways corroborate with the use of holistic nanosystem (nanomaterial inhibition molecule) could possibly be an encouraging direction for future cancer therapy.
Similar content being viewed by others


Introduction
Cancer remains one of the deadliest diseases affecting large number of people worldwide every year. Even after profound cancer treatments, cancer relapse and drug resistance are reported. In the past decade, underlying cause discovered to be associated with tumor recurrence, metastasis and chemoresistance are a relatively small population of stem cells inhabiting each adult tissue called as the cancer stem cells (CSCs). These stem cells in the long run have the opportunity to accumulate the mutations required for malignant transformation owing to their unlimited division potential. These cells were first identified by Bonnet and Dick (1997)1 in acute myeloid leukemia and following their findings many other groups have identified these cells in various solid tumors of brain,2 breast,3 pancreas,4 prostate5, 6 to name a few. CSCs display certain properties such as high expression of drug efflux transporters, abnormal cellular metabolism, deregulated SRPs, acquisition of epithelial-mesenchymal transition and extensive DNA-repair mechanisms.
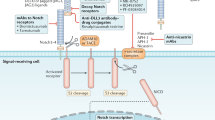
Self-renewal is one of the important properties employed by the CSCs to maintain the proliferating capacities. As genetic and epigenetic changes might have a role in the unrestrained growth, invasion and acquired resistance in cancer cells, it is implicated that epigenesis may accord deregulation of self-renewal pathways (SRPs) in CSCs. There are number of signaling pathways functioning in the normal stem cells, which have assigned roles in the early embryogenesis-like cell proliferation, cell differentiation, cell fate, cell polarity and so on and are under strict regulation. In CSCs, these SRPs when deregulated lead to extensive cell proliferation and may be considered an early event in the process of carcinogenesis. Extensive experimental evidences have revealed Hedgehog (Hh), Wnt, Notch and B-cell-specific moloney murine leukemia virus integration site 1 (BMI1) pathways to be the key players in maintaining the proliferating capacity of CSCs and activated in most of the solid tumors.7 Among other signaling proteins such as phosphatase and tensin homolog,8 bone morphogenetic protein and transforming growth factor beta are also of specific interest as they too control self-renewal and cell differentiation in various tissues and are additionally implicated in tumorigenesis. Recent investigations of targeting the signaling pathways in CSCs have found to be of prime interest. This review focuses on several aspects of major SRPs, which are found to be upregulated in CSCs and certain novel strategies to target these pathways by nanodrug-delivery platforms for the prevention of tumor relapse and chemoresistance (Figure 1).

Targeting strategies in self-renewal pathways in CSCs including their pharmacological antagonists and different nanoparticles used for formulation. (1) Hh ligand Inhibitors (2) GLI Antagonists (3) SMO Inhibitors (4) Anti-DLL4 Antibodies (5) γ –Secretase Inhibitors (6) MAML Inhibitors (7) Anti-FZD Antibodies (8) Wnt Ligand inhibitors (9) Wnt Transcription Complex Inhibitors (10) HDAC Inhibitors.
Self-renewal pathways in CSCs
CSCs make up a minor fraction of the tumor tissues. It acquires a heterogeneous phenotype and can maintain tumor formation at a high degree. Apparently, it is seen that the CSCs share common attributes with the normal stem cells, for instance, self-renewal and differentiation capacity. However, there exist fine-drawn differences between CSCs and normal stem cells for using the same pathways. The molecular mechanisms underlying these phenomena of CSCs hijacking the SRPs of normal stem cells for its own maintenance though remains vague. In the following sections, we are going to review the potential pathways, which are implicated in the CSCs self-renewal activity and tumor initiation through immense experimental findings.
Hh pathway
It is known that the Hh pathway helps in controlling cell growth, tissue patterning, morphogenesis9 in animal development. The Hh family of proteins has at least three Drosophila Hh gene homologs in vertebrates: Sonic Hh (SHh), Desert Hh and Indian Hh, among which SHh is the most widely used one. The Hh is a 400–460 amino-acid long precursor protein. The (HhN) amino-terminal domain works as a signaling molecule, whereas the carboxy-terminal domain (HhC) has an auto-catalyzing Hint module. The signaling cell releases the Hh protein through a committed transmembrane receptor called the Dispatched. This happens only after the amino terminal of the Hh protein is being palmitoylated by Rasp/Skinny located in endoplasmic reticulum.10 The modified Hh protein binds to its 12 transmembrane receptor known as the Patched (Ptc) and initiates the signaling process. In Hh pathway, a seven-pass transmembrane receptor named Smoothened (Smo) activation is necessary for further signaling process. In the absence of Hh, the Ptc prevents Smo from being located to the primary cilium and its catalytic activity. However, when Ptc is bound by Hh ligand, the inhibitory effect of Ptc on Smo is rendered inactive. Smo now activates the Gli family of transcription factors to carry out the downstream signaling process. Without Smo activation, Gli is maintained in a complex with Suppressor of Fused, which is a negative regulator of Hh signaling. Upon Smo activation, Gli is dissociated from Suppressor of Fused-Gli complex for nuclear translocation to promote the transcription of Hh targeted genes namely patched, cyclin (D/E). In mammals, there are three types of Gli transcription factors Gli1, Gli2 and Gli3 of which Gli1 and Gli2 are activators and Gli3 acts as a repressor. The loss of Suppressor of Fused results in the activation of Hh signaling, which indicates its central role in the repression of the pathway.11
The Hh signal transduction pathway components tightly control embryonic development, and also expressed in postnatal and adult tissues, where these components have assigned roles in the maintenance of stem cells, tissue repair and regeneration. Hence, defects in Hh signaling may affect at the embryonic and later stages of life in humans.12 Many human congenital diseases have been associated with Hh signaling defects such as holoprosencephaly in which there is loss of one copy of SHh.13 Mutations in Ptc1 result in a rare autosomal genetic form of basal cell carcinoma also known as the Gorlin syndrome.14, 15 Increasing evidence have widely supported the fact that dysregulated Hh signaling is present in majority of the human cancers today, which includes brain tumors, melanomas, leukemia’s, gastro-intestinal, malignancies of the breast, ovary, prostate and pancreas.16 However, in most of these cancers mutation of Hh pathway components is not the only basis for its aberrant activation, but rather has been caused by high expression of Hh ligands.17, 18 Experimental evidences in the past have confirmed the presence of CSCs in most of the human tumors and the self-renewal property of these cells has been attributed to Hh signaling.19, 20, 21, 22, 23 Hh signaling maintains the self-renewal capacity of the malignant clone, which was demonstrated in mouse models of chronic myeloid leukemia.20, 23 Hh signaling is also under epigenetic regulation in CSCs mainly the Gli transcription factors. As Gli1 and Gli2 are acetylated, their deacetylation mediated by Histone deacetylase (HDAC) complex promotes Hh pathway activation. Downregulation of Gli1 is mediated by miR-324-5p, and subsequent loss of miR-324-5p have led to neoplastic transformation into medulloblastoma.24 Ptc and Gli1 proteins were seen to be highly expressed in ovarian cancer patients as reported by Liao et al.25 The authors in this study also observed that there was a significant overexpression of SHh mRNA in the patient’s tumor tissues. It is also affirmed that Hh signaling has an active role in the progression of prostate cancer; however, there is paucity of the precise mechanism involved in its abnormal signaling. Sheng et al.26 have reported a loss-of-function mutations in Suppressor of Fused, in most of the prostatic tumor tissues. Other independent studies carried out by groups have presented with data that there is a ligand-dependent paracrine or autocrine Hh signaling in prostate tumors.27, 28 Hh signaling is also found to regulate self-renewal in normal and mammary CSCs acting in concert with BMI pathway as investigated by Liu et al.29 in their in vitro and in vivo studies.
Notch pathway
Notch signaling is a developmental pathway in multicellular organisms involved in cell fate decisions and pattern formation during embryogenesis.30 Post-translational modifications result in the formation of a heterodimeric NECD (notch receptor comprising of an extracellular domain) and TM-NICD (transmembrane-intracellular domain) inserted in the plasma membrane of a signal-receiving cell. Once a ligand for example, Delta (DLL1, DLL3, DLL4) and Jagged (jag1, jag2) binds to the notch receptor, the TNF-alpha ADAM metalloprotease-converting enzyme mediates the cleavage of NECD from TM-NICD. The NECD-ligand complex is endocytosed/recycled in the signal-sending cell by Mind Bomb ubiquitination, whereas in the signal-receiving cell the γ-secretase enzyme cleaves TM-NICD complex, releasing NICD. It further proceeds into the nucleus and associate with the CSL (centromere-binding factor 1/Suppressor of hairless/Lag1) transcription complex. This CSL-NICD complex now subsequently activates the notch target genes: Hairy and enhancer of split family, p21 and Myc.
Apart from regulating cellular communication in embryogenesis, it also helps in stem cell growth and differentiation. Studies have elucidated the pathological role of notch pathway in human malignancies going from T-cell acute lymphoblastic leukemia (T-ALL)31 to breast cancer32, 33 and others where inappropriate activation of the pathway that led to uncontrolled proliferation, restricted differentiation and prevents apoptosis in the cancer cells. Of late, a mere reason of focusing on notch pathway in recent years is due to the identification of a distinct cellular hierarchy in human acute myeloid leukemia1 and other solid tumors.2, 3 This cellular hierarchy is the CSCs, which maintains the tumor and recapitulates the features of normal stem cells. Notch pathway is one of the developmental pathways active in this subset of CSCs, which maintains the self-replication and differentiation decisions. A significant evidence of the Notch pathway, that it is related for the survival of CSCs, came from the independent studies conducted by Farnie and Clarke;34 Sansone et al.35 Farnie and Clarke reported the role of aberrant notch signaling as one of the factors involved in early breast cancer. Studies by Gustafasson et al.36 have indicated that the notch and hypoxia response factor HIFα interacts with each other to assist the outset of a stem cell phenotype and its survival in hypoxic environment. Based on these findings, Sansone et al.35 carried out various studies to report that the expression of notch-3 is being controlled by the 66k-Da isoform of the Src homology 2 domain-containing gene (p66Shc), which gets induced in a breast cancer cell line when exposed to a hypoxic environment also leading to the survival of mammary gland progenitor cells. Notch signaling also has an oncogenic role in T-ALL where Notch 1 was identified to be involved in t (7; 9)(q34;q34.3) chromosomal translocation to bring out the disease outcome.37 Subsequent studies have brought newer insights to the role of Notch in human T-ALLs, with discovery of two types activating mutations within Notch 1.38 One mutation was in the extracellular hetero-dimerization domain, a change in the amino-acid sequence leading to ligand-independent metalloproteinase cleavage site S2, whereas the second involved Notch 1 proline, glutamic acid, serine, threonine sequence domain. These mutations were reported to be present in 50% of human T-ALLs.38 Notch 1 is also shown to have an elevated expression in pancreatic CSCs compared with non-pancreatic CSCs.39 In pancreatic cancer, notch pathway maintains the epithelial cells in a progenitor state, acquiring epithelial-mesenchymal transition phenotype leading to tumor growth, invasiveness and metastasis.40, 41 Emerging evidences show that the resistance of pancreatic cancers toward several chemotherapeutic measures is due to activated Notch signaling, although underlying mechanism still remains elusive.41, 42 These studies provides the rationale to develop targeted therapies, which will interfere with notch signaling in human malignancies.
WNT pathway
The Wnt signaling pathway is an ancient and evolutionary conserved developmental pathway, which controls stem cells and determines cellular fate during development. The Wnt family is a group of 19 glycoproteins in humans involving a complex mechanism of signaling phenomena, with salient functional and biological outcome.43 It may lead to much serious pleiotropic pathology when these tightly controlled mechanisms go awry. The Wnt ligand binds to a transmembrane receptor Frizzled and displaces the GSK-3β (glycogen synthase kinase 3 beta) from the adenomatous polyposis coli (APC)/Axin/GSK-3β regulatory complex. However, the absence of Wnt ligand marks the degradation of β-catenin a cell adhesion protein and transcription regulator in APC/Axin/GSK-3β and casein kinase1 destruction complex44, 45 through the beta transducing repeat containing E3 ubiquitin protein ligase pathway. Once Wnt ligand binds to its receptor the pathway is turned on and brings the co receptor low-density lipoprotein receptor related protein 5/6 to the vicinity of the Wnt bound Frizzled complex. This activates downstream component Disheveled by sequential phosphorylation, polyubiquitination, polymerization and finally stabilizing β-catenin.46 β-catenin now translocate to the nucleus where it associates with T-cell factor/lymphoid-enhanced factor family of transcription factors, and recruits other co-activators such as cAMP response element-binding protein, p300,47, 48 Bcl949 and Pygopus.50 This ultimately leads to transcription of target Wnt genes: survivin, cyclin D and c-myc.
The relevance of Wnt signaling in human cancers was perhaps best well known for its role in colon cancer where the healthy colonic epithelia accumulates mutation in specific genes such as APC, β-catenin, K-ras and p53.51 Morin PJ et al.52 had carried out genetic studies in four different kinds of APC mutants and analyzed that the presence of APC mutations in colorectal cancer also leads to defective downregulation of β-catenin and Tcf-4 transcriptional activity. There are numerous mechanisms that can drive the aberrant Wnt/β-catenin signaling, leading to cancer formation in a mutually exclusive manner. In certain colorectal cancers, there is a probability of finding an exclusive catenin (cadherin-associated protein) beta 1 mutation when APC mutations are lacking.53, 54 This was also supported by the conclusive evidence, which came from the studies of Mirabelli-Primadehl et al.55 regarding the role of β-catenin mutations in colorectal cancers. Hepatocellular carcinoma56 and endometrial ovarian tumors57, 58 were also found to possess catenin (cadherin-associated protein) beta 1 mutations, which led to aberrant nuclear accumulation of β-catenin. A vast majority of the colorectal tumors harbor APC mutations, which may lead to the constitutive activation of β-catenin59, 60, 61 Like Hh and Notch, Wnt/β-catenin signaling too has an important role in embryogenesis and regulates cell proliferation and lineage differentiation in many tissues.62 In adults, Wnt signals are basically involved in stem cell renewal especially in intestinal crypts,63 hair follicles64 and bone growth plate.65 As Wnt signaling has a notable role to play in stem cell proliferation and differentiation, its disruptions will certainly affect stem cell function with serious implications for malignancy. Consistent findings have shed light to the fact that β-catenin is present in a variety of CSCs settings66, 67, 68 including colon,69 cutaneous CSC70 and also HSC.71 Among all these CSCs, colon CSCs were found to have a very high concentration of β-catenin, which contributes to its stemness, in part orchestrated by the microenvironment finally giving rise to drug resistance and also metastasis.69 Wnt signaling has been also shown to be responsible for epithelial-mesenchymal transition72 in tumors as a result of high concentration of β-catenin in the nucleus.73 This leads to the arrest of tumor cell division and acquiring mesenchymal markers like fibronectin74 while retaining the self-renewal capacity, a characteristic feature employed by the CSCs.
BMI1 pathway
The BMI1 pathway is one of the proto-oncogenic signaling pathways like Hh, Notch and Wnt involved in the differentiation and self-renewal mechanisms of stem cells persistently.75 The BMI1 belongs to the Polycomb group of gene family, well-known epigenetic gene silencers, targeting the p16 and p19Arf locus76 both of which suppresses cell proliferation. Human BMI1 gene comprises of 10 exons and is localized on chromosome 10.77 BMI1 gene encodes a 324 amino-acid long protein with a predominant nuclear localization comprising of a N-terminal RING finger domain and a central helix turn helix motif.78 BMI1 affects morphogenesis during embryonic development and in hematopoiesis as reported by van Der Lugt et al.79 in 1994 with a pervasive expression in almost all tissues. Extensive studies have also reported the association of BMI1 in the initiation of various cancers where BMI1 can cooperate with c-myc and initiate the disease.80 Its expression was found to be highly upregulated in acute myeloid leukemia,81 cancers of the lung,82 ovaries,83 breast84 and neuroblastoma.85 It is noted that CSCs are highly enriched with BMI1, and seen to be co-expressive with stem cell markers, CD133 and CD44, in most of the tumor CSC population.86, 87, 88 Zhang et al.89 in their study asserted that epithelial ovarian cancers arise from a population of tumor-initiating cells with the CD44- and CD117-positive marker phenotype along with the expression of BMI1 and others such as Notch 1, ATP-binding cassette sub-family G member 2, Nanog, Nestin and Oct-4. The expression of these markers led to chemoresistance and exacerbated the disease condition. Cui H et al.85 reported BMI1 to be overexpressed in human neuroblastoma primary tumors and cell lines, cooperating with MYCN gene in transforming the benign S-type neuroblastoma cells. Prostate cancer cells too have a heightened expression of BMI1 in tumors with Gleason scores of 8 or higher.90 Glinsky and colleagues91 carried out a microarray analysis in 11 different types of cancer specimens and indicated that the conserved BMI1 driven pathway is engaged in a metastatic behavior of human malignancies along with a stem cell-like expression profile ultimately leading to disease recurrence after therapy. These studies indicate that the overexpression of BMI1 is critical for the maintenance of CSCs in most of the human tumors.
Targeting strategies to inhibit self-renewal pathways in CSCs
Conventional cancer treatment of chemotherapy and radiotherapy can target only the bulk of sensitive tumor cells, which are in rapidly dividing phase. This therapeutic intervention induces many tumor cells to undergo apoptosis and die, whereas the CSCs survive this process by remaining in G0 phase and give rise to 'second-line tumors' with acquired resistance.92, 93, 94 Henceforth, current cancer research is focused toward targeting these CSCs and it has become essential to develop novel therapeutic approaches to prevent cancer recurrence and emergence of drug resistance. Even though tremendous research has been carried out to eliminate the CSCs, but efficient modalities to target the SRPs in CSCs have been gaining prime focus in recent years. During and after the treatment period CSCs maintain their self-renewal and differentiation capacities by activating the embryonic signaling pathways. The Hh, Notch, Wnt and BMI1 maintains the proper functionality in normal stem cells but a deregulated behavior in these pathways, owing to some alterations in the genes encoding the signaling molecules is observed in CSCs and also have been found in human tumor samples clearly stating their role in tumor development and maintenance.95, 96 As normal stem cells and CSCs share similarities in the signaling pathways, it would be extremely important while designing drugs to understand the complex biology of these pathways to destroy the CSCs and selectively sparing the normal stem cells.
Drugs targeting self-renewal pathways
Cyclopamine, a plant derived teratogen binds and deactivate Smo which is otherwise being suppressed by Ptc. Targeting the Hh pathway using cyclopamine was shown by Taipale et al.97 where they suggested that Hh pathway related tumors associated with Ptc mutations might respond well to treatment with cyclopamine. As cyclopamine is a steroidal compound, it affects the activity of Ptc by blocking its sterol-sensing domain.98, 99 Bar EE et al.100 conducted a study on cyclopamine-mediated inhibition of Hh pathway in glioblastoma CSCs, and observed a significant 40–60% decrease in growth of adherent glioma cell lines with high Gli1 expression and no new neurospheres formed. Apart from cyclopamine, another synthetic small molecule inhibitors of Smo, GDC-0449 identified by Genentech was shown to inhibit the Hh pathway activity in metastatic basal cell carcinoma (ClinicalTrials.govnumber, NCT00607724).101 Oral administration of GDC-0449 was given to 33 patients with advanced basal cell carcinoma for a median duration of 9.8 months and reported two complete responses and 16 partial responses.101 GDC-0449 was also shown to have its inhibitory effect in medulloblastoma, pancreatic cancer but its effect is more prominent in advanced basal cell carcinoma. Several other small molecule Smo antagonists, which are investigated clinically include IPI-926,102 BMS-833923 (Clinical trials.govnumber, NCT00884546), PF-04449913 (Clinical trials.govnumber, NCT00953758), LDE-225.103, 104 However, there may be resistance to these molecules over a period of time due to point mutations in Smo. Hence, targeting the SHh ligand and the downstream components such as Gli transcription factors by small molecules namely Robotnikinin105 and HPIs 1-4,106 GANT58,107 GANT61,107 respectively, is a promising approach to prevent tumor relapse and metastasis. In addition to chemical compounds used for the treatment of human cancer, researchers have also considered the use of dietary chemopreventive agents known as nutraceuticals for targeting the Hh signaling such as Resveratrol,108 Curcumin109 and epigallocatechin-3-gallate,110 which have been experimentally shown to inhibit Hh signaling in prostate cancer, medulloblastoma and chondrosarcoma, respectively.
Most of the agents that have been developed to inhibit notch signaling are designed to target notch ligands, notch receptors, ligand receptor binding, γ-secretase-mediated cleavage and transcriptional nuclear complex. γ-secretase inhibitors are small molecule agents, which are widely studied, as notch activation largely depends on γ-secretase activity and is a promising target. A number of clinical trials on γ-secretase inhibitors is well indicated to inhibit notch signaling in many cancers, for example, T-ALL, central nervous system malignancies,111 breast cancer.112 MK0752, one of the potent γ-secretase inhibitors in clinical development was shown to inhibit notch signaling in majority of human T-ALL.113 Another γ-secretase inhibitor PF-03084014 was shown to inhibit Notch activity in T-ALL cell lines by Wei P et al.114 Apart from targeting the γ-secretase activity, notch ligand-inhibiting agents specially DLL4 monoclonal antibodies, for example, OMP-21M18 are in clinical development, designed for patients diagnosed with colon cancer, pancreatic cancer and small cell lung cancer.115 DLL4, specific notch ligand for embryonic vascular development and arteriogenesis116, 117 when blocked by a selective antibody-impeded tumor growth in several solid tumor models.118 Other agents that inhibit notch signaling in cancer include mastermind-like peptide inhibitors, which interferes with the notch nuclear co-activator mastermind-like protein, a part of the Notch transcriptional complex119 and notch soluble receptor decoys.120 Also, the use of natural compounds such as genistein,121 sulforaphane,122 quercetin123 owing to their relative low toxicity was seen to inhibit notch activity in tumor cells or in CSCs.
Agents that can inhibit Wnt signaling, currently under investigations, employ strategies to target receptor/ligand interactions, cytosolic and nuclear signaling components. One of the approaches to inhibit receptor ligand interactions is to target the Frizzled family of receptors by using antibodies. Studies have been carried out using a humanized antibody against Frizzled 10 for patients with synovial sarcoma.124 In vitro studies revealed that synovial sarcoma cells were suppressed by the polyclonal antibody in mediating antibody dependent cell-mediated cytotoxicity against the Frizzled 10 receptor overexpressed cells.124 Monoclonal antibodies targeting the Wnt (1–2) ligands have also disclosed the inhibition of Wnt signaling in colon cancer125 and human melanoma.126 Disheveled protein is one of the key cytosolic signaling components in the Wnt pathway that associates extracellular signals to its downstream components. Disheveled could be a therapeutic intervention in inhibiting the Wnt pathway for cancer therapy. Compounds that have been preclinically tested in this direction include FJ9127 and NSC668036.128 One of the critical steps in the activation of Wnt signaling is the interaction of β-catenin with the T-cell factor/lymphoid-enhanced factor transcription factors, and recruits a myriad of co-activators such as cAMP response element-binding protein, p300 to name a few.47 These co-activators represent potential targets to interfere with the β-catenin/transcription factor stabilization complex. ICG-001 a small molecule inhibitor129, 130 (Institute for chemical genomics) was developed in this direction to target these co-activators.
BMI1 has no enzymatic function hence traditional drug discovery approaches to target this protein remains a challenge. However, the use of HDAC inhibitors to suppress the expression of BMI1 and its downstream components was recently shown by Bommi et al.131 in human breast cancer. The HDAC inhibitors such as sodium butyrate and valproic acid were investigated in the study where the compounds seem to inhibit BMI1 activity through a transcriptional mechanism repressing the polycomb complexes. Another drug artemisinin and its derivatives having antimalarial activity were shown to have inhibition on cancer cell growth and angiogenesis. This drug was investigated to check its inhibitory role in regulating BMI1 expression both in protein and transcript levels in nasopharyngeal carcinoma cells.132 To date, no small molecules have been reported to inhibit BMI1 with competent specificities, although experimental evidences cited above using HDAC inhibitors and artemisinin bring a rationale to develop more agents for therapeutic targeting of BMI 1.
Prospects of nanodrug targeting
In current cancer treatment strategies, targeted drug delivery is one of the safest ways to target the tumor. To address this issue, nanoparticles have had an important role in delivery of drugs specifically at the designated site at the required concentration, evading immune response without having any off targets within the safety margins. Nanoparticles in the past have received quite unprecedented success as drug-delivery vectors in cancer therapy and diagnosis because of their biophysiological properties and the ability to interact with cells due to the similarity of their size with cellular components.133, 134, 135 They can carry multiple payloads owing to their large surface area, multi-functionalized with targeting moieties and controlled drug release.136, 137 Taking into account about the multiple advantages of nanoparticles, they can be harnessed to the best of their ability to target the drug-resistant CSCs. Independent studies conducted by researchers have applied nanoparticles to target CSCs in diverse overlapping areas. Lee et al.138 and Swaminathan et al.139 in their distinctive studies have made use of nanoparticles as 'beacons' to label CSCs as a diagnostic measure. Nanoparticles were also successfully used to deliver non-druggable anticancer agents to kill the drug-resistant CSCs.140 Moreover, nanoparticles in the form of stealthy liposomes were used as therapeutic intervention by Liu et al.141 to wipe out CSCs and non CSCs selectively. Many groups have recently targeted the CSCs effectively through the use of combination therapy of antibodies and conventional chemotherapeutic drugs against the CSC surface markers CD133+142 and drug efflux transporters.143 Yu et al.144 in their study eliminated CD133+ osteosarcoma CSCs through salinomycin delivery via CD133 aptamer-conjugated PEGylated PLGA nanoparticles. These approaches though have received encouraging results, but still leave plenty of room for improvement. Another approach to target the CSCs, which is the main focus of this review, and have received a lot of attention over the years is the targeting of the SRPs, which are implicated to maintain the self-renewal capacity of the CSCs and involved in tumorigenesis. Till date, SRPs as discussed in the above sections are being targeted directly by the use of small molecule inhibitors, monoclonal antibodies and natural compounds. Although these agents have shown promising results in inhibiting the deregulated pathways in CSCs145, 146 there have been certain drawbacks associated such as toxicity, poor water solubility and poor specificity. Hence, nanoformulation of these compounds along with the combination of conventional chemotherapeutic drugs is a holistic approach to inhibit the SRPs in CSCSs.
Chenna et al.147 recently have engineered a polymeric nanoparticle encapsulating a small molecule inhibitor, HPI-1 (Hh pathway inhibitor), which was shown to bypass the secondary mutational resistance toward Smoothened antagonists. Hh signaling is seen to be aberrantly active in most of the human cancers, and Smo secondary mutation abrogates the binding of most of the Hh inhibitors. The group addressed this issue by nanoformulating HPI-1 (NanoHHI) that is a potent antagonist of Gli1 and reported that NanoHHI markedly inhibits the growth of mouse medulloblastoma allografts, which harbor a SmoD477G-binding site mutation, accompanied by significant downregulation of Gli1 mRNA. Nanoformulation of HPI-1 improved its aqueous solubility and also systemic bioavailability.147 The same group further confirmed their studies by using NanoHHI to check the inhibition of Hh signaling in hepatocellular carcinoma (HCC) in an orthotopic model. NanoHHI markedly reduced systemic metastases in HCC cell lines both in vitro and in vivo settings. Moreover, it also decreased the population of CD133+-expressing HCC cells, considered to be the tumor-initiating cells.148 Lim K et al. revealed that polymeric nanoparticle formulation of curcumin suppressed the growth of multiple brain tumor cell lines. The authors observed that NanoCurc when administered to brain tumor cell lines in a dose-dependent manner, it led to programmed cell death in addition to depleting CSCs. In their study, microarray analyses disclosed that when medulloblastoma DAOY cells treated with 20 μM curcumin showed 2.4-fold downregulation of Gli1 expression, which is a key effector in Hh signaling. However, notch activity was not seen to be much affected by curcumin treatment in DAOY cells.149 A liquid–lipid nanoparticle delivery system has been harnessed in a recent study by You et al.150 to deliver the Smo antagonist CPA-LLP (cyclopamine) in 4T1 murine breast cancer and Miapaca-2 human pancreatic carcinoma models (Figures 2a and b). The group used a combination strategy of CPA-LLP and core-cross-linked polymeric micelles bound lutetium-177 in the carcinoma models and reported slow tumor growth. Pancreatic ductal adenocarcinoma is characterized with desmoplasia, aberrant Hh signaling and downregulation of tumor suppressor miR-let7b. Desmoplastic environment provides the niche for CSCs. Mahato et al.151 carried out synergistic treatment of pancreatic ductal adenocarcinoma through co-delivery of Hh inhibitor GDC-0449 and miRNA (miR-let7b) into micelles using methoxy poly (ethylene glycol)-block-poly (2-methyl- 2-carboxyl-propylenecarbonate-graft-dodecanol-graft-tetraethylene-pentamine) (mPEG-b-PCC-g-DC-g-TEPA). It was observed that the combination therapy of GDC-0449 and miR-let7b micelles led to reduced cell viability in the different pancreatic cell lines (HPAF-II, Capan-I, T3M4, MIA-PaCa-I) even at low dose concentration of the formulation (Figures 2c–f).

(a) Schematic illustration of study design. Radioactive polymeric micelles containing 177Lu were injected intratumorally, and CPA-loaded lipid nanoparticles were injected intravenously. (b) Transmission electron microscopy images of CPA- LLP (negative staining). (Reproduced with permission from You J et al. 2015). (c–f) Effect of GDC-0449 and miR-let7b on cell viability in human pancreatic cancer cell line by micelles. HPAF-II, Capan-1, T3M4 and MIAPaCa-2 cells (5000/well) were treated with micelles containing (blue bars) GDC-0449 (0, 1, 5 and 10 μM), (green bars) GDC-0449 and scrambled miRNA, (red bars) GDC-0449 and miR-let7b (10 pmol), (peach bars) miR-let7b alone, and (purple bars) blank for 48 h. Cell viability was measured by MTT assay at the end of incubation period. (Reproduced with permission from Kumar V et al. 2015).
Notch signaling is mostly targeted by the use of gamma-secretase inhibitors but its clinical use is hindered by acute after-effects and hence the need for an alternative strategy. A novel approach of delivering the gamma-secretase inhibitors to block Notch signaling was presented by Mamaeva and colleagues using imagable mesoporous silica nanoparticles, which were found to be biocompatible, biodegradable and delivered gamma-secretase inhibitors without any toxic side-effects (Figures 3a–c). The group designed a drug-loaded mesoporous silica nanoparticles of average size centered ~200–350 nm and surface modified with folate (FA) to the outer polyethylenimine layer of the particles. In vitro analyses were screened using different breast cancer cell lines (MCF7 (FR-positive), MDA-MB-231, T47D, SK-BR-3, MDA-MB-468). The study revealed the mesoporous silica nanoparticles-mediated delivery of gamma-secretase inhibitors was specific toward the cells and also inhibited Notch signaling. MCF7 cells were reported to have the highest FA-mediated endocytosis due to its surface functionalization. Moreover, in vivo studies also supported that targeted gamma-secretase inhibitors delivery-enhanced tumor penetration and retainment at the tumor site as compared with free drug.152 Recently Lo et al. have designed a small interfering RNA-delivery approach against the enhancer of zeste homolog 2 and Oct-4 genes upregulated in head and neck squamous cell carcinoma using polyurethane-short branch polyethylenimine. The small interfering RNA polyethylenimine constructs used was able to repress epithelial-mesenchymal transition and radioresistance in aldehyde dehydrogenase 1+/CD44+ CSC-like cells, in addition to inhibiting Wnt signaling, which may be involved in the CSCs.153

Mesoporous silica nanoparticles (MsnPs) accumulate in the tumors, are biocompatible biodegradable and eliminated through renal excretion. (a) In vivo imaging of mice injected peritumoral with PEI-MSNPs or folate (FA)-MSNPs. Images of the abdominal area demonstrate accumulation of fluorescence in the bladder, and imaging of the dorsal area show accumulation of fluorescence in the tumors. Time lapse imaging of the abdominal area shows elimination of fluorescence within 48 h after injections (number of animals per group, n=4, two tumors per animal). (b) Ex vivo analyses (left) and quantification of fluorescence intensity (right) in organs from mice injected intravenous (i.v.) with FA-MSNPs. Mice were killed 196 h after injection (n=4). Please note the occasional signal from brain tissue, which most likely represents background fluorescence, as it is present also in untreated control animals. (c) Histological analysis of the brain, kidney, spleen, liver and lungs of untreated mice and FA-MSNPs-treated mice showed no morphological changes. Mice were killed 192 h after i.v.injection. (Reproduced with permission from Mamaeva V et al. 2011.)
Although these experimental findings are encouraging to target the SRPs through nanoparticle-mediated delivery. However, it is imperative to extend more research in combining the SRPs-targeting therapeutics with nanotechnology-based platforms for a robust cancer treatment strategy for clinical applications.
Conclusion and future direction
In this review, we have tried to render a picture of the heterogeneous CSCs being implicated to be a cause of cancer relapse, chemo and radioresistance in recent times. Understanding the complex biology behind the survival mechanism of CSCs in solid tumors, deregulation in the SRPs is seen to be one of the prominent reasons for their inevitable existence even after treatment. Despite the availability of small molecule inhibitors used to target the SRPs, a small fraction of them only has been put to clinical application owing to their non-specific toxicity and solubility issues. This could be solved by nanoformulating these compounds, which will overcome their barriers and specifically deliver these molecules to the designated sites. Nanoparticles as mentioned above have been used in recent times to target the CSCs in solid tumors; hence, nanotechnology could also be extended to target the SRPs active in CSCs. As there occurs crosstalks between the different signaling pathways in cancer development and progression, inhibition of one could lead to the downregulation of the others. Nanoparticles could provide a platform to carry multiple pathway inhibitors along with a conventional chemotherapeutic to target the pathways. Although there have been very few reports cited in literature in this direction, comprehending the biology of the pathways combined with the use of wide range of nanoparticles in dispose is a challenging area of research and leaves a futuristic hope for cancer treatment in killing the CSCs.
References
Bonnet D, Dick JE . Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–737.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003; 63: 5821–5828.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF . Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–3988.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V et al. Identification of pancreatic cancer stem cells. Cancer Res 2007; 67: 1030–1037.
Maitland NJ, Collins AT . Prostate cancer stem cells: a new target for therapy. J Clin Oncol 2008; 26: 2862–2870.
Lang SH, Frame FM, Collins AT . Prostate cancer stem cells. J Pathol 2009; 217: 299–306.
Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004; 351: 657–667.
Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/β-catenin signaling. PLoS Biol 2009; 7: e1000121.
Ingham PW, McMahon AP . Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001; 15: 3059–3087.
Micchelli CA, The I, Selva E, Mogila V, Perrimon N . Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 2002; 129: 843–851.
Svärd J, Henricson KH, Persson-Lek M, Rozell B, Lauth M, Bergström Å et al. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell 2006; 10: 187–197.
Ruiz iAltaba A, Sánchez P, Dahmane N . Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2002; 2: 361–372.
Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 1996; 14: 357–360.
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85: 841–851.
Johnson RL, Rothman AL, Xie J, Goodrich L V, Bare JW, Bonifas JM et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996; 272: 1668–1671.
Kiesslich T, Berr F, Alinger B, Kemmerling R, Pichler M, Ocker M et al. Current status of therapeutic targeting of developmental signalling pathways in oncology. Curr Pharm Biotechnol 2012; 13: 2184–2220.
Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425: 846–851.
Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB . Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003; 422: 313–317.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i, Altaba A . HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 2007; 17: 165–172.
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008; 14: 238–249.
Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67: 2187–2196.
Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA 2007; 104: 4048–4053.
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009; 458: 776–779.
Ferretti E, De Smaele E, Miele E, Laneve P, Po A, Pelloni M et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J 2008; 27: 2616–2627.
Liao X, Siu MKY, Au CWH, Wong ESY, Chan HY, Ip PPC et al. Aberrant activation of hedgehog signaling pathway in ovarian cancers: effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009; 30: 131–140.
Sheng T, Li C, Zhang X, Chi S, He N, Chen K et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer 2004; 3: 29.
Fan L, Pepicelli C V, Dibble CC, Catbagan W, Zarycki JL, Laciak R et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004; 145: 3961–3970.
Sanchez P, Hernández AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA 2004; 101: 12561–12566.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66: 6063–6071.
Artavanis-Tsakonas S . Notch signaling: cell fate control and signal integration in development. Science 1999; 284: 770–776.
Roy M, Pear WS, Aster JC . The multifaceted role of Notch in cancer. Curr Opin Genet Dev 2007; 17: 52–59.
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 2005; 65: 8530–8537.
Dickson BC, Mulligan AM, Zhang H, Lockwood G, O’Malley FP, Egan SE et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol 2007; 20: 685–693.
Farnie G, Clarke RB . Mammary stem cells and breast cancer–role of Notch signalling. Stem Cell Rev 2007; 3: 169–175.
Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M et al. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007; 25: 807–815.
Gustafsson M V, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J et al. Hypoxia requires Notch signaling to maintain the undifferentiated cell state. Dev Cell 2005; 9: 617–628.
Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991; 66: 649–661.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306: 269–271.
Wang YH, Li F, Luo B, Wang XH, Sun HC, Liu S et al. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma 2009; 56: 371–378.
Castellanos JA, Merchant NB, Nagathihalli NS . Emerging targets in pancreatic cancer: and cancer stem cells. Onco Targets Ther 2013; 6: 1261–1267.
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009; 69: 2400–2407.
Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets 2011; 15: 817–828.
Komiya Y, Habas R . Wnt signal transduction pathways. Organogenesis 2008; 4: 68–75.
MacDonald BT, Tamai K, He X . Wnt/??-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Gordon MD, Nusse R . Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem 2006; 281: 22429–22433.
Bilic J, Huang Y-L, Davidson G, Zimmermann T, Cruciat C-M, Bienz M et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007; 316: 1619–1622.
Mosimann C, Hausmann G, Basler K . Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol 2009; 10: 276–286.
Willert K, Jones KA . Wnt signaling: is the party in the nucleus? Genes Dev 2006; 20: 1394–1404.
Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S et al. Wnt/Wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell 2002; 109: 47–60.
Jessen S, Gu B, Dai X . Pygopus and the Wnt signaling pathway: a diverse set of connections. Bioessays 2008; 30: 448–456.
Fearon ER, Vogelstein B . A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997; 275: 1787–1790.
Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T et al. Activation of the ??-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res 1998; 58: 1021–1026.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW . Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58: 1130–1134.
Mirabelli-Primdahl L, Gryfe R, Kim H, Millar A, Luceri C, Dale D et al. ??-Catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res 1999; 59: 3346–3351.
Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA . Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol 1999; 155: 703–710.
Palacios J, Gamallo C . Mutations in the β-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res 1998; 58: 1344–1347.
Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suárez A, Armas A . beta-catenin expression pattern in stage I and II ovarian carcinomas : relationship with beta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol 1999; 155: 527–536.
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66: 589–600.
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB et al. Identification of FAP locus genes from chromosome 5q21. Science 1991; 253: 661–665.
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997; 275: 1784–1787.
Clevers H . Wnt/beta-catenin signaling in development and disease. Cell 2006; 127: 469–480.
Pinto D, Gregorieff A, Begthel H, Clevers H . Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 2003; 17: 1709–1713.
Van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L et al. Development of several organs that require inductive epithelial- mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev 1994; 8: 2691–2703.
Andrade AC, Nilsson O, Barnes KM, Baron J . Wnt gene expression in the post-natal growth plate: regulation with chondrocyte differentiation. Bone 2007; 40: 1361–1369.
Eaves CJ, Humphries RK . Acute myeloid leukemia and the Wnt pathway. N Engl J Med 2010; 362: 2326–2327.
Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol 2008; 73: 59–66.
Reya T, Clevers H . Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–850.
Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12: 468–476.
Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008; 452: 650–653.
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003; 423: 409–414.
Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 2001; 98: 10356–10361.
Jung A, Schrauder M, Oswald U, Knoll C, Sellberg P, Palmqvist R et al. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am J Pathol 2001; 159: 1613–1617.
Kirchner T, Brabletz T . Patterning and nuclear beta-catenin expression in the colonic adenoma-carcinoma sequence. Analogies with embryonic gastrulation. Am J Pathol 2000; 157: 1113–1121.
Gil J, Bernard D, Peters G . Role of polycomb group proteins in stem cell self-renewal and cancer. DNA Cell Biol 2005; 24: 117–125.
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M . The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397: 164–168.
Alkema MJ, Wiegant J, Raap AK, Berns A, van Lohuizen M . Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum Mol Genet 1993; 2: 1597–1603.
Itahana K, Zou Y, Itahana Y, Martinez J-L, Beausejour C, Jacobs JJL et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol 2003; 23: 389–401.
Van Der Lugt NMT, Domen J, Linders K, Van Roon M, Robanus-Maandag E, Te Riele H et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev 1994; 8: 757–769.
Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM . Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991; 65: 753–763.
Sawa M, Yamamoto K, Yokozawa T, Kiyoi H, Hishida A, Kajiguchi T et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int J Hematol 2005; 82: 42–47.
Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer 2001; 84: 1372–1376.
Zhang F, Sui L, Xin T . Correlations of Bmi-1 expression and telomerase activity in ovarian cancer tissues. Exp Oncol 2008; 30: 70–74.
Dimri GP, Martinez JL, Jacobs JJL, Keblusek P, Itahana K, Van Lohuizen M et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res 2002; 62: 4736–4745.
Cui H, Hu B, Li T, Ma J, Alam G, Gunning WT et al. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol 2007; 170: 1370–1378.
Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA 2009; 106: 16281–16286.
Yin T, Wei H, Gou S, Shi P, Yang Z, Zhao G et al. Cancer stem-like cells enriched in Panc-1 spheres possess increased migration ability and resistance to gemcitabine. Int J Mol Sci 2011; 12: 1595–1604.
Raaphorst FM . Deregulated expression of Polycomb-group oncogenes in human malignant lymphomas and epithelial tumors. Hum Mol Genet 2005; 14: R93–R100.
Zhang S, Balch C, Chan MW, Lai H-C, Matei D, Schilder JM et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008; 68: 4311–4320.
Van Leenders GJLH, Dukers D, Hessels D, van den Kieboom SWM, Hulsbergen CA, Witjes JA et al. Polycombgroup oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol 2007; 52: 455–463.
Glinsky G V, Berezovska O, Glinskii AB . Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005; 115: 1503–1521.
Neuzil J, Stantic M, Zobalova R, Chladova J, Wang X, Prochazka L et al. Tumour-initiating cells vs. cancer ‘stem’ cells and CD133: what’s in the name? Biochem Biophys Res Commun 2007; 355: 855–859.
Visvader JE, Lindeman GJ . Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8: 755–768.
McDermott SP, Wicha MS . Targeting breast cancer stem cells. Mol Oncol 2010; 4: 404–419.
Lobo NA, Shimono Y, Qian D, Clarke MF . The biology of cancer stem cells. Annu Rev Cell Dev Biol 2007; 23: 675–699.
Sánchez-García I, Vicente-Dueñas C, Cobaleda C . The theoretical basis of cancer-stem-cell-based therapeutics of cancer: can it be put into practice? Bioessays 2007; 29: 1269–1280.
Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000; 406: 1005–1009.
Goodrich L V, Scott MP . Hedgehog and patched in neural development and disease. Neuron 1998; 21: 1243–1257.
Beachy PA, Cooper MK, Young KE, Von Kessler DP, Park WJ, Hall TMT et al. Multiple roles of cholesterol in hedgehog protein biogenesis and signaling. Cold Spring Harb Symp Quant Biol 1997; 62: 191–204.
Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007; 25: 2524–2533.
Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009; 361: 1164–1172.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324: 1457–1461.
Skvara H, Kalthoff F, Meingassner JG, Wolff-Winiski B, Aschauer H, Kelleher JF et al. Topical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol 2011; 131: 1735–1744.
Stuetz A, de Rie MA, Skvara H, Mickel L, Schuster C, Stary G et al. FC24 LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin's syndrome). Melanoma Res 2010; 20: e40.
Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat Chem Biol 2009; 5: 154–156.
Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci USA 2009; 106: 14132–14137.
Lauth M, Bergström A, Shimokawa T, Toftgård R . Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA 2007; 104: 8455–8460.
Ślusarz A, Shenouda NS, Sakla MS, Drenkhahn SK, Narula AS, MacDonald RS et al. Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer Res 2010; 70: 3382–3390.
Elamin MH, Shinwari Z, Hendrayani SF, Al-Hindi H, Al-Shail E, Khafaga Y et al. Curcumin inhibits the Sonic Hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol Carcinog 2010; 49: 302–314.
Tang GQ, Yan TQ, Guo W, Ren TT, Peng CL, Zhao H et al. (-)-Epigallocatechin-3-gallate induces apoptosis and suppresses proliferation by inhibiting the human Indian Hedgehog pathway in human chondrosarcoma cells. J Cancer Res Clin Oncol 2010; 136: 1179–1185.
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 2011; 29: 3529–3534.
Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer 2011; 105: 796–806.
Deangelo DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol 2006; 24: 6585.
Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther 2010; 9: 1618–1628.
Oncomed Pharmaceuticals. A Phase 1 Dose Escalation Study of OMP-21M18 in Subjects With Solid Tumors. ClinicalTrials.gov 2012, available at https://clinicaltrials.gov/show/NCT00744562.
Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA 2004; 101: 15949–15954.
Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev 2004; 18: 2474–2478.
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang W-C, Chanthery Y et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006; 444: 1083–1087.
Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC et al. Direct inhibition of the NOTCH transcription factor complex. Nature 2009; 462: 182–188.
Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res 2008; 68: 4727–4735.
Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH . Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther 2006; 5: 483–493.
Kallifatidis G, Labsch S, Rausch V, Mattern J, Gladkich J, Moldenhauer G et al. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol Ther 2011; 19: 188–195.
Kawahara T, Kawaguchi-Ihara N, Okuhashi Y, Itoh M, Nara N, Tohda S . Cyclopamine and quercetin suppress the growth of leukemia and lymphoma cells. Anticancer Res 2009; 29: 4629–4632.
Nagayama S, Fukukawa C, Katagiri T, Okamoto T, Aoyama T, Oyaizu N et al. Therapeutic potential of antibodies against FZD 10, a cell-surface protein, for synovial sarcomas. Oncogene 2005; 24: 6201–6212.
He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005; 24: 3054–3058.
You L, He B, Xu Z, Uematsu K, Mazieres J, Fujii N et al. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res 2004; 64: 5385–5389.
Fujii N, You L, Xu Z, Uematsu K, Shan J, He B et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res 2007; 67: 573–579.
Shan J, Shi D-L, Wang J, Zheng J . Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry 2005; 44: 15495–15503.
Takahashi-Yanaga F, Kahn M . Targeting Wnt signaling: can we safely eradicate cancer stem cells? Cancer Res 2010; 16: 3153–3162.
Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci USA 2004; 101: 12682–12687.
Bommi P V, Dimri M, Sahasrabuddhe AA, Khandekar JD, Dimri GP . The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle 2010; 9: 2663–2673.
Wu J, Hu D, Yang G, Zhou J, Yang C, Gao Y et al. Down-regulation of BMI-1 cooperates with artemisinin on growth inhibition of nasopharyngeal carcinoma cells. J Cell Biochem 2011; 112: 1938–1948.
Veeranarayanan S, Poulose AC, Mohamed MS, Varghese SH, Nagaoka Y, Yoshida Y et al. Synergistic targeting of cancer and associated angiogenesis using triple-targeted dual-drug silica nanoformulations for theragnostics. Small 2012; 8: 3476–3489.
Raveendran S, Poulose AC, Yoshida Y, Maekawa T, Kumar DS . Bacterial exopolysaccharide based nanoparticles for sustained drug delivery, cancer chemotherapy and bioimaging. Carbohydr Polym 2013; 91: 22–32.
Raveendran S, Chauhan N, Palaninathan V, Nagaoka Y, Yoshida Y, Maekawa T et al. Extremophilic polysaccharide for biosynthesis and passivation of gold nanoparticles and photothermal ablation of cancer cells. Part Part Syst Charact 2015; 32: 54–64.
Sivakumar B, Aswathy RG, Nagaoka Y, Iwai S, Venugopal K, Kato K et al. Aptamer conjugated theragnostic multifunctional magnetic nanoparticles as a nanoplatform for pancreatic cancer therapy. RSC Adv 2013; 3: 20579.
Raveendran S, Palaninathan V, Nagaoka Y, Fukuda T, Iwai S, Higashi T et al. Extremophilic polysaccharide nanoparticles for cancer nanotherapy and evaluation of antioxidant properties. Int J Biol Macromol 2015; 76: 310–319.
Lee K, Drachev VP, Irudayaraj J . DNA-gold nanoparticle reversible networks grown on cell surface marker sites: application in diagnostics. ACS Nano 2011; 5: 2109–2117.
Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, Panyam J . CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release 2013; 171: 280–287.
Wei X, Senanayake TH, Warren G, Vinogradov S V . Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of cd44-positive and drug-resistant tumors. Bioconjug Chem 2013; 24: 658–668.
Liu Y, Lu WL, Guo J, Du J, Li T, Wu JW et al. A potential target associated with both cancer and cancer stem cells: a combination therapy for eradication of breast cancer using vinorelbine stealthy liposomes plus parthenolide stealthy liposomes. J Control Release 2008; 129: 18–25.
Bostad M, Berg K, Høgset A, Skarpen E, Stenmark H, Selbo PK . Photochemical internalization (PCI) of immunotoxins targeting CD133 is specific and highly potent at femtomolar levels in cells with cancer stem cell properties. J Control Release 2013; 168: 317–326.
Yang C, Xiong F, Wang J, Dou J, Chen J, Chen D et al. Anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles against cancer stem-like cell activity in multiple myeloma. Nanomedicine (Lond) 2013; 9: 45–60.
Yu Z, Ni M, Xiong M, Zhang X, Cai G, Chen H et al. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int J Nanomedicine 2015; 10: 2537–2554.
Takebe N, Harris PJ, Warren RQ, Ivy SP . Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011; 8: 97–106.
Maugeri-Saccà M, Zeuner A, De Maria R . Therapeutic targeting of cancer stem cells. Front Oncol 2011; 1: 10.
Chenna V, Hu C, Pramanik D, Aftab BT, Karikari C, Campbell NR et al. A polymeric nanoparticle encapsulated small-molecule inhibitor of Hedgehog signaling (NanoHHI) bypasses secondary mutational resistance to smoothened antagonists. Mol Cancer Ther 2012; 11: 165–173.
Xu Y, Chenna V, Hu C, Sun HX, Khan M, Bai H et al. Polymeric nanoparticle-encapsulated hedgehog pathway inhibitor HPI-1 (NanoHHI) inhibits systemic metastases in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res 2012; 18: 1291–1302.
Lim KJ, Bisht S, Bar EE, Maitra A, Eberhart CG . A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol Ther 2011; 11: 464–473.
You J, Zhao J, Wen X, Wu C, Huang Q, Guan F et al. Chemoradiation therapy using cyclopamine-loaded liquid–lipid nanoparticles and lutetium-177-labeled core-crosslinked polymeric micelles. J Control Release 2015; 202: 40–48.
Kumar V, Mondal G, Slavik P, Rachagani S, Batra SK, Mahato RI . Codelivery of small molecule Hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol Pharm 2015; 12: 1289–1298.
Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A et al. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Mol Ther 2011; 19: 1538–1546.
Lo WL, Chien Y, Chiou GY, Tseng LM, Hsu HS, Chang YL et al. Nuclear localization signal-enhanced RNA interference of EZH2 and Oct4 in the eradication of head and neck squamous cell carcinoma-derived cancer stem cells. Biomaterials 2012; 33: 3693–3709.
Acknowledgements
Ankita Borah and Ankit Rochani would like to acknowledge their sincere gratitude to the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan for the financial support under the Monbukagakusho fellowship during the research. Also, part of this study has been supported by a grant for the program of the strategic research foundation at private universities S1101017, organized by the MEXT, Japan since April 2012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Borah, A., Raveendran, S., Rochani, A. et al. Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis 4, e177 (2015). https://doi.org/10.1038/oncsis.2015.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2015.35
This article is cited by
-
Downregulation of circ-Foxo3 in breast cancer stem-like cells
BMC Research Notes (2023)
-
Development and application of nanomaterials, nanotechnology and nanomedicine for treating hematological malignancies
Journal of Hematology & Oncology (2023)
-
Pretreatment of prostate cancer cells with salinomycin and Wnt inhibitor increases the efficacy of cabazitaxel by inducing apoptosis and decreasing cancer stem cells
Medical Oncology (2023)
-
RNA-binding protein complex LIN28/MSI2 enhances cancer stem cell-like properties by modulating Hippo-YAP1 signaling and independently of Let-7
Oncogene (2022)
-
Spelling Out CICs: A Multi-Organ Examination of the Contributions of Cancer Initiating Cells’ Role in Tumor Progression
Stem Cell Reviews and Reports (2022)
