
Key Points
-
Glioblastoma is the paradigm of tumour-associated immunosuppression
-
Several glioma-specific peptide vaccines, with or without dendritic cell support, are in late clinical development
-
Vaccines can be combined with agents that nonspecifically boost immune responses, such as immune checkpoint inhibitors or TGFβ pathway inhibitors
-
Standardization of clinical trial conduct might facilitate progress in this challenging field of oncology
Abstract
Astrocytic and oligodendroglial gliomas are intrinsic brain tumours characterized by infiltrative growth and resistance to classic cancer therapies, which renders them inevitably lethal. Glioblastoma, the most common type of glioma, also exhibits neoangiogenesis and profound immunosuppressive properties. Accordingly, strategies to revert glioma-associated immunosuppression and promote tumour-directed immune responses have been extensively explored in rodent models and in large clinical trials of tumour immunotherapy. This Review describes vaccination approaches investigated for the treatment of glioma. Several strategies have reached phase III clinical trials, including vaccines targeting epidermal growth factor receptor variant III, and the use of either immunogenic peptides or tumour lysates to stimulate autologous dendritic cells. Other approaches in early phases of clinical development employ multipeptide vaccines such as IMA-950, cytomegalovirus-derived peptides, or tumour-derived peptides such as heat shock protein-96 peptide complexes and the Arg132His mutant form of isocitrate dehydrogenase. However, some preclinical trial data suggest that addition of immunomodulatory reagents such as immune checkpoint inhibitors, transforming growth factor-β inhibitors, signal transducer and activator of transcription 3 inhibitors, or modifiers of tryptophan metabolism could augment the therapeutic activity of vaccination and overcome glioma-associated immunosuppression.
Similar content being viewed by others


Main
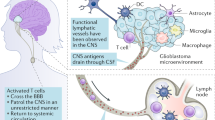
Throughout the past century, pivotal research issues in oncology have included the extent to which immunosuppression is a risk factor for the development of cancer, and the extent to which cancer induces immunosuppression. Accordingly, immunosuppression has been extensively studied in brain tumours such as glioma, which commonly kills affected patients by locally destructive growth rather than by systemic metastasis. Multiple cellular interactions and pathways that might potentially mediate glioma-associated immunosuppression have been identified1,2 (Fig. 1).

Membrane-bound and soluble mediators of glioma-associated immunosuppression both attract and promote the expansion of populations of immunosuppressive cells, and interfere with the generation of antitumour immunity at the T cell–antigen-presenting cell (APC) interface. CTLA4, cytotoxic T cell antigen 4; GDF15, growth and differentiation factor 15; IDO, indoleamine 2,3-dioxygenase; Kyn, kynurenine; LLT1, lectin-like transcript 1; MDSC, myeloid-derived suppressor cell; NK, natural killer; PD1, programmed cell death protein 1; PDL1, programmed cell death 1 ligand 1; PGE, prostaglandin E2, STAT3, signal transducer and activator of transcription 3; TDO, tryptophan 2,3-dioxygenase; TGFβ, transforming growth factor β; Treg cell, regulatory T cell; Trp, tryptophan.
Several decades ago, researchers also recognized that gliomas in the brain promote systemic immunosuppression to a certain degree3, although such immunosuppression is not associated with an increased risk of opportunistic infections. Tumours growing in essentially immunocompromised environments such as the brain would not be expected to derive an advantage from inducing additional immunosuppression. Moreover, even glioblastomas — the most common and most malignant gliomas — seem to be largely incapable of seeding outside the CNS4. This observation has been attributed, at least partially, to immune defence mechanisms that operate only outside the brain. However, experimental or clinical data supporting the existence of efficient anti-glioma immune responses in the periphery, but not inside the CNS, has remained very challenging to obtain.
One of the most remarkable features of gliomas is the observation that these tumours develop much more frequently in elderly individuals (aged ≥60 years), and that outcome is substantially worse in this age group than in younger individuals5. Molecular profiling has enabled the characterization of distinct subtypes of glioblastoma that are most common in children and adults up to 40 years of age6. In the population of adults with glioblastoma, the small prognostically favourable subgroup of tumours with isocitrate dehydrogenase (IDH) gene mutations is virtually absent in patients aged ≥60 years7. By contrast, high-throughput studies of the three most common IDH wild-type glioblastoma subtypes in adults (namely, receptor tyrosine kinase (RTK) 1, RTK 2, and mesenchymal) show few differences at the genomic, transcriptomic or epigenetic level between tumours from elderly individuals (>65 years) and those from younger patients6,8. These data suggest that the status of the host, rather than the molecular make-up of the tumour, might to a large extent determine the outcome of these types of cancer.
In summary, the clinical relevance of glioma-associated immunosuppression remains debated. Nevertheless, the past 5 years have seen the clinical translation of various cancer immunotherapies, some of which are glioma-specific. Several of these strategies involve innovative vaccination strategies that are challenging, in terms of both conceptional design and logistical conduct, to implement in the clinic (Fig. 2). Immediate conceptual challenges include the choice of antigen to be targeted, the selection of patients on the basis of tumour biomarkers and immune markers (such as HLA status), and the timing of assessment of the status of such markers. Further conceptual challenges involve the integration of immunotherapy into post-surgical treatment schedules (such as tapering of corticosteroids), the wound healing process, and the initiation of radiotherapy and chemotherapy, which all occur within a few weeks. Accordingly, logistical challenges include the need for rapid testing of biomarkers (which might require shipment of biological samples), generation of the vaccine at a remote location, and delivery of the vaccine back to the hospital setting where the patient will be treated.

Patients undergo apheresis to enable generation of a dendritic cell-based vaccine using peptides derived from tumour-associated antigens. Multiple doses of the vaccine can be produced and used for the patient's treatment. PBMC, peripheral blood mononuclear cell.
In this article, we review the experimental evidence supporting the development of vaccine-based immunotherapies for patients with glioma and the clinical experience gathered so far with several such approaches. We also outline current ideas about how to improve the clinical results achieved with glioma vaccines by overcoming specific limitations of current immunotherapies, including efforts to antagonize glioma-associated immunosuppression.
Data from animal models
The use of vaccination to induce immune responses against gliomas has been assessed extensively in rodent models. However, the few available syngeneic mouse glioma models do not fully reflect the biology of human glioma tumours because cell-line-based models do not reproduce the heterogeneity typical of the human disease, and genetically engineered models are, by their nature, a simplification. Furthermore, important differences between mouse and human immune systems must be considered when findings from preclinical models are translated to patients in the clinic9. Another important consideration in experimental studies is the timing of vaccination in relation to that of tumour cell inoculation: the longer the gap between initiation of vaccination and tumour cell inoculation, the more potentially relevant are the resulting observations for patients with unresected tumours. Conversely, vaccination soon after tumour cell inoculation (as is typical in rodent studies) might resemble the clinical scenario in patients with recently resected tumours limited to microscopic disease. Despite their limitations, however, preclinical models do provide important information that helps to predict whether glioma vaccines, alone or in combination with other drugs, will have antitumour activity in the clinical setting.
One of the most frequently used mouse glioma models is based on the GL-261 cell line, which was generated by treatment with the chemical carcinogen methylcholanthrene. These cells are usually implanted stereotactically into the brains of syngeneic C57BL/6 mice, where they produce immunogenic tumours10. A spontaneous glioma that developed in a VM/Dk mouse has been used to generate several syngeneic mouse glioma cell lines11,12, which are increasingly used for immunotherapy studies and have the potential advantage over GL-261 that cancer was not chemically induced13. A popular genetic approach uses the RCAS-TVA retroviral gene-transfer system, which can induce syngeneic tumours even in outbred mice14. A popular transgenic model was created by inactivation of p53 in conjunction with loss of the Nf1 gene (encoding neurofibromin)15.
One simple approach to tumour vaccine development is to modify glioma cell lines ex vivo and to use the modified cells as a vaccine against the parent tumour in the mouse brain. For example, the growth of intracerebral SMA-560 gliomas was inhibited by peripheral vaccination using irradiated SMA-560 cells engineered to express MICA (MHC class I polypeptide-related sequence A)16. MICA is a ligand for NKG2D (NKG2-D type II integral membrane protein), an immunoreceptor expressed on T cells and natural killer (NK) cells that stimulates cellular immune responses17. Other studies have employed cytokine-transduced glioma cells as a vaccine, most using granulocyte–macrophage colony-stimulating factor (GM-CSF)18,19. The efficacy of vaccines derived from glioma cells might be enhanced by culturing the source cells under hypoxic conditions: vaccination with lysates from GL-261 cells cultured in 5% O2 caused an increase in cytotoxic T-cell proliferation, tumoricidal function, and trafficking to the tumour site20, although the mechanisms underlying this effect remained obscure.
Vaccines based on dendritic cells (DCs) have been assessed for approximately two decades in glioma-bearing mice, with variable success21. Effective vaccines have been prepared using DCs pulsed with tumour-specific peptides, tumour lysate, or vectors encoding putative tumour antigens22,23,24,25. DC-based vaccination might hold particular promise when the tumour stem cell compartment is used as a source of antigen: in terms of mounting immune responses against orthotopic gliomas, vaccines based on DCs pulsed with a lysate derived from mouse glioma cells were more effective if these glioma cells had stem cell properties26.
Peptide-based vaccines, alone or in combination with therapies aimed at overcoming glioma-associated immunosuppression, have also been investigated in immunocompetent mouse models. For example, an antibody to transforming growth factor-β (TGFβ) augmented the efficacy of vaccination with two peptides, GARC-177–85 and EphA2671–679, derived from candidate glioma-associated antigens27.
Some evidence suggests that vaccines are more effective when given in combination with molecularly targeted therapies. For example, administration of a vaccine derived from DCs pulsed with autologous tumour lysate in combination with antibodies targeting programmed cell death protein 1 (PD1) prolonged the survival of glioma-bearing mice, whereas no such effect was observed with either treatment alone28. Similarly, the efficacy of a vaccine consisting of irradiated GL-261 cells expressing GM-CSF was improved by co-treatment with antibodies targeting cytotoxic T cell antigen 4 (CTLA4)29.
The vast majority of preclinical studies focusing on vaccination in glioblastoma have limitations that impede translation of their findings to the clinic. Specifically, treatment was started very early in relation to the time of tumour cell inoculation, suggesting that vaccine treatment was initiated during immunological priming rather than during the chronic immune homeostasis stage, when treatment is typically initiated in patients with glioblastoma. Moreover, highly immunogenic tumours such as GL-261 were studied, and subcutaneous instead of intracranial tumour cell inoculation was often used, perhaps because researchers erroneously assumed that these two methods would be equally informative30. Finally, the age (and thus immune status) of vaccinated mice might not be comparable to that of most patients with glioblastoma. Nonetheless, despite these limitations, mouse models of glioma can lead to a more comprehensive understanding of immune system function, as well as the pitfalls associated with vaccination attempts.
Data from clinical trials
A summary of completed clinical trials of vaccination therapy for gliomas is provided in Table 1. Ongoing or planned trials are summarized in Table 2. Current approaches to vaccine-based immunotherapy for glioma exhibit variable degrees of complexity, primarily resulting from selection of the target antigen(s) and the decision whether or not to use autologous, patient-derived immune cells to generate the vaccine (which commonly requires monocyte apheresis and ex vivo maturation into DCs). Selection of the target antigen ranges from approaches targeting a single tumour-specific mutant protein such as isocitrate dehydrogenase (IDH) Arg132His (IDHR132H) or epidermal growth factor receptor (EGFR) variant III (EGFRvIII)31,32, to approaches targeting a predefined panel of tumour-associated antigens (such as ICT-107)33 or a personalized panel of tumour-associated antigens selected by genome profiling from a limited pool of targets (as used in the GAPVAC trial)34. Unbiased antigen-selection approaches using undefined tumour-derived peptides (HSPPC-96) or whole-tumour-cell lysates (DCVax) have also been used35,36.
Vaccines that target defined peptides (as opposed to those targeting uncharacterized proteins or using unbiased antigen selection) are probably less prone to unexpected adverse events, such as induction of tolerance rather than stimulation of immune responses, because the effects of targeting known antigens can be modelled in mice and studied in human cell culture systems, enabling adverse effects to be detected. However, tumour-associated antigens are less likely than tumour-specific antigens to induce immune responses, as high-affinity T cells that respond to tumour-associated antigens are selected against during thymic T-cell development37. Compared with peptides derived from normal proteins that are overexpressed in gliomas, peptides from altered proteins that are only expressed in tumours, such as EGFRvIII or IDHR132H, have the theoretical advantage of tumour specificity and a reduced risk of inducing autoimmunity. Few such antigens have been defined in glioblastoma. Thus, vaccination trials in patients with glioma, and the potential integration of vaccines into standard of care in this setting, might have to overcome obstacles not previously encountered in the clinical management of patients with brain tumours (Figs 2,3).

Administration of the vaccine along with an appropriate adjuvant, such as granulocyte–macrophage colony-stimulating factor (GM-CSF), results in antigen-presenting cell (APC) activation and subsequent priming of T helper cells. These processes ultimately induce CD8+ T cell antitumour immune responses.
Vaccines targeting EGFRvIII. The EGFR gene is amplified in approximately 40% of IDH wild-type glioblastomas38,39. More than half of such EGFR-amplified tumours exhibit a deletion mutation that results in expression of a truncated protein referred to as EGFRvIII40. This protein exhibits loss of the ligand-binding domain, resulting in constitutive kinase activity41. Moreover, the truncated protein exhibits a novel amino acid sequence, which has been identified as potentially immunogenic31. A vaccine known as rindopepimut employs this peptide sequence to evoke immune responses, and its efficacy against glioma was explored in three uncontrolled phase II clinical trials, which consistently demonstrated encouraging progression-free and overall survival data42,43,44. Admittedly, the patients vaccinated with rindopepimut in these trials were highly selected, in that the inclusion criteria required gross total tumour resection and an absence of progression at the first scan after completion of chemoradiotherapy42,43,44. The favourable results from these three trials provided the rationale for a pivotal phase III placebo-controlled trial of rindopepimut in patients with newly diagnosed glioblastoma (ACT-IV)45. Both groups of patients concurrently received standard maintenance temozolomide. However, an interim analysis conducted in early 2016 concluded that the ACT-IV trial should be terminated because the primary end point of improved overall survival was unlikely to be met45.
Despite the negative outcome of this phase III trial, at least three important lessons can be learned from the results of ACT-IV: that the generation of strong humoral immune responses to the vaccine did not translate into a survival benefit; that EGFRvIII expression was lost in approximately half of the patients in each arm of the study, indicating that EGFRvIII expression is not a stable feature of EGFR-amplified glioblastoma; and that a trend towards long-term survival benefit in vaccinated patients was detectable only in those with residual disease45. The latter observation contradicts the prevailing hypothesis that minimal residual disease (and, thus, absence of an extensive immunosuppressive micromilieu) is required for immunotherapy to be effective45.
In parallel to ACT-IV, a smaller randomized clinical trial (ReACT) conducted in the USA compared rindopepimut with placebo in patients with recurrent glioblastoma46. Both groups of patients also received bevacizumab, the standard of care for this indication in the USA. Data are available in abstract form only, but overall survival seemed to favour the experimental arm (median 11.3 months; 95% CI 9.9–16.2 months) over the control arm (median 9.3 months; 95% CI7.1–11.4 months; HR 0.53, P = 0.0177 in the per-protocol analysis46). The primary end point of improved progression-free survival at 6 months was not met, although a beneficial trend in the proportion of patients reaching this end point also favoured the experimental treatment: 28% of patients in the vaccine arm versus 16% of patients in the control arm (P = 0.1163) as ascertained by independent review46.
Why vaccine therapy would show efficacy in patients with recurrent glioblastoma (who have been previously exposed to steroids, radiotherapy and temozolomide chemotherapy) while seeming not to improve outcome in those with newly diagnosed glioblastoma (who should be relatively immunocompetent) remains challenging to understand. One might assume that previous exposure of the tumour cells to genotoxic stress exerted by radiotherapy and alkylating agent chemotherapy would make tumours more immunogenic; also, a transcriptomic profiling study indicated that recurrent tumours are more likely to exhibit a mesenchymal profile, which is characterized by expression of immune and inflammatory genes47. Furthermore, the patients in ReACT probably had more-extensive disease than those in ACT-IV, although we must assume, on the basis of data from ACT-IV45, that only approximately half of the patients in the ReACT study would have had tumours that were still expressing EGFRvIII by the time of vaccination, as enrolment was based on EGFRvIII expression in the tissue obtained at initial surgery46.
Although experimental data support the combination of vascular endothelial growth factor (VEGF) antagonism and immunotherapy48,49, this combination might not be explored further as clinical development of bevacizumab in glioblastoma has been halted. Two trials reported no improvement in overall survival with bevacizumab therapy in newly diagnosed patients37,38, and a further trial in patients with recurrent glioblastoma also reported negative results for bevacizumab treatment, albeit in combination with lomustine50,51,52.
EGFRvIII has also been used as a target for chimeric antigen receptor (CAR)-based T-cell therapy53, although as this strategy is not based on vaccination it is outside the scope of the present Review and will not be discussed further. Future trials will show whether CAR T-cell therapy exerts clinically meaningful antitumour activity in patients with glioblastoma.
Multipeptide vaccines. Multipeptide vaccines have gained increasing interest in the past few years. A vaccine consisting of three peptides derived from glioma-associated antigens has been explored in 26 HLA-A2-positive children with newly diagnosed diffuse intrinsic pontine glioma, anaplastic astrocytoma or glioblastoma54. Vaccination was safe and resulted in a measurable immune response54, but no conclusions on efficacy can be derived from this uncontrolled study, which pooled several different disease entities. A similar vaccination strategy was also used in a phase I study of patients with WHO grade II glioma55. No dose-limiting toxicity was noted, and immune responses against at least three epitopes were observed in most patients55.
Tumour-associated peptides derived from non-mutated proteins are shared to a high degree between different glioblastomas, and their presentation is thought to mainly reflect the presence of deregulated signalling pathways56. Patient-specific selection of tumour-associated peptides might enable the development of an individualized therapeutic cancer vaccine targeting antigens that are not abundantly present in the majority of glioblastomas, but show exceptionally high expression and potential immunogenicity in a given patient's tumour, thereby maximizing the chance of successful induction of relevant immune responses in that individual. IMA950 is one such multipeptide therapeutic vaccine developed for patients with glioblastoma. The IMA950 vaccine includes 11 tumour-associated peptides (nine HLA-A*02 class I peptides, an elongated class I peptide and one class II peptide), as well as the synthetic hepatitis B virus marker peptide IMA-HBV-001. A phase I trial of IMA950 in 45 patients with newly diagnosed glioblastoma receiving maintenance temozolomide has been completed. Treatment-related adverse events were generally mild, but two patients experienced dose-limiting fatigue or anaphylaxis57. Ultimately, 36 of 40 evaluable patients were characterized as single-peptide responders, and 20 patients were characterized as multipeptide responders (according to the vaccine-specific T-cell response criteria defined in the trial protocol). However, progression-free survival was only 74% at 6 months and 31% at 9 months, and median overall survival was 15.3 months57.
Taking the concept of personalized vaccines to the next level of individualization, the Glioma Actively Personalized Vaccines Consortium (GAPVAC) initiated a phase I trial of vaccines based on individualized selection of both tumour-associated peptides and tumour-specific peptides34. The term actively personalized vaccine (APVAC) had been previously coined by the Regulatory Research Group of the Association of Cancer Immunotherapy58. In the GAPVAC-101 trial, both APVACs were integrated into standard care (surgery and chemoradiotherapy followed by maintenance temozolomide) in newly diagnosed patients with glioblastoma34. APVAC 1 vaccines contained 5–10 peptides selected from a library of proteins obtained by expression profiling of the patient's tumour; the peptides most strongly associated with the tumour were selected for each patient to maximize the number of effective antitumour immune responses. APVAC 2 vaccines contained 1–2 custom-made synthetic mutated peptides. Next-generation sequencing and mass spectrometry were employed to compare the tumour and patient genomes and identify suitable mutated peptides for this purpose.
Another novel multipeptide vaccination strategy utilizes neoepitopes derived from mutant peptides expressed on an individual patient's tumour. A feasibility trial evaluating the administration of up to 20 neoepitopes per patient has been initiated as an individualized tumour vaccine strategy in patients with newly diagnosed glioblastoma59. Results of these approaches are eagerly awaited.
Dendritic-cell-based vaccines. DCs have been used for many years to generate vaccines for use in both paediatric and adult patients with glioma. Most clinical reports describe single-centre experiences, however, and many open questions remain with regard to the precise conduct of the treatment60,61.
In an early phase I trial in adults with glioblastoma, autologous DCs were pulsed with acid-eluted autologous tumour peptides to yield the vaccine62. The researchers concluded that the absence of bulky, progressive disease and low expression of TGFβ2 defined a subgroup of patients who might be most suitable for further studies of vaccine efficacy62. An ensuing phase I study in 23 such patients combined this vaccination approach with Toll-like receptor (TLR) agonist treatment63. The results led to the hypothesis that glioblastomas with a mesenchymal gene-expression profile exhibit increased immune cell infiltration associated with increased immunogenicity, and are, therefore, more amenable to immunotherapy than are tumours with other profiles63. DCVax is an ongoing phase III trial of a DC vaccine generated with autologous tumour lysate, and was based on the experience summarized above, but is not recruiting patients at present64.
Another phase I–II trial assessed the activity of a DC-based multipeptide vaccine derived from glioma-associated antigens in 22 patients: 13 with glioblastoma, five with anaplastic astrocytoma, three with anaplastic oligodendroglioma, and one with anaplastic oligoastrocytoma65. This trial yielded findings suggestive of clinical efficacy. In total, nine vaccinated patients (41%) — four with glioblastoma and five with anaplastic glioma — remained progression-free for ≥12 months65. The clinically most advanced DC-based vaccine to date is ICT-107, which is generated by exposing autologous patient-derived DCs to peptides derived from six proteins predicted to be abundant in glioblastoma and thought to be linked to the glioma stem cell signature33,66. These six proteins are glycoprotein 100 (gp100), melanoma-associated antigen 1 (MAGE1), interferon-inducible protein AIM2 (also known as absent in melanoma 2), tyrosine kinase-type cell surface receptor HER2 (also known as proto-oncogene Neu or receptor tyrosine-protein kinase erbB2), IL-13Rα2 (IL-13 receptor subunit α2), and tyrosinase related protein-2 (TRP2). The MAGE1 and AIM2 peptides were predicted to be HLA-A1-associated whereas the other four epitopes were predicted to be HLA-A2-associated66. The safety of the ICT-107 vaccine was confirmed in a phase I trial that enrolled 21 patients (17 with newly diagnosed glioblastoma, three with recurrent glioblastoma and one with brainstem glioma)33. Median progression-free survival in the newly diagnosed patients was 16.9 months, and median overall survival was 38.4 months, data which the researchers interpreted as encouraging. An increased duration of both overall survival and progression-free survival correlated with gene expression related to four of the six target proteins in the newly diagnosed glioblastoma cohort33.
The ensuing randomized phase II trial of ICT-107 did not meet the primary end point of improved overall survival66. However, post hoc analyses revealed that a potential benefit was probably restricted to the subgroup of HLA-A2-positive individuals, who accounted for 77 of the 124 (62%) patients who underwent randomization. Moreover, the lack of an MRI scan to rule out progression at the time of randomization (that is, after completion of concomitant temozolomide and radiotherapy) might have accounted for the inclusion of patients with early progression in the group without promoter methylation of MGMT (methylated-DNA — protein-cysteine methyltransferase), which could have contributed to the failure to reach the primary end point66. On the basis of this phase II experience, a pivotal phase III trial of ICT-107 has been initiated, which mandates confirmation of the absence of progression after completion of chemoradiotherapy and limits enrolment to HLA-A2-positive patients67.
IDHR132H-specific vaccines. Evidence for the therapeutic efficacy of IDHR132H-specific vaccines stems from preclinical studies in a humanized mouse sarcoma model32 and an orthotopic syngeneic mouse glioma model68. IDH1 is mutated in more than 70% of diffuse and anaplastic gliomas, but only approximately 5% of glioblastomas69. The vast majority of IDH1 mutations result in a protein with an arginine to histidine amino acid substitution at position 132 (IDHR132H)70. In addition to the metabolic and epigenetic consequences of this mutation with regard to gliomagenesis and tumour behaviour71, IDHR132H harbours a neoepitope that, similarly to many other mutated antigens72, is presented by professional antigen-presenting cells or MHC class II-expressing glioma cells73, thereby stimulating mutation-specific CD4+ T-cell responses74. Indeed, spontaneous T-cell and antibody responses to IDHR132H are observed in a fraction of patients with glioma32. The IDHR132H neoepitope also seems to be capable of presentation by multiple MHC class II allelotypes.
From an immunological perspective, the IDHR132H mutation represents an interesting target for immunotherapy as it is not only tumour-specific but also expressed in all tumour cells and, thus, represents a clonal neoantigen with high uniformity and penetrance. In human immune responses to IDHR132H, only mutation-specific CD4+ T cells have been observed32. In the absence of mutation-specific CD8+ T effector cells, the cellular mechanisms of the efferent arm of the therapeutic response remain unclear, but the preclinical data suggest that B cells are required75.
The ongoing NOA-16 trial76 is a phase I safety, tolerability and immunogenicity multicentre study evaluating a 20-mer IDHR132H peptide in patients with treatment-naive WHO grade III–IV IDH1-mutated gliomas. Patient enrolment is not confined to a specific MHC class II haplotype, but the trial population is enriched for an unfavourable prognosis by restricting enrolment to patients whose tumours have an astrocytic molecular phenotype. Eight vaccines are integrated in the primary therapy, which in most patients comprises radiochemotherapy combined with temozolomide. The trial is accompanied by a translational research programme that aims to identify key biomarkers for predicting and monitoring response to the vaccine76. This important initiative will characterize the immunological mechanism of the (primarily T-helper-cell-driven) antitumour immune response, and promote the development of rational combination therapies. However, concerns regarding efforts to target mutant IDHR132H have been expressed, as this mutated protein negatively regulates the growth of gliomas when exogenously transduced77. However, given that a vaccine would target cells expressing IDHR132H rather than the mutant protein itself, these concerns are unlikely to reflect the scenario of the therapeutic approach.
HSPPC-96. Heat-shock proteins (HSPs) are involved in cellular responses to stressors such as heat, from which this protein family derived its name. Notably, HSP-96 can bind tumour-associated antigens, and HSP-96 — peptide complex (HSPPC-96) can be taken up by antigen-presenting cells, potentially triggering specific antitumour responses78,79. HSPPC-96 has, therefore, been used to generate vaccines that aim to boost antitumour immune responses80.
HSPPC-96 vaccination of patients with recurrent glioblastoma resulted in specific immune responses in the blood as well at the tumour site81. A subsequent phase II trial enrolled 41 patients with recurrent glioblastoma who had undergone complete resection of the tumour. Survival was 90.2% at 6 months and 29.3% at 12 months35. In the absence of a control group in this trial, however, no statement on vaccine efficacy can be made35. Current limitations on the use of HSPPC-96-based vaccines include the necessity of prior tumour resection, as 7 g of tumour tissue is needed to prepare at least four 25 μg doses of vaccine. A randomized phase II trial of HSPPC-96 vaccination in patients with newly diagnosed glioblastoma is ongoing82 (Table 2).
Cytomegalovirus proteins. Several groups have demonstrated that human cytomegalovirus (CMV) proteins are expressed in >90% of glioblastomas83,84,85, although other researchers have failed to detect human CMV protein or DNA in glioblastoma samples86,87. Expression of CMV protein has not been detected in normal brain tissue surrounding virus-positive glioblastomas83,84,88,89, suggesting that viral antigens could be subverted as tumour-specific targets. Subclinical reactivation of latent CMV infection is frequent in critically ill and immunocompromised patients90, and DCs pulsed with CMV antigens are potent inducers of virus-specific immune responses91,92,93,94,95.
A study published in 2014 demonstrated that CMV-specific T-cell immune responses can recognize and effectively kill autologous glioblastoma cells expressing the immunodominant pp65 viral antigen at endogenous levels96, supporting the development of CMV-directed immunotherapy. Moreover, CMV-reactive T cells might recognize glioblastoma cells independently of CMV antigen expression97. In a small randomized pilot trial, patients who received CMV pp65-specific DCs in combination with vaccine-site preconditioning using tetanus–diphtheria toxoid showed better than predicted progression-free survival (median 10.8 months) and overall survival (median 18.5 months) from diagnosis98.
Boosting immune responses to glioma
As summarized in Figure 1, multiple mechanisms (including cell-surface-based and paracrine pathways) mediate glioma-associated immunosuppression49,99,100,101,102,103,104,105,106,107, which is likely to limit the efficacy of active immunotherapy in the absence of effective countermeasures. Accordingly, this situation offers a strong rationale for combining specific immunological targeting via a glioma-specific vaccine with necessarily nonspecific approaches to reduce local and systemic immunosuppression, as described below.
Immune checkpoint modulators. The most straightforward approach to increase general immune responsiveness is administration of immune checkpoint modulators. Immune checkpoint inhibitors are therapeutic monoclonal antibodies that intercept receptor–ligand interactions involved in regulating immune cell activity. These drugs might, therefore, relieve glioma-associated immunosuppression and facilitate tumour cell destruction by immune cells108 (Fig. 4). Immune checkpoint inhibitors such as ipilimumab (which targets CTLA4), nivolumab and pembrolizumab (which target PD1), and atezolizumab and durvalumab (which target PDL1) have shown remarkable and sometimes lasting activity against several cancer types, including melanoma, lung cancer, renal cell cancer, bladder cancer, head and neck cancer, and Hodgkin lymphoma109,110,111.

Immune checkpoint inhibition using therapeutic monoclonal antibodies, such as those directed against PD1 (programmed cell death protein 1) or PDL1 (programmed cell death 1 ligand 1), might counteract glioma-associated immunosuppression in the tumour microenvironment or peripheral immune sites such as regional lymph nodes. Relief of tumour-associated immunosuppression could boost the activity of glioma-specific cytotoxic T lymphocytes (CTL) generated by vaccination.
High expression levels and immunosuppressive activity of the PD1–PDL1 axis have been documented in human glioblastoma112,113,114. Moreover, inhibition of this pathway results in substantial antitumour activity in rodent models of glioma28,115, and PD1 inhibition in combination with DC vaccination prolonged the survival of glioma-bearing mice to a greater extent than treatment with either agent alone. Immune checkpoint inhibitors have also proven active against brain metastases in clinical trials7, and a report published in 2016 documented clinically significant responses to the PD1 inhibitor nivolumab in two patients with recurrent glioblastoma who also had germline biallelic mismatch repair deficiency, providing proof of the concept that this therapeutic strategy is feasible in human CNS tumours108,116. Large clinical trials are currently evaluating the efficacy of immune checkpoint inhibitors in patients with newly diagnosed and recurrent glioblastoma117.
Current preclinical research is not only evaluating additional immune checkpoint inhibitors, but also identifying checkpoint receptors that activate immune responses and so could be targeted using agonistic antibodies. Early preclinical data indicate that the co-stimulatory molecule OX40 (also known as CD134 or tumour necrosis factor receptor superfamily member 4) and its ligand OX40L (also known as CD252 or tumour necrosis factor ligand superfamily member 4) are involved in T-cell activation in glioblastoma118. Future research will show whether immune checkpoint modulators are capable of synergistically increasing the antitumour response of vaccination strategies against glioma in the clinical setting.
TGFβ inhibitors. Neutralizing the biological activity of TGFβ, the master immunosuppressive cytokine associated with glioma, also presents an option to improve the activity of vaccines for glioma treatment. The utility of this approach has been demonstrated in mouse models of glioma, in which treatment with TGFβ inhibitors enhanced the therapeutic efficacy of peptide vaccines and promoted CD70-mediated tumour rejection27,119.
In humans, inhibition of TGFβ activity alone, using receptor tyrosine kinase inhibitors such as galunisertib, showed little activity in patients with glioma at first relapse120. Continuous dosing of such drugs is poorly tolerated, which necessitated a 2 weeks on, 2 weeks off dosing regimen in the clinical trial setting120 that might be insufficient to inhibit the TGFβ–SMAD pathway to a relevant extent. Combinations of TGFβ inhibitors and a specific immunotherapy approach might still be worth exploring, however. For instance, the dosing of TGFβ inhibitors could be limited to specific weeks of active vaccine treatment.
Immunometabolic pathway inhibitors. The immunosuppressive microenvironment surrounding glioma is further maintained by metabolization of tryptophan, an essential amino acid. Tryptophan is cleaved by dioxygenases such as indoleamine-2,3-dioxygenase (IDO) or tryptophan-2,3-dioxygenase (TDO), resulting in several metabolites, but mainly kynurenine. Depletion of tryptophan and high levels of kynurenine both lead to impaired T-cell activation in vitro121. Hence, the addition of inhibitors of IDO, TDO or the aryl hydrocarbon receptor (AHR; a receptor for kynurenine that transduces its immunosuppressive signals to T cells and myeloid cells), could help to increase the activity of immune responses following vaccination122,123. Similarly, inhibition of the activity of signal transducer and activator of transcription 3 (STAT3), which is a central regulator of various glioma-derived immunosuppressive mechanisms, might represent a therapeutic strategy to overcome the immunosuppressive tumour microenvironment124. Such concepts are only at an early stage of clinical development; however, a phase I study in patients with recurrent malignant glioma or brain metastasis from melanoma is ongoing125.
Targeting regulatory T cells. Patients with glioblastoma are profoundly immunosuppressed, at least partly as a result of an excessive number of immunosuppressive myeloid-derived suppressor cells (MDSC) and regulatory T cells (Treg cells)126,127,128,129. Key features of this immunosuppressive phenotype can be reversed by eliminating Treg cells126. When Treg cells are depleted in vitro, T-cell proliferation and cytokine secretion return to normal levels126; in vivo depletion of Treg cells 1 week after tumour implantation prolonged the survival of mice inoculated with glioma cells, demonstrating not only that Treg-mediated immunosuppression is reversible, but also that Treg impairment positively influences antitumour immunity130.
Previous attempts to eliminate Treg cells have had mixed outcomes131,132, possibly because Treg depletion was not employed in the unique host environment that exists after therapeutic temozolomide-induced lymphodepletion. These strategies might also have failed because they employed the IL-2 moiety itself to target Treg cells. This method would result in indiscriminate targeting of low-affinity IL-2βγ receptors, which are expressed on a broad subset of immune cells, including memory T cells. Alternative strategies that employ IL-2Rα-targeted immunotoxins result in rapid and indiscriminate killing of all IL-2Rα-expressing cells, which might include recently activated, vaccine-induced effector T cells. In marked contrast, IL-2Rα-specific antibodies lacking the bound immunotoxin eliminate Treg cells while having no effect on effector T cells in lymphopenic mice133.
Conclusions and future perspectives
Glioblastoma remains one of the most-studied tumours in the context of cancer-associated immunosuppression. Numerous soluble mediators and cell-based pathways of immunosuppression were first delineated in glioblastoma, and these observations collectively support further efforts at establishing efficacious antitumour immunotherapies. Although proof of efficacy is not yet available for any of the glioma-specific peptide vaccines currently in clinical development, the addition of immune checkpoint inhibitors or other approaches that boost immune responses in vaccinated patients might ultimately be able to demonstrate that active immunotherapy can control the growth of human intracranial neoplasms.
Meanwhile, the clinical development of immunotherapy for glioblastoma could be aided by collective efforts to introduce measures of quality control and standardization of inclusion and exclusion criteria, as well as standardized response and other efficacy criteria in immunotherapy trials. The iRANO (Immunotherapy Response Assessment in Neuro-Oncology) criteria, which essentially caution against the premature assumption of inefficacy or treatment failure in early phase clinical trials of immunotherapy134, are just a first step in this direction. The identification of tumour or serum biomarkers that predict response or progression of glioblastoma is needed to improve the conduct and efficiency of clinical trials. Such efforts will require international collaboration involving all the major organizations and disciplines involved in the orchestration of care for patients with brain tumours.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Nduom, E. K., Weller, M. & Heimberger, A. B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 17 (Suppl. 7), vii9–vii14 (2015).
Mangani, D., Weller, M. & Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 130, 1–9 (2017).
Roszman, T., Elliott, L. & Brooks, W. Modulation of T-cell function by gliomas. Immunol. Today 12, 370–374 (1991).
Schweitzer, T., Vince, G. H., Herbold, C., Roosen, K. & Tonn, J. C. Extraneural metastases of primary brain tumors. J. Neurooncol. 53, 107–114 (2001).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 17 (Suppl. 4), iv1–iv62 (2015).
Sturm, D. et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 (2012).
Hartmann, C. et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 120, 707–718 (2010).
Bozdag, S. et al. Age-specific signatures of glioblastoma at the genomic, genetic, and epigenetic levels. PLoS ONE 8, e62982 (2013).
Oh, T. et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 12, 107 (2014).
Jacobs, V. L., Valdes, P. A., Hickey, W. F. & De Leo, J. A. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN Neuro 3, e00063 (2011).
Serano, R. D., Pegram, C. N. & Bigner, D. D. Tumorigenic cell culture lines from a spontaneous VM/Dk murine astrocytoma (SMA). Acta Neuropathol. 51, 53–64 (1980).
Sampson, J. H. et al. Characterization of a spontaneous murine astrocytoma and abrogation of its tumorigenicity by cytokine secretion. Neurosurgery 41, 1365–1372 (1997).
Ahmad, M. et al. How stemlike are sphere cultures from long-term cancer cell lines? Lessons from mouse glioma models. J. Neuropathol. Exp. Neurol. 73, 1062–1077 (2014).
Fisher, G. H. et al. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene 18, 5253–5260 (1999).
Zhu, Y. et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8, 119–130 (2005).
Friese, M. A. et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 63, 8996–9006 (2003).
Ullrich, E., Koch, J., Cerwenka, A. & Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2, e26097 (2013).
Herrlinger, U. et al. MIP-1α antagonizes the effect of a GM-CSF-enhanced subcutaneous vaccine in a mouse glioma model. J. Neurooncol. 66, 147–154 (2004).
Herrlinger, U. et al. Vaccination for experimental gliomas using GM-CSF-transduced glioma cells. Cancer Gene Ther. 4, 345–352 (1997).
Olin, M. R. et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin. Cancer Res. 16, 4800–4808 (2010).
Heimberger, A. B. et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J. Neuroimmunol. 103, 16–25 (2000).
Grauer, O. M. et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int. J. Cancer 122, 1794–1802 (2008).
Jouanneau, E. et al. Dendritic cells are essential for priming but inefficient for boosting antitumour immune response in an orthotopic murine glioma model. Cancer Immunol. Immunother. 55, 254–267 (2006).
Yamanaka, R. et al. Marked enhancement of antitumor immune responses in mouse brain tumor models by genetically modified dendritic cells producing Semliki Forest virus-mediated interleukin-12. J. Neurosurg. 97, 611–618 (2002).
Prins, R. M., Odesa, S. K. & Liau, L. M. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 63, 8487–8491 (2003).
Pellegatta, S. et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 66, 10247–10252 (2006).
Ueda, R. et al. Systemic inhibition of transforming growth factor-β in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin. Cancer Res. 15, 6551–6559 (2009).
Antonios, J. P. et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 1, e87059 (2016).
Agarwalla, P., Barnard, Z., Fecci, P., Dranoff, G. & Curry, W. T. Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J. Immunother. 35, 385–389 (2012).
Huszthy P. C. et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol. 14, 979–993 (2012).
Heimberger, A. B. et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 9, 4247–4254 (2003).
Schumacher, T. et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512, 324–327 (2014).
Phuphanich, S. et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 62, 125–135 (2013).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02149225 (2016).
Bloch, O. et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. 16, 274–279 (2014).
Chiang, C. L., Coukos, G. & Kandalaft, L. E. Whole tumor antigen vaccines: where are we? Vaccines (Basel) 3, 344–372 (2015).
Mohme, M., Neidert, M. C., Regli, L., Weller, M. & Martin, R. Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat. Rev. 40, 248–258 (2014).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Weller, M. et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J. Clin. Oncol. 27, 5743–5750 (2009).
Weller, M. et al. Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int. J. Cancer 134, 2437–2447 (2014).
Batra, S. K. et al. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 6, 1251–1259 (1995).
Sampson, J. H. et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 28, 4722–4729 (2010).
Sampson, J. H. et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 13, 324–333 (2011).
Schuster, J. et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 17, 854–861 (2015).
Weller, M. et al. ACT IV: an international, double-blind, phase 3 trial of rindopepimut in newly diagnosed, EGFRvIII-expressing glioblastoma. Neuro Oncol. 18, (Suppl. 6), vi17–vi18 (2016).
Reardon, D. A. et al. ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma [abstract]. J. Clin. Oncol. 33 (Suppl.), 2009 (2015).
Gill, B. J. et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl Acad. Sci. USA 111, 12550–12555 (2014).
Johnson, B. F., Clay, T. M., Hobeika, A. C., Lyerly, H. K. & Morse, M. A. Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Exp. Opin. Biol. Ther. 7, 449–460 (2007).
Voron, T. et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 212, 139–148 (2015).
Chinot, O. L. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 709–722 (2014).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708 (2014).
Wick, W. et al. EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine in patients with first progression of a glioblastoma [abstract]. J. Clin. Oncol. 34 (Suppl.), 2001 (2016).
Johnson, L. A. et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra22 (2015).
Pollack, I. F. et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol. 32, 2050–2058 (2014).
Okada, H. et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clin. Cancer Res. 21, 286–294 (2015).
Dutoit, V. et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 135, 1042–1054 (2012).
Rampling, R. et al. A Cancer Research UK first time in human phase I trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin. Cancer Res. 22, 4776–4785 (2016).
Britten, C. M. et al. The regulatory landscape for actively personalized cancer immunotherapies. Nat. Biotechnol. 31, 880–882 (2013).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02287428 (2016).
Ardon, H. et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 54, 519–525 (2010).
Ardon, H. et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 61, 2033–2044 (2012).
Liau, L. M. et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 11, 5515–5525 (2005).
Prins, R. M. et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 17, 1603–1615 (2011).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00045968 (2016).
Okada, H. et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 29, 330–336 (2011).
Wen, P. et al. A randomized double blind placebo-controlled phase 2 trial of dendritic cell (DC) vaccine ICT-107 following standard treatment in newly diagnosed patients with GBM. Neuro Oncol. 16 (Suppl. 5), v22 (2014).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02546102 (2017).
Pellegatta, S. et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 3, 4 (2015).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Hartmann, C. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 118, 469–474 (2009).
Waitkus, M. S., Diplas, B. H. & Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 18, 16–26 (2016).
Platten, M. & Offringa, R. Cancer immunotherapy: exploiting neoepitopes. Cell Res. 25, 887–888 (2015).
Bunse, L. et al. Proximity ligation assay evaluates IDH1 R132H presentation in gliomas. J. Clin. Invest. 125, 593–606 (2015).
Melief, C. J. Mutation-specific T cells for immunotherapy of gliomas. N. Engl. J. Med. 372, 1956–1958 (2015).
Schumacher, T., Bunse, L., Wick, W. & Platten, M. Mutant IDH1: an immunotherapeutic target in tumors. Oncoimmunology 3, e974392 (2014).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02454634 (2016).
Li, S. et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 15, 57–68 (2013).
Suto, R. & Srivastava, P. K. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 269, 1585–1588 (1995).
Tamura, Y., Peng, P., Liu, K., Daou, M. & Srivastava, P. K. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 278, 117–120 (1997).
Ampie, L. et al. Heat shock protein vaccines against glioblastoma: from bench to bedside. J. Neurooncol. 123, 441–448 (2015).
Crane, C. A. et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 kD chaperone protein. Clin. Cancer Res. 19, 205–214 (2013).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03018288 (2017).
Mitchell, D. A. et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 10, 10–18 (2008).
Cobbs, C. S. et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 62, 3347–3350 (2002).
Prins, R. M., Cloughesy, T. F. & Liau, L. M. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N. Engl. J. Med. 359, 539–541 (2008).
Baumgarten, P. et al. Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics. Neuro Oncol. 16, 1469–1477 (2014).
Tang, K. W., Hellstrand, K. & Larsson, E. Absence of cytomegalovirus in high-coverage DNA sequencing of human glioblastoma multiforme. Int. J. Cancer 136, 977–981 (2015).
Dziurzynski, K. et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 14, 246–255 (2012).
Ranganathan, P., Clark, P. A., Kuo, J. S., Salamat, M. S. & Kalejta, R. F. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86, 854–864 (2012).
Limaye, A. P. et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 300, 413–422 (2008).
Kleihauer, A. et al. Ex vivo generation of human cytomegalovirus-specific cytotoxic T cells by peptide-pulsed dendritic cells. Br. J. Haematol. 113, 231–239 (2001).
Cho, H. I., Han, H., Kim, C. C. & Kim, T. G. Generation of cytotoxic T lymphocytes specific for human cytomegalovirus using dendritic cells in vitro. J. Immunother. 24, 242–249 (2001).
Raftery, M. J., Schwab, M., Diesner, S., Egerer, G. & Schonrich, G. Dendritic cells cross-presenting viral antigens derived from autologous cells as a sensitive tool for visualization of human cytomegalovirus-reactive CD8+ T cells. Transplantation 73, 998–1002 (2002).
Peggs, K. S. & Mackinnon, S. Clinical trials with CMV-specific T cells. Cytotherapy 4, 21–28 (2002).
Szmania, S. et al. Isolation and expansion of cytomegalovirus-specific cytotoxic T lymphocytes to clinical scale from a single blood draw using dendritic cells and HLA-tetramers. Blood 98, 505–512 (2001).
Nair, S. K. et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin. Cancer Res. 20, 2684–2694 (2014).
Knight, A. et al. CMV-independent lysis of glioblastoma by ex vivo expanded/activated Vδ1+ γδ T cells. PLoS ONE 8, e68729 (2013).
Mitchell, D. A. et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369 (2015).
Roth, P. et al. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res. 16, 3851–3859 (2010).
Wu, A. et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 12, 1113–1125 (2010).
Hishii, M. et al. Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery 37, 1160–1166 (1995).
Wolpert, F. et al. HLA-E contributes to an immune-inhibitory phenotype of glioblastoma stem-like cells. J. Neuroimmunol. 250, 27–34 (2012).
Lemke, D. et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin. Cancer Res. 18, 105–117 (2012).
Codo, P. et al. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget 5, 7651–7662 (2014).
Lauro, G. M., Di Lorenzo, N., Grossi, M., Maleci, A. & Guidetti, B. Prostaglandin E2 as an immunomodulating factor released in vitro by human glioma cells. Acta Neuropathol. 69, 278–282 (1986).
Ichinose, M., Masuoka, J., Shiraishi, T., Mineta, T. & Tabuchi, K. Fas ligand expression and depletion of T-cell infiltration in astrocytic tumors. Brain Tumor Pathol. 18, 37–42 (2001).
Roth, P. et al. Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res. 67, 3540–3544 (2007).
Preusser, M., Lim, M., Hafler, D. A., Reardon, D. A. & Sampson, J. H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 11, 504–514 (2015).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Rizvi, N. A. et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 16, 257–265 (2015).
Berghoff, A. S. et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 17, 1064–1075 (2015).
Nduom, E. K. et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 18, 195–205 (2016).
Wintterle, S. et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 63, 7462–7467 (2003).
Reardon, D. A. et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res. 4, 124–135 (2016).
Bouffet, E. et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 34, 2206–2211 (2016).
Weiss, T., Weller, M. & Roth, P. Immunotherapy for glioblastoma: concepts and challenges. Curr. Opin. Neurol. 28, 639–646 (2015).
Shibahara, I. et al. OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: a clue for successful immunotherapy. Mol. Cancer 14, 41 (2015).
Aulwurm, S., Wischhusen, J., Friese, M., Borst, J. & Weller, M. Immune stimulatory effects of CD70 override CD70-mediated immune cell apoptosis in rodent glioma models and confer long-lasting antiglioma immunity in vivo. Int. J. Cancer 118, 1728–1735 (2006).
Brandes, A. A. et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 18, 1146–1156 (2016).
Platten, M., Weller, M. & Wick, W. Shaping the glioma immune microenvironment through tryptophan metabolism. CNS Oncol. 1, 99–106 (2012).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Wainwright, D. A. et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 20, 5290–5301 (2014).
Ferguson, S. D., Srinivasan, V. M. & Heimberger, A. B. The role of STAT3 in tumor-mediated immune suppression. J. Neurooncol. 123, 385–394 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01904123 (2016).
Fecci, P. E. et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 66, 3294–3302 (2006).
El Andaloussi, A. & Lesniak, M. S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. J. Neurooncol. 8, 234–243 (2006).
Grauer, O. M. et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int. J. Cancer. 121, 95–105 (2007).
Raychaudhuri, B. et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 13, 591–599 (2011).
El Andaloussi, A., Han, Y. & Lesniak, M. S. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J. Neurosurg. 105, 430–437 (2006).
Morse, M. A. et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 112, 610–618 (2008).
Jacobs, J. F. et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin. Cancer Res. 16, 5067–5078 (2010).
Attia, P. et al. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J. Immunother. 29, 208–214 (2006).
Okada, H. et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 16, e534–e542 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01480479 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01498328 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00905060 (2014).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00293423 (2014).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01213407 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01222221 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02465268 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01814813 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02455557 (2017).
Acknowledgements
The authors' research work is supported by grants from the Canton of Zurich HSM-2 (Hochspezialisierte Medizin 2) programme, the German Research Fund (Deutsche Forschungsgemeinschaft), the Swiss National Science Foundation and the Swiss Cancer League (all to M.W. and P.R.).
Author information
Authors and Affiliations
Contributions
All authors wrote the manuscript, researched data for the article, undertook review or editing of the manuscript before submission and contributed substantially to discussions of the article content.
Corresponding author
Ethics declarations
Competing interests
The authors declare that M.W. has received research grants from Acceleron, Actelion, Bayer, Isarna, Merck Sharp & Dohme, Merck EMD (Emanuel Merck, Darmstadt), Novocure, Piqur and Roche and honoraria for lectures, advisory board participation or consulting from Bristol-Myers Squibb, Celldex, Immunocellular Therapeutics, Isarna, Magforce, Merck Sharp & Dohme, Merck EMD, Northwest Biotherapeutics, Novocure, Pfizer, Roche, Teva and Tocagen. M. Preusser has received research support from Böhringer-Ingelheim, GlaxoSmithKline, Merck Sharp & Dohme and Roche, as well as honoraria for lectures, advisory board participation or consulting from Bristol-Myers Squibb, CMC Contrast, Gerson Lehrman Group, GlaxoSmithKline, Mundipharma, Novartis and Roche. D.A.R. has received research grants from Celldex Therapeutics, Incyte and Midatech, as well as honoraria for lectures, advisory board participation or consulting from Abbvie, Amgen, Bristol-Myers Squibb, Cavion, Celldex Therapeutics, EMD Serono, Genentech (Roche), Inovio, Juno Pharmaceuticals, Merck & Co, Midatech, Momenta Pharmaceuticals, Novartis, Novocure, Oxigene, Regeneron, and Stemline Therapeutics. M. Platten has received research support from Merck and Novartis, as well as honoraria for lectures, consultation or advisory board participation from Alexion, Bayer, Genentech (Roche), Merck & Co, Medac, Miltenyi Biotec, Novartis and Teva. M. Platten also holds patents on isocitrate dehydrogenase vaccines and aryl hydrocarbon receptor inhibition. P.R. has received research support from Merck Sharp & Dohme and honoraria for lectures or advisory board participation from Merck Sharp & Dohme, Molecular Partners, Novartis and Roche. W.W. has received research funding from Apogenix, Böhringer-Ingelheim, Genentech (Roche), Merck Sharp & Dohme and Pfizer, as well as honoraria for participating in a speaker's bureau for Merck Sharp & Dohme and consulting for Genentech (Roche). J.H.S. declares that he is an employee of Annias, a shareholder in Annias and Istari, and has received honoraria for consulting from Celldex, BrainLAB and Medicenna, as well as licensing fees from Celldex.
Rights and permissions
About this article

Cite this article
Weller, M., Roth, P., Preusser, M. et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat Rev Neurol 13, 363–374 (2017). https://doi.org/10.1038/nrneurol.2017.64
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2017.64
This article is cited by
-
mRNA-based precision targeting of neoantigens and tumor-associated antigens in malignant brain tumors
Genome Medicine (2024)
-
NCAPH serves as a prognostic factor and promotes the tumor progression in glioma through PI3K/AKT signaling pathway
Molecular and Cellular Biochemistry (2024)
-
Polyploidy, EZH2 upregulation, and transformation in cytomegalovirus-infected human ovarian epithelial cells
Oncogene (2023)
-
Decreased eukaryotic initiation factors expression upon temozolomide treatment—potential novel implications for eIFs in glioma therapy
Journal of Neuro-Oncology (2023)
-
Multimodal targeting of glioma with functionalized nanoparticles
Cancer Cell International (2022)
