
Key Points
-
Viruses are exceptionally diverse and are grouped by genome replication and encapsidation strategies into seven distinct classes: two classes of DNA viruses (encapsidating single-stranded (ss)DNA or double-stranded (ds)DNA), three classes of RNA viruses (encapsidating mRNA-sense ssRNA, antisense ssRNA or dsRNA) and two classes of reverse-transcribing viruses (encapsidating RNA or DNA).
-
Despite substantial life-cycle differences, positive-strand RNA ((+)RNA) viruses, dsRNA viruses and reverse-transcribing viruses share multiple similarities in genome replication. All replicate their genomes through RNA intermediates that also serve as mRNAs. Moreover, the intracellular RNA-replication complexes of (+)RNA viruses share similarities in structure, assembly and function with the polymerase-containing virion cores of dsRNA and reverse transcribing viruses.
-
Brome mosaic virus (BMV) RNA-replication factors 1a and 2apol and cis-acting template-recruitment signals parallel retrovirus Gag, Pol and RNA-packaging signals in virion assembly: 1a localizes to specific membranes, self-interacts and induces ∼60-nm membrane invaginations to which it recruits 2apol and viral RNAs for replication. Therefore, like retroviruses and dsRNA viruses, BMV sequesters its genomic RNA and polymerase in a virus-induced compartment for replication.
-
BMV and some other alphavirus-like (+)RNA viruses also parallel retroviruses in using tRNA-related sequences to initiate genome replication, and share with dsRNA reoviruses aspects of the function and interaction of their RNA polymerase and RNA-capping enzymes. Emerging results indicate that the genome-replication machineries of these viruses might share other mechanistic features.
-
Whereas (+)RNA alphavirus-like viruses, dsRNA reoviruses and retroviruses are linked by the above similarities, (+)RNA picornaviruses, dsRNA birnaviruses and reverse-transcribing hepadnaviruses share some distinct features, including protein-primed nucleic-acid synthesis. Such parallels suggest that at least some (+)RNA viruses, dsRNA viruses and reverse-transcribing viruses might have evolved from common ancestors. The transitions required for such evolution can be readily envisioned and some have precedents.
-
These underlying parallels in genome replication by four of the seven main virus classes might provide a basis for more generalizable or broader-spectrum approaches for virus control.
Abstract
Viruses are divided into seven classes on the basis of differing strategies for storing and replicating their genomes through RNA and/or DNA intermediates. Despite major differences among these classes, recent results reveal that the non-virion, intracellular RNA-replication complexes of some positive-strand RNA viruses share parallels with the structure, assembly and function of the replicative cores of extracellular virions of reverse-transcribing viruses and double-stranded RNA viruses. Therefore, at least four of seven principal virus classes share several underlying features in genome replication and might have emerged from common ancestors. This has implications for virus function, evolution and control.
Similar content being viewed by others


Main
Despite continuing advances, established and emerging viruses remain major causes of human disease, with dramatic costs in mortality, morbidity and economic terms. In addition to acute diseases, viruses cause at least 15–20% of human cancers1,2 and are implicated in neurological and other chronic disorders. One of many challenges in controlling viruses and virus-mediated diseases is that viruses show an amazing diversity in basic characteristics and life cycles, including differences in virion structure, replication strategies, genetic organization, gene expression and many other fundamental processes. Therefore, even the very processes against which antivirals are targeted often differ radically among virus classes. Inherent in this remarkable variety are intriguing issues about the multiplicity of virus origins and the functional and evolutionary relations of existing viruses. Such issues have practical as well as academic importance, as underlying similarities among virus classes might serve as a foundation for broader-spectrum antiviral strategies.
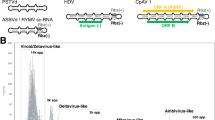
One of the most elemental differences among viruses is their diversity in genome replication and encapsidation strategies, which define seven major classes (Fig. 1). Some viruses replicate their genomes solely through DNA intermediates, packaging these genomes in infectious virions either as double-stranded (ds)DNA or single-stranded (ss)DNA. By contrast, most viruses replicate their genomes solely through RNA intermediates. Such RNA viruses are divided into three classes based on whether their virions package the genome as mRNA-sense (positive-strand) ssRNA, antisense (negative-strand) ssRNA or dsRNA. Other viruses replicate by interconverting their genomes between RNA and DNA. The virions of such reverse-transcribing viruses always initially package the RNA forms of their genomes, and either might (for example, hepadnaviruses and foamy retroviruses) or might not (for example, orthoretroviruses) reverse-transcribe the RNA into DNA before the virion exits the initially infected producer cell.

The bracket highlights the four virus classes emphasized in this review. (+)RNA, positive-strand RNA, which is single-stranded RNA of the same polarity as viral mRNA; (-)RNA, negative-strand RNA, which is single-stranded RNA of anti-mRNA polarity; dsRNA, double-stranded RNA; SARS, severe acute respiratory syndrome.
Viruses in each of these seven classes tend to share additional features, such as gene-expression strategies and so on, that further cluster and differentiate their members from the other classes, showing that these classes represent meaningful, functionally distinct groupings and probable evolutionary lineages. Some of these variations arise because the type of nucleic acid delivered by the virion to a target cell dictates early infection and gene-expression steps. For example, to initiate viral gene expression, dsRNA virus virions and negative-strand RNA ((−)RNA) virus virions contain viral polymerases that transcribe the genome into translatable mRNA, and reverse-transcribing-virus virions contain polymerases that copy the genome into cell-transcribable DNA (Box 1). Positive-strand RNA ((+)RNA) viruses, the virions of which deliver immediately translatable messenger-sense RNAs, encapsidate their RNA without a polymerase and form strictly intracellular RNA-replication and mRNAtranscription complexes (Box 1).
Despite these and other differences, recent results have revealed fundamental parallels in the genome-replication processes of certain (+)RNA viruses, dsRNA viruses and reverse-transcribing viruses. In particular, the intracellular RNA-replication complexes of some, if not many, (+)RNA viruses share several similarities with the replicative cores of virions from both dsRNA viruses and reverse-transcribing viruses. This review outlines these similarities and their potential implications for virus function and evolution. As primary examples, we review similarities among (+)RNA viruses in the alphavirus-like superfamily, the dsRNA reoviruses and the retroviruses. Other shared characteristics with similar evolutionary implications have been recognized recently among certain other (+)RNA viruses, the dsRNA birnaviruses and the reverse-transcribing hepadnaviruses, including parallels in viral RNA-polymerase structure, capsid proteins and protein priming of genome replication3,4,5,6,7.
Negative-strand RNA ((−)RNA) viruses (Fig. 1) also share similarities with some of the basic features reviewed here, suggesting that the functional and evolutionary parallels discussed below might be extended further. These possibilities are not discussed here in detail for reasons of space. In addition, whereas (+)RNA viruses, dsRNA viruses and reverse-transcribing viruses each use identical (+)RNA molecules as genome-replication intermediates and mRNAs, (−)RNA viruses are distinguished by using different forms of (+)RNA for these functions.
(+)RNA virus and retrovirus parallels
tRNAs and genome replication. One of the first similarities recognized between the replication of retroviruses and (+)RNA viruses was the role of tRNAs in initiating retroviral reverse transcription and of tRNA-like elements in initiating RNA replication by a subset of (+)RNA viruses such as the bromoviruses8,9 (Fig. 2). Bromoviruses, discussed further below, have three genomic RNAs with highly conserved, structured, tRNA-like 3′ ends (Fig. 2b). These 3′ ends terminate in 3′-CCAOH sequences that are completed by tRNA-nucleotidyl transferase, they are specifically aminoacylated in vitro and in vivo with tyrosine, and they contain the promoter for (−)RNA synthesis10,11,12,13.

a | Schematic of the genomic RNA of a simple retrovirus and encoded virion proteins Gag and Gag–Pol. b | Schematic of bromovirus genomic RNAs 1, 2 and 3 and encoded RNA-replication factors 1a and 2apol. CA, capsid; Env, envelope-protein gene; ER, endoplasmic reticulum; MA, matrix; NC, nucleocapsid; RE, RNA template recruiting element for genomic RNA replication; TLS, 3′ tRNA-like sequence, which contains the promoter for negative-strand RNA synthesis; tRNA, host tRNA primer for negative-strand cDNA synthesis; Ψ, RNA-packaging signal.
tRNA-related sequences initiate negative-strand synthesis for genomic RNA replication in both retroviruses and (+)RNA viruses, but the mechanisms are distinct. For retroviruses, a cellular tRNA covalently primes negative-strand cDNA synthesis, whereas for the relevant (+)RNA viruses, a viral tRNA-like element serves as a recognition site and template for (−)RNA synthesis that is initiated de novo, without a primer. A natural intermediate and potential evolutionary link between these processes was identified by Lambowitz and colleagues, who showed that a Neurospora crassa mitochondrial retroplasmid initiates reverse transcription without a primer at the tRNA-like 3′ end of its genomic RNA, paralleling negative-strand initiation by (+)RNA viruses14.
Membrane-associated RNA-replication complexes. As noted above, (+)RNA viruses differ from retroviruses and other RNA viruses in that they do not encapsidate their polymerases in extracellular virions. Nevertheless, emerging results show that similarities between (+)RNA-virus RNA replication and retrovirus reverse transcription are not limited to aspects of negative-strand initiation and the general similarities of RNA and DNA polymerases. Instead, as detailed below, (+)RNA-virus RNA replication occurs in virus-induced compartments which have many similarities to the replicative cores or capsids of retrovirus virions.
(+)RNA-virus replication is invariably localized to intracellular membranes. Different (+)RNA viruses target distinct but usually specific membranes, such as those of the endoplasmic reticulum (ER)15,16,17,18,19, endosomes20,21, mitochondria22 or chloroplasts23. RNA replication is usually associated with rearrangements of these target membranes, often giving rise to membrane invaginations, single- or double-membrane vesicles, membrane-bound vesicle packets and other structures.
For many (+)RNA viruses, RNA synthesis localizes to membranes bearing 50–70-nm vesicular compartments that are invaginated away from the cytoplasm into the lumen of the affected secretory compartment or organelle. Such invaginations, termed spherules, were first visualized in early electron microscopy (EM) studies of alphavirus-infected cells20,21. There are many other examples of such invaginations in and beyond the alphavirus-like superfamily, such as those associated with bromoviruses24,25, nodaviruses22 and tymoviruses23. As illustrated in Fig. 3a–c, for nodaviruses and bromoviruses, such spherules frequently occur in contiguous clusters and are often light-bulb-shaped structures, the interiors of which are connected to the cytoplasm through membranous necks.

a | Mitochondria in a flock-house-nodavirus-infected Drosophila cell, showing the typical 50–70-nm, light-bulb-shaped spherular invaginations of the outer mitochondrial membrane into the expanded lumen between the inner and outer membranes. Reproduced with permission from Ref. 22 © (2001) American Society for Microbiology. b | Mitochondrion in a flock-house-nodavirus-infected Drosophila cell that has been sectioned perpendicular to the axis of the spherule necks, rather than parallel to these axes as in panel a. This view reveals a 'vesicle packet' appearance (B. Kopek and P.A., unpublished results). Note that invagination into the lumen of any closed membrane compartment such as the endoplasmic reticulum (ER) or mitochondrial envelope creates a spherule interior that remains connected to the cytoplasm, but that in the section shown in b, the spherule appears separated from the cytoplasm by two or more bounding membranes. c | Similar 50–70-nm spherular vesicles invaginated from the outer perinuclear ER membrane into the ER lumen, in a yeast cell expressing brome mosaic virus (BMV) replication factor 1a in the absence of other viral components. Indistinguishable spherules occur in cells expressing 1a and BMV 2apol in a 20/1 ratio and replicating BMV RNA3. Reproduced with permission from Ref. 45 © (2002) Elsevier. d | Karmellae-like layering of the outer perinuclear ER membrane in cells expressing BMV 1a plus elevated levels of BMV 2apol, and replicating BMV RNA3. Note at top and bottom left that the ∼60-nm intermembrane space is contiguous with the cytoplasm. Reproduced with permission from Ref. 84 © (2004) National Academy of Sciences, USA.
Parallels with retrovirus capsids. Retroviruses package their reverse transcriptase (also designated Pol) and their genomic RNA templates into membrane-enveloped capsid shells assembled by the major capsid protein, Gag26,27,28 (Fig. 4a). For most retroviruses, these capsids bud from the cell together with viral envelope proteins and are delivered to new cells by infection, giving rise to intracellular complexes in which reverse transcription occurs29,30,31. For foamy retroviruses and retrovirus-like LTR (long terminal repeat) retrotransposons, reverse transcription occurs in such capsids without leaving the producer cell32,33,34. Such capsids contain hundreds35 to thousands36 of Gag proteins and approximately 10–20-fold fewer Gag–Pol fusion proteins28,37. Retrovirus genomic RNAs are selectively packaged in these capsids by Gag interaction with specific cis-acting signals, often designated as Ψ38,39.

Highly simplified schematics are shown in each case. a | Assembly of a retrovirus capsid includes the interaction of membrane-associated Gag and Gag–Pol. Gag-dependent genomic RNA encapsidation takes place through packaging signal Ψ, and this is followed by budding. To emphasize similarities with panels b and c, synthesis of negative-strand cDNA (dashed lines) is shown prior to budding, as occurs for foamy retroviruses. b | Assembly and function of a bromovirus RNA-replication complex at the outer endoplasmic-reticulum membrane includes interaction of membrane-associated 1a and 2apol. 1a-dependent genomic RNA encapsidation takes place through the recruitment element (RE) template recruitment signal. This is followed by synthesis and retention of negative-strand RNA (dashed black lines), and asymmetric synthesis and export of positive-strand progeny RNA (red lines), which for at least some positive-strand RNA ((+)RNA) viruses proceeds by a semi-conservative mechanism as shown143. c | Assembly and function of the capsid core of a generalized double-stranded (ds)RNA virus includes encapsidation of genomic RNAs by a packaging signal (PS), synthesis and retention of negative-strand RNA (dashed black lines) and subsequent asymmetric synthesis and export of positive-strand progeny RNA (red lines). (+)RNA synthesis by dsRNA viruses can be either semi-conservative144, as shown, or conservative90,145.
Recently, links between the intracellular spherules of (+)RNA viruses and retrovirus capsids emerged from studies of brome mosaic virus (BMV) RNA replication. BMV is the type member of the bromoviruses and a representative member of the alphavirus-like superfamily of (+)RNA viruses. This superfamily includes many viruses that infect animals or plants, all of which share three conserved protein domains that are involved in RNA replication40,41. In BMV, these domains are distributed across two replication proteins, 1a and 2apol (Fig. 2b), that co-localize on ER membranes at the sites of viral RNA synthesis15,16. 1a contains an RNA-helicase-like domain and an RNA-capping domain with m7G methyltransferase and covalent m7G binding activities required for capping of viral RNAs in vivo42,43,44. 2apol contains a central polymerase domain.
Protein 1a is a multifunctional protein with central roles in the genesis and operation of BMV RNA-replication complexes (Fig. 4b). In the absence of other viral factors, 1a localizes to ER membranes16 and induces invagination of the spherular replication compartments45. 1a also recruits nascent or full-length 2apol to ER membranes by an interaction between the 1a helicase-like domain and the N-terminal extension that precedes the 2apol polymerase domain46,47,48.
Protein 1a also transfers BMV genomic RNA-replication templates to a new state, which was first recognized because the stability and accumulation of BMV genomic RNAs was dramatically increased by 1a co-expression in yeast49,50,51. Subsequent work showed that 1a induces the transfer of BMV genomic RNAs to a novel, membrane-associated, nuclease-resistant state45. The location of the nuclease-resistant RNA and the site of RNA synthesis seem to be the spherule interior, as (+)RNA and (−)RNA templates and nascent BMV RNAs show identical membrane association and nuclease resistance, and immunogold EM localizes 5-bromo-UTP (BrUTP)-labelled nascent RNAs to the spherule interior45.
Recruitment of BMV genomic RNAs to the RNA-replication complex by 1a is controlled in cis by internal (RNA3) or 5′ proximal (RNA1 and RNA2) recruitment elements (REs) on each genomic RNA (Fig. 2b), which are necessary and sufficient for 1a responsiveness51,52. Mutational studies show that the RE activity of BMV RNA derivatives in 1a-induced recruitment is closely linked to their relative template activity in (−)RNA synthesis and full RNA replication51,52. Therefore, 1a-responsive RNA-template recruitment seems to be a crucial step that precedes replication.
As shown in Fig. 4a,b, the roles of 1a, 2apol and the cis-acting REs parallel the roles of Gag, Pol and Ψ in retrovirus replication. Similar to Gag, 1a localizes to the cytoplasmic face of membranes as a peripheral membrane protein53, self-interacts54, and is the sole viral factor required to induce membrane invagination45. Immunogold EM labelling and biochemical analyses show that each spherule contains hundreds of 1a proteins45. The resulting spherules (Fig. 3c) are remarkably similar to the necked vesicles that result when retrovirus budding is arrested by mutations in the Gag late domain that recruits host factors required for membrane breakage and fusion55. Moreover, the high multiplicity of 1a in spherules, its strong membrane association and self-interaction, and the dependence of endocytic and secretory vesicle formation and enveloped-virion budding on protein coats or shells56,57 indicate that 1a might induce membrane invagination by forming a capsid-like shell similar to that of Gag. Similarly, the 1a–RE interaction in RNA-template recruitment parallels the Gag–Ψ interaction in retrovirus RNA packaging. Like retrovirus Pol, BMV 2apol is not required for spherule formation or RNA recruitment. However, when expressed, 2apol is incorporated into the replication complex in a 1a/2apol ratio of ∼25, similar to the Gag/Pol ratio of 10/20 (Refs 37,45).
Similarities with other (+)RNA viruses
The universal association of (+)RNA-virus RNA replication with modified intracellular membranes, often in association with membrane invaginations, and other shared features imply that the RNA-replication complexes of a wide variety of (+)RNA viruses might use principles that are similar to those illustrated in Fig. 4b. Such parallels include the following: both alphavirus- and cucumovirus-induced membrane spherules contain dsRNA25,58; BrUTP labelling implies that the interiors of alphavirus spherules are the sites of viral RNA synthesis59; hepatitis C virus and coronavirus (−)RNA-replication templates are in a membrane-associated, nuclease-resistant state60,61,62; hepatitis C virus replication proteins are present in active replication complexes at >100-fold excess over viral RNA and are sequestered in a protease-resistant state61,62; poliovirus polymerase and possibly other replication factors self-oligomerize in extended lattices63; and tombusvirus genomic RNA possesses an internal cis signal for initial template recruitment and separate 3′ terminal sequences for negative-strand initiation64.
Furthermore, just as for retrovirus Gag and BMV 1a, the membrane structures associated with genome replication by many (+)RNA viruses can be induced by a subset of viral RNA-replication proteins that do not include the polymerase. Examples include the RNA-replication-associated membrane structures induced by alphavirus nsP123 (Ref. 65), arterivirus nsp2 and nsp3 (Ref. 66), hepatitis C virus nsP4B (Refs 19,67) and picornavirus 2BC (Refs 18,68,69).
As explained further below, the parallels among (+)RNA-virus RNA-replication complexes, retrovirus capsids and dsRNA-virus capsids (Fig. 4) imply possible mechanistic explanations for several common features of (+)RNA-virus RNA replication, such as the ability of the replication complex to retain (−)RNA templates for positive-strand synthesis, differential regulation of (−)RNA and (+)RNA synthesis, and frequent downregulation of polymerase expression.
Pol regulation in retroviruses and (+)RNA viruses. In retrovirus genomes, the Pol open reading frame (ORF) follows that of Gag and is expressed by a rare translational frameshift or translational readthrough event, generating an ∼20-fold ratio of Gag/Gag–Pol (Fig. 2a) that is incorporated into the final virion (Fig. 4a). This highly asymmetric ratio regulates the free volume and other parameters of the capsid, and is functionally important, as increasing the level of Gag–Pol relative to Gag inhibits retrovirus virion assembly, release and infectivity37,70,71.
Similarly, many (+)RNA viruses downregulate expression of their polymerase relative to RNA-replication proteins related in sequence and/or functions to 1a. The finding that BMV 1a parallels Gag in acting at high multiplicity to induce the RNA-replication compartment (Fig. 4a,b) implies that such polymerase downregulation might satisfy functional requirements similar to those governing the Gag/Gag–Pol ratio in retrovirus capsids. Like retroviruses, many (+)RNA viruses use translational readthrough or frameshift to downregulate polymerase expression. Well-studied examples include the animal alphaviruses and arteriviruses and plant tobamoviruses and tombusviruses. In each case, the ORF upstream to that of polymerase encodes a protein(s) with parallels to 1a. Specifically, alphavirus nsP123 and tobamovirus p130 are homologous to 1a (Ref. 40), and nsP123 is sufficient to induce membrane spherules65; arterivirus ORF1a proteins induce membrane rearrangements associated with RNA replication66; and tombusvirus p33 self-interacts and directs membrane association of itself, viral RNA and polymerase72. As with Gag and Pol, increasing expression of the polymerase-containing fusion proteins at the expense of the upstream membrane-interacting proteins inhibits tobamovirus and alphavirus replication73,74.
Unlike the above examples, bromoviruses express 1a and 2apol from separate genomic RNAs (Fig. 2b). In this regard, they parallel the foamy virus genus of retroviruses, which express Gag and Pol as independent proteins from separate mRNAs34. Although expressed separately, direct in vivo interaction between the C-terminal 1a helicase domain and N-terminal sequences of nascent 2apol results in a 1a–2apol complex with a polarity that is reminiscent of orthoretrovirus Gag–Pol46,47,48 (Fig. 2). Moreover, whereas translation from separate mRNAs precludes regulation by frameshift or readthrough, BMV downregulates 2apol at translation initiation75 and by degradation of 2apol that is not complexed with 1a (Ref. 76). 1a–2apol interaction itself is downregulated by competing 1a–1a interaction54 and 2apol phosphorylation77. Therefore, like alphaviruses78, bromoviruses have evolved several mechanisms to reduce polymerase accumulation and incorporation to achieve the retrovirus-like 1a to 2apol ratio of 25/1 in RNA-replication complexes45.
Some other (+)RNA viruses, such as hepatitis C virus and picornaviruses, use polyprotein expression strategies that translate all viral ORFs, including polymerase, at equimolar levels. In at least some of these cases, only a fraction of polymerase sequences might be active owing to production of processing intermediates that lack polymerase sequences or activity, polymerase-active and -inactive conformers, polymerase sequestration in the nucleus and/or inclusion bodies, and other effects62,79. Some retrotransposons that produce equimolar levels of Gag and Pol appear to use related strategies to regulate polymerase incorporation or activity80,81.
Alternative membrane rearrangements. Whereas RNA replication by many (+)RNA viruses induces spherular membrane invaginations similar to those shown in Fig. 3a–c, some (+)RNA viruses induce alternative membrane rearrangements. In many cases, the topologies of these rearranged membranes remain under investigation and, as discussed below, some superficially distinct membrane structures might share underlying parallels in topology, assembly and function. Similarly, depending on the conditions, retrovirus Gag not only assembles normal capsids but also sheets, tubes and other structures82.
Flavivirus RNA replication localizes to packets in which an outer bounding membrane surrounds 50–100-nm vesicles that label with antibodies to viral RNA-replication proteins and dsRNA83. These vesicle packets are similar to EM views of BMV spherules invaginated into the ER lumen84 or nodavirus spherules invaginated into the lumen between the inner and outer mitochondrial membranes22, when sectioned perpendicular to the direction of invagination (Fig. 3b). BrUTP-labelled RNA synthesis by the related hepatitis C virus localizes to potentially similar clusters of ∼85-nm vesicles surrounded by undulating membranes, termed the membranous web19,67. Recently, models with strong parallels to bromovirus replication complexes have been proposed for RNA-replication complexes of hepatitis C virus62,85.
RNA replication by picornaviruses, arteriviruses and coronaviruses occurs in conjunction with double-membrane vesicles, that is, vesicles bearing two closely appressed bounding membranes17,18,86. Similar to spherules, arterivirus double-membrane vesicles are thought to form by invagination of appressed ER membranes17. Some EM sections of poliovirus double-membrane vesicles display a narrow neck that connects the inner and outer membranes to each other, and that also connects the vesicle interior to the cytoplasm87, indicating possible genesis by invagination, continuing connection with the cytoplasm, or both.
Interconversion of membrane rearrangements. Further evidence linking seemingly distinct membrane rearrangements in RNA replication is that altering the relative levels and interactions of BMV replication factors 1a and 2apolshifts the associated membrane rearrangements between two dramatically distinct forms84. Expressing 1a plus low levels of 2apol (1a/2apol≈20) induces spherular replication complexes matching those of natural bromovirus24,25 and alphavirus21 infections (Fig. 3c). By contrast, expressing 1a plus higher levels of 2apol induces stacks of appressed double membranes that support RNA replication to levels as high as spherules84 (Fig. 3d). The spaces between these double membrane layers parallel spherule interiors in being 50–60 nm wide, containing 1a and 2apol, and being directly connected to the cytoplasm. Therefore, BMV-induced spherules and double membrane layers seem to be functionally and topologically equivalent forms that are built from the same protein–protein and protein–membrane interactions, but in altered proportions.
Similar stacked, double-membrane ER layers and other ordered ER membrane arrays are induced by overexpression of membrane-associated picornavirus replication factors 2B and 2BC (Refs 68,88). Moreover, the double-membrane vesicles associated with picornavirus RNA replication, the spherule-bearing mitochondrial membranes associated with nodavirus RNA replication and the spherule-bearing chloroplast membranes associated with tymovirus RNA replication all cluster by interaction of surface membranes bearing viral replication factors22,23,63,89. Therefore, a continuum of vesicle-forming and planar membrane interactions seems to be a normal part of RNA replication by many (+)RNA viruses.
(+)RNA virus and dsRNA virus parallels
Many of the above parallels between (+)RNA viruses and retroviruses extend to dsRNA viruses, which also sequester genomic RNA templates, viral polymerase and, often, RNA-capping functions in a protein shell for genome replication90,91 (Fig. 4c). Possible connections between certain (+)RNA and dsRNA viruses were recognized previously, based on conservation of polymerase5,92 and, in one case, on additional replication-factor sequences93.
Similarities with Reoviridae cores. BMV RNA-replication factors and replication complexes share several similarities with the icosahedrally symmetric replicative cores of the dsRNA-virus family Reoviridae (Fig. 4). Reoviridae virions consist of one or more outer-protein shells that are shed during cell entry, surrounding an RNA-containing core that synthesizes RNA in vivo and in vitro90. For the Orthoreovirus genus of the Reoviridae, the transcriptionally active, dsRNA-containing core is bound by an ∼60-nm icosahedral shell made from 60 dimers of protein λ1 (Ref. 94). At each of the twelve 5-fold axes of the core, one copy of the λ3 polymerase resides inside the shell95, whereas on the shell exterior, a pentamer turret of the λ2 capping proteins surrounds the 5-fold axis94. The λ3 polymerase copies the genomic dsRNA templates into new (+)RNA progeny, exporting the nascent RNAs to the cytoplasm through the λ2 pentamer turret, which adds m7G caps to the 5′ ends.
BMV 1a shows many parallels in function and protein–protein interactions with reovirus core proteins λ1 and λ2, implying potentially similar roles in RNA synthesis (Fig. 5). 1a parallels λ1 as the self-interacting, high-copy-number inducer of the viral RNA-replication compartment, yielding the spherular BMV replication complexes. The 50–70-nm intra-membrane diameter of these spherules is similar to the reovirus λ1 core-protein shell. The C-terminal 1a NTPase/helicase domain and its alphavirus homologues have NTPase, RNA 5′-triphosphatase and helicase activities96,97,98,99, matching λ1 (Refs 100,101). The 1a NTPase/helicase domain also anchors BMV RNA polymerase 2apol to the replication complex by direct interaction102, whereas λ1 performs the same role for reovirus RNA polymerase λ3 (Ref. 95). The resulting twelve λ3 polymerases per core are similar to the estimated 10–15 2apol per BMV RNA-replication complex45. Continuing these parallels, λ1 and the 1a NTPase/helicase domain each anchor RNA-capping functions to the RNA-synthesis complex in the form of reovirus λ2 protein and the BMV 1a N-terminal domain, respectively. λ2 (Refs 103,104) and the 1a N-terminal domain42,43,44,105 both possess guanylyltransferase and methyltransferase activities that add m7G caps to the 5′ ends of the (+)RNA products of their respective RNA-synthesis complexes.

The schematics illustrate similarities between interaction and function of reovirus core shell-forming protein λ1, RNA-capping protein λ2 and RNA-dependent polymerase λ3, and the interactions and functions of the brome mosaic virus (BMV) 1a C-proximal NTPase/helicase domain (1aC), 1a N-proximal RNA-capping domain (1aN) and 2apol RNA-dependent RNA polymerase. The reovirus λ1–λ2–λ3 interactions shown in the schematic occur at each of the twelve 5-fold axes of the reovirus core.
RNA packaging, synthesis and export. Recent results indicate possible parallels between the recruitment of (+)RNA templates to BMV RNA-replication complexes and the packaging of (+)RNAs in dsRNA viruses. For dsRNA bacteriophage φ6, viral (+)RNAs are translocated into a pre-formed, empty core by a hexameric viral NTPase/helicase, P4 (Refs 106,107). Similarly, recruitment of BMV genomic RNAs from a membrane-bound, nuclease-sensitive state into the nuclease-resistant spherule replication complex, but not formation of the membrane-invaginated spherules themselves, is blocked by single amino-acid substitutions that inhibit the NTPase activity of the 1a NTPase/helicase domain99. Also, like φ6 P4, the 1a-homologous NTPase/helicase domain of protein p126 of tobacco mosaic virus forms hexamers108. For the Reoviridae, packaging viral (+)RNAs for replication also involves a viral protein with NTPase, RNA-binding and helix-destabilizing activities — the octameric NSP2 for the Rotavirus genus109,110 and, potentially, μ2 for the Orthoreovirus genus90,110,111. Like φ6 P4 (Ref. 106) and BMV 1a (Ref. 112), μ2 also seems to be a co-factor for (+)RNA synthesis111.
For dsRNA viruses, packaging of (+)RNAs is associated with, or followed by, (−)RNA synthesis, yielding dsRNAs that are protected in cores or core-like precursors90,91. Similarly, BMV (−)RNA synthesis depends on 1a-mediated (+)RNA recruitment51,52, and the resulting (−)RNA products are exclusively retained in the nuclease-resistant, membrane-associated RNA-replication compartment45. This retention in a virus-induced RNA-replication compartment explains the efficient, repetitive in vivo use of (−)RNAs as templates for (+)RNA synthesis, and the lack of (−)RNA exchange between RNA-replication complexes in the same cell113. Compartmentalization of RNA templates also seems to be important in protecting dsRNA-virus genomes and the potentially dsRNA-replication intermediates of (+)RNA viruses25,58 from dsRNA-induced host defences, including interferon responses and RNA interference90,91,114,115,116.
A reovirus-like pathway for (+)RNA replication (Fig. 4b,c) would also explain the common early shutoff of (−)RNA synthesis in BMV and other (+)RNA viruses, while (+)RNA synthesis continues unabated112,117,118, and the dependence of (−)RNA synthesis on continuing protein synthesis118. In such a model, (−)RNAs might be synthesized primarily during or immediately after replication-complex assembly from newly synthesized proteins, forming dsRNAs that would preferentially or exclusively be templates for (+)RNA synthesis in the resulting stable replication complexes. Replication-complex assembly and negative-strand synthesis seem to cease on exhaustion of a limiting host factor, possibly the target membrane for replication-complex formation15,118. Pre-formed replication complexes, however, remain active in (+)RNA synthesis112,118.
For both the Reoviridae90 and the alphavirus-like superfamily119,120, (−)RNAs are not capped. By contrast, (+)RNA products are modified with 5′ m7G caps and exported to the cytoplasm from both dsRNA virus cores94,95,121 and BMV spherules43,44,45. This and the parallels noted above between the capping functions and polymerase interactions of the responsible reovirus proteins and 1a suggest that the transcription/capping/export complexes at the 5-fold axes of dsRNA-virus cores might provide models for at least some aspects of the corresponding processes at the spherule necks of BMV and other (+)RNA-virus replication complexes.
Possible evolutionary relationships
The above results show that reverse-transcribing viruses, dsRNA viruses and many, if not all, (+)RNA viruses share central features of genome replication (Fig. 6). Most fundamentally, all replicate their genome through an RNA intermediate that also functions as an mRNA. In all three cases, viral proteins that are translated from these mRNAs capture the mRNA template with its polymerase in a multi-subunit core, sequestering viral RNA from competing processes such as translation and degradation, and sequestering polymerase from competing templates. The polymerase then copies the mRNA into a complementary template from which more mRNA is produced.

All three classes of virus share a similar replication cycle for their genomic RNA (central cyclical steps) but derive their infectious virions from different intermediates in that cycle (radial arrows). See text for further details. (+)RNA, positive-strand RNA; dsRNA, double-stranded RNA.
The variations on this theme that distinguish the three virus classes primarily relate to possessing an RNA-dependent RNA or DNA polymerase, and to deriving infectious virions from different intermediates in the common replication cycle. Orthoretroviruses, such as HIV, export the replicative core prior to copying the mRNA intermediate (Fig. 6, bottom right). By contrast, foamy viruses, hepadnaviruses and dsRNA viruses export the replicative core after copying the mRNA intermediate (Fig. 6, bottom left). For (+)RNA viruses, the virion is separate from the replication complex, and capsid proteins that are not involved in RNA replication package and export the mRNA intermediate before it is sequestered with polymerase (Fig. 6, top). These distinct virion-assembly choices have important consequences, as noted in the introduction, but from some perspectives could be considered temporary extracellular excursions because, as soon as the next cell is infected, each virus re-enters the central, shared replication pathway.
Although no set of shared features can distinguish divergent from convergent evolution, the many parallels summarized here suggest that (+)RNA viruses, dsRNA viruses and reverse-transcribing viruses might have arisen from common ancestors. The transitions required for such evolution can be readily envisioned, and in some cases have precedents. RNA-dependent RNA and DNA polymerases, for example, have related structures122,123, and a shift between the two has generally been postulated as the basis for the emergence of DNA-based biology from the RNA world9. In keeping with the potential for such transitions, BMV RNA-dependent RNA polymerase can copy DNA templates124, and reverse transcriptase can, with single point mutations, incorporate ribonucleotide triphosphates (rNTPs) to produce RNA125.
Established and emerging results further indicate that the intracellular RNA-replication complexes of (+)RNA viruses might evolve into infectious extracellular virions, similar to those of dsRNA or reverse-transcribing viruses, by relatively simple modifications. The spherule RNA-replication complexes of alphaviruses, for example, occur on endosomal membranes and, in lesser numbers, at the plasma membrane, where their appearance closely mimics budding virions59. Such spherules are released into the medium at low frequency59, which might explain the novel infectious particles released from cells replicating alphavirus derivatives with all natural virion proteins deleted and replaced by envelope protein G of vesicular stomatitis virus126. As G protein is readily incorporated into many particles that bud from the plasma membrane and confers the ability to attach to, and fuse membranes with, many cells, incorporation of G protein into spherule membranes might confer infectivity by allowing released replication complexes or their RNAs to enter new cells.
Once acquired, infectivity might be enhanced and optimized through subsequently selected events. For retroviruses and other enveloped viruses, efficient virion budding requires recruiting functions from the host-cell multivesicular-body pathway57. This host machinery can be recruited by incorporating into viral proteins any of a range of short protein-interaction motifs or L domains, which function in a largely position-independent manner55. RNA-recombination events that are suitable to acquire such L domains, favourable envelope-protein genes or other relevant functions from cellular or other viral RNAs are rampant in (+)RNA viruses and retroviruses, representing a major force in virus evolution127,128,129.
Additional pathways for evolutionary transitions between the intracellular RNA-replication complexes of (+)RNA viruses and dsRNA virions have recently been suggested6,130. As the replication complexes of (+)RNA viruses are invariably membrane-associated, it is notable that virions of the Rotavirus genus of the dsRNA Reoviridae bud through membranes during assembly and exit from cells131. Moreover, rotavirus transmembrane protein nsP4, which mediates virion-core association with ER membranes, is required for correct assembly of the viroplasms in which core assembly and replicative RNA synthesis occur, indicating linkage of these processes with membranes132,133. dsRNA bacteriophage φ6 virions also become membrane-enveloped during exit134.
Whereas most views of virus relationships have tended to emphasize the features and classes depicted in Fig. 1, it is intriguing that several alternative, orthogonal virus groupings are defined and mutually distinguished by features that are more strongly conserved across two or more of the classes in Fig. 1. Selected examples include the conservation of an unusual polymerase-sequence rearrangement between (+)RNA tetraviruses and dsRNA birnaviruses5, or the use of protein-primed genome replication by the (+)RNA picornaviruses, dsRNA birnaviruses and reverse-transcribing hepadnaviruses3,4,7, in contrast to the use of tRNA-like elements by bromoviruses and retroviruses. Such relationships suggest that transitions between the virus classes depicted in Fig. 1 might have evolved on more than one occasion, and that links between viruses within a class might not always be as strong as links between classes.
Implications for virus control
The emergence of common underlying principles in the central replication processes of (+)RNA viruses, reverse-transcribing viruses and dsRNA viruses suggests that some of these shared features might provide useful targets for broader-spectrum or generalizable approaches for virus control. One approach might be to modulate membrane lipid composition, as recent results show that assembly and function of membraneassociated (+)RNA-virus replication complexes and retrovirus caspids are highly sensitive to the lipid composition of their target membranes, which in at least some cases can be manipulated by small-molecule therapeutics135,136,137,138. Another general approach might be to interfere with oligomerization or function of oligomerizing replication factors such as retrovirus Gag and BMV 1a by dominant negative mutants43,99, inhibition of required chaperones139,140, targeted incorporation of destructive ligands or other approaches. Further interventions might be based on interfering with viral RNA interactions that are essential for genome packaging, replication and other steps, using nucleic-acid aptamers or other inhibitors141. Other approaches might interfere with the trafficking of viral replication factors and RNAs to their required intracellular sites of assembly142. Opportunities for such interference will continue to increase as further aspects of these processes are understood in greater detail.
Concluding remarks
An exciting aspect of current virology is that advancing results have not only continued to enrich our understanding of individual viruses, but also to reveal unifying principles that link many aspects of virus infection, replication and host interactions across a surprisingly wide breadth of virus–host systems. The resulting insights display a fundamental order within the vast and sometimes apparently chaotic diversity of known viruses, with important ramifications for virus function and evolution. As the full extent of this tapestry of relations is still emerging, ongoing studies will continue to extend and refine the underlying mechanistic connections. In association with their mechanistic importance, the results should have a growing impact on our abilities to limit the continuing toll and emerging threat of viral diseases, and to develop the beneficial uses of viruses.

References
Butel, J. S. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis 21, 405–426 (2000).
Talbot, S. J. & Crawford, D. H. Viruses and tumours — an update. Eur. J. Cancer 40, 1998–2005 (2004).
Paul, A. V., Rieder, E., Kim, D. W., van Boom, J. H. & Wimmer, E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 74, 10359–10370 (2000).
Murray, K. E. & Barton, D. J. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J. Virol. 77, 4739–4750 (2003). References 3 and 4 reveal and refine parallels in protein-primed genome replication by RNA picornaviruses and reverse-transcribing hepatitis B virus.
Gorbalenya, A. E. et al. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 324, 47–62 (2002). Reveals that an unusual re-ordering of polymerase domains is shared by certain (+)RNA viruses and dsRNA viruses.
Coulibaly, F. et al. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120, 761–772 (2005).
Ahlquist, P. Virus evolution: fitting lifestyles to a T. Curr. Biol. 15, R465–R467 (2005).
Weiner, A. M. & Maizels, N. tRNA-like structures tag the 3′ ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proc. Natl Acad. Sci. USA 84, 7383–7387 (1987). Proposes evolutionary relationships among tRNA-like 3′ ends of certain (+)RNA-virus genomic RNAs, retroviral tRNA priming and chromosomal telomeres.
Maizels, N. & Weiner, A. M. in The RNA World (eds Gesteland, R. F., Cech, T. R. & Atkins, J. F.) 79–111 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1999).
Miller, W. A., Bujarski, J. J., Dreher, T. W. & Hall, T. C. Minus-strand initiation by brome mosaic virus replicase within the 3′ tRNA-like structure of native and modified RNA templates. J. Mol. Biol. 187, 537–546 (1986).
Dreher, T. W. & Hall, T. C. Mutational analysis of the tRNA mimicry of brome mosaic virus RNA. Sequence and structural requirements for aminoacylation and 3′-adenylation. J. Mol. Biol. 201, 41–55 (1988).
Dreher, T. W. & Hall, T. C. Mutational analysis of the sequence and structural requirements in brome mosaic virus RNA for minus strand promoter activity. J. Mol. Biol. 201, 31–40 (1988).
Choi, S. K., Hema, M., Gopinath, K., Santos, J. & Kao, C. Replicase-binding sites on plus- and minus-strand brome mosaic virus RNAs and their roles in RNA replication in plant cells. J. Virol. 78, 13420–13429 (2004).
Wang, H. & Lambowitz, A. M. The Mauriceville plasmid reverse transcriptase can initiate cDNA synthesis de novo and may be related to reverse transcriptase and DNA polymerase progenitor. Cell 75, 1071–1081 (1993). Identifies a natural intermediate between tRNA-primed retrovirus reverse transcription and de novo RNA synthesis on tRNA-like 3′ ends of some RNA viruses.
Restrepo-Hartwig, M. & Ahlquist, P. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol. 70, 8908–8916 (1996).
Restrepo-Hartwig, M. & Ahlquist, P. Brome mosaic virus RNA replication proteins 1a and 2a colocalize and 1a independently localizes on the yeast endoplasmic reticulum. J. Virol. 73, 10303–10309 (1999).
Pedersen, K. W., van der Meer, Y., Roos, N. & Snijder, E. J. Open reading frame 1-a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 73, 2016–2026 (1999).
Suhy, D. A., Giddings, T. H., Jr & Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74, 8953–8965 (2000).
Gosert, R. et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77, 5487–5492 (2003).
Grimley, P. M., Berezesky, I. & Friedman, R. M. Cytoplasmic structures associated with an arbovirus infection: loci of viral ribonucleic acid synthesis. J. Virol. 2, 1326–1338 (1968).
Froshauer, S., Kartenbeck, J. & Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 107, 2075–2086 (1988). References 20 and 21 are classic papers showing association of alphavirus RNA synthesis with modified intracellular membranes.
Miller, D. J., Schwartz, M. D. & Ahlquist, P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 75, 11664–11676 (2001).
Prod'homme, D., Le Panse, S., Drugeon, G. & Jupin, I. Detection and subcellular localization of the turnip yellow mosaic virus 66K replication protein in infected cells. Virology 281, 88–101 (2001).
Kim, K. S. An ultrastructural study of inclusions and disease in plant cells infected by cowpea chlorotic mottle virus. J. Gen. Virol. 35, 535–543 (1977).
Hatta, T. & Francki, R. I. B. Cytopathic structures associated with tonoplasts of plant cells infected with cucumber mosaic and tomato aspermy viruses. J. Gen. Virol. 53, 343–346 (1981).
Morikawa, Y. HIV capsid assembly. Curr. HIV Res. 1, 1–14 (2003).
Scarlata, S. & Carter, C. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 1614, 62–72 (2003).
Swanstrom, R. & Wills, J. W. in Retroviruses (eds Coffin, J. M., Hughes, S. H. & Varmus, H. E.) 263–334 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1997).
Ansari-Lari, M. A. & Gibbs, R. A. Expression of human immunodeficiency virus type 1 reverse transcriptase in trans during virion release and after infection. J. Virol. 70, 3870–3805 (1996).
Yuan, B., Fassati, A., Yueh, A. & Goff, S. P. Characterization of Moloney murine leukemia virus p12 mutants blocked during early events of infection. J. Virol. 76, 10801–10810 (2002).
Nermut, M. V. & Fassati, A. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J. Virol. 77, 8196–8206 (2003).
Garfinkel, D. J., Boeke, J. D. & Fink, G. R. Ty element transposition: reverse transcriptase and virus-like particles. Cell 42, 507–517 (1985).
Eichinger, D. J. & Boeke, J. D. The DNA intermediate in yeast Ty1 element transposition copurifies with virus-like particles: cell-free Ty1 transposition. Cell 54, 955–966 (1988).
Linial, M. L. Foamy viruses are unconventional retroviruses. J. Virol. 73, 1747–1755 (1999).
Palmer, K. J. et al. Cryo-electron microscopy structure of yeast Ty retrotransposon virus-like particles. J. Virol. 71, 6863–6868 (1997).
Briggs, J. A. et al. The stoichiometry of Gag protein in HIV-1. Nature Struct. Mol. Biol. 11, 672–675 (2004).
Shehu-Xhilaga, M., Crowe, S. M. & Mak, J. Maintenance of the Gag/Gag–Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75, 1834–1841 (2001).
Berkowitz, R., Fisher, J. & Goff, S. P. RNA packaging. Curr. Top. Microbiol. Immunol. 214, 177–218 (1996).
D'Souza, V. & Summers, M. F. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature 431, 586–590 (2004).
Ahlquist, P. et al. Sindbis virus proteins nsP1 and nsP2 contain homology to nonstructural proteins from several RNA plant viruses. J. Virol. 53, 536–542 (1985).
van Regenmortel, M. H. V. (ed.) Virus Taxonomy (Academic Press, San Diego, 2000).
Ahola, T. & Ahlquist, P. Putative RNA capping activities encoded by brome mosaic virus: methylation and covalent binding of guanylate by replicase protein 1a. J. Virol. 73, 10061–10069 (1999).
Ahola, T., den Boon, J. A. & Ahlquist, P. Helicase and capping enzyme active site mutations in brome mosaic virus protein 1a cause defects in template recruitment, negative-strand RNA synthesis, and viral RNA capping. J. Virol. 74, 8803–8811 (2000).
Kong, F., Sivakumaran, K. & Kao, C. The N-terminal half of the brome mosaic virus 1a protein has RNA capping-associated activities: specificity for GTP and S-adenosylmethionine. Virology 259, 200–210 (1999).
Schwartz, M. et al. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9, 505–514 (2002). Reveals parallels between the structure, assembly and function of bromovirus RNA-replication complexes and retrovirus virions.
Kao, C. C. & Ahlquist, P. Identification of the domains required for direct interaction of the helicase-like and polymerase-like RNA replication proteins of brome mosaic virus. J. Virol. 66, 7293–7302 (1992).
Chen, J. & Ahlquist, P. Brome mosaic virus polymerase-like protein 2a is directed to the endoplasmic reticulum by helicase-like viral protein 1a. J. Virol. 74, 4310–4318 (2000).
Chen, J., Noueiry, A. & Ahlquist, P. An alternate pathway for recruiting template RNA to the brome mosaic virus RNA replication complex. J. Virol. 77, 2568–2577 (2003).
Janda, M. & Ahlquist, P. Brome mosaic virus RNA replication protein 1a dramatically increases in vivo stability but not translation of viral genomic RNA3. Proc. Natl Acad. Sci. USA 95, 2227–2232 (1998).
Sullivan, M. & Ahlquist, P. Cis-acting signals in bromovirus RNA replication and gene expression: networking with viral proteins and host factors. Semin. Virol. 8, 221–230 (1997).
Chen, J., Noueiry, A. & Ahlquist, P. Brome mosaic virus protein 1a recruits viral RNA2 to RNA replication through a 5′ proximal RNA2 signal. J. Virol. 75, 3207–3219 (2001).
Sullivan, M. & Ahlquist, P. A brome mosaic virus intergenic RNA3 replication signal functions with viral replication protein 1a to dramatically stabilize RNA in vivo. J. Virol. 73, 2622–2632 (1999).
den Boon, J., Chen, J. & Ahlquist, P. Identification of sequences in brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J. Virol. 75, 12370–12381 (2001).
O'Reilly, E. K., Wang, Z., French, R. & Kao, C. C. Interactions between the structural domains of the RNA replication proteins of plant-infecting RNA viruses. J. Virol. 72, 7160–7169 (1998).
Freed, E. O. Viral late domains. J. Virol. 76, 4679–4687 (2002).
McMahon, H. T. & Mills, I. G. COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr. Opin. Cell Biol. 16, 379–391 (2004).
Morita, E. & Sundquist, W. I. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20, 395–425 (2004).
Lee, J.-Y., Marshall, J. A. & Bowden, D. S. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 200, 307–312 (1994).
Kujala, P. et al. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 75, 3873–3884 (2001).
Sethna, P. B. & Brian, D. A. Coronavirus genomic and subgenomic minus-strand RNAs copartition in membrane-protected replication complexes. J. Virol. 71, 7744–7749 (1997).
Miyanari, Y. et al. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278, 50301–50308 (2003).
Quinkert, D., Bartenschlager, R. & Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79, 13594–13605 (2005).
Lyle, J. M., Bullitt, E., Bienz, K. & Kirkegaard, K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science 296, 2218–2222 (2002). Shows that poliovirus RNA polymerase can self-interact in extended networks consistent with RNA replication on membrane surfaces.
Panaviene, Z., Panavas, T. & Nagy, P. D. Role of an internal and two 3′-terminal RNA elements in assembly of tombusvirus replicase. J. Virol. 79, 10608–10618 (2005).
Salonen, A. et al. Properly folded nonstructural polyprotein directs the Semliki Forest virus replication complex to the endosomal compartment. J. Virol. 77, 1691–1702 (2003).
Snijder, E. J., van Tol, H., Roos, N. & Pedersen, K. W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 82, 985–994 (2001).
Egger, D. et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984 (2002).
Cho, M. W., Teterina, N., Egger, D., Bienz, K. & Ehrenfeld, E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202, 129–145 (1994).
Teterina, N. L. et al. Requirements for assembly of poliovirus replication complexes and negative-strand RNA synthesis. J. Virol. 75, 3841–3850 (2001).
Felsenstein, K. M. & Goff, S. P. Expression of the Gag–Pol fusion protein of Moloney murine leukemia virus without Gag protein does not induce virion formation or proteolytic processing. J. Virol. 62, 2179–2182 (1988).
Karacostas, V., Wolffe, E. J., Nagashima, K., Gonda, M. A. & Moss, B. Overexpression of the HIV-1 Gag–Pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193, 661–671 (1993).
Panavas, T., Hawkins, C. M., Panaviene, Z. & Nagy, P. D. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology 338, 81–95 (2005).
Ishikawa, M., Meshi, T., Motoyoshi, F., Takamatsu, N. & Okada, Y. In vitro mutagenesis of the putative replicase genes of tobacco mosaic virus. Nucleic Acids Res. 14, 8291–8305 (1986).
Li, G. & Rice, C. M. Mutagenesis of the in-frame opal termination codon preceding nsP4 of Sindbis virus: studies of translational readthrough and its effect on virus replication. J. Virol. 63, 1326–1337 (1989).
Noueiry, A. O., Chen, J. & Ahlquist, P. A mutant allele of essential, general translation initiation factor DED1 selectively inhibits translation of a viral mRNA. Proc. Natl Acad. Sci. USA 97, 12985–12990 (2000).
Ishikawa, M., Diez, J., Restrepo-Hartwig, M. & Ahlquist, P. Yeast mutations in multiple complementation groups inhibit brome mosaic virus RNA replication and transcription and perturb regulated expression of the viral polymerase-like gene. Proc. Natl Acad. Sci. USA 94, 13810–13815 (1997).
Kim, S. H., Palukaitis, P. & Park, Y. I. Phosphorylation of cucumber mosaic virus RNA polymerase 2a protein inhibits formation of replicase complex. EMBO J. 21, 2292–2300 (2002).
deGroot, R. J., Rümenapf, T., Kuhn, R. J., Strauss, E. G. & Strauss, J. H. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Biochemistry 88, 8967–8971 (1991).
Hajimorad, M. R. et al. Nla and Nlb of peanut stripe potyvirus are present in the nucleus of infected cells, but do not form inclusions. Virology 224, 368–379 (1996).
Levin, H. L., Weaver, D. C. & Boeke, J. D. Novel gene expression mechanism in a fission yeast retroelement: Tf1 proteins are derived from a single primary translation product. EMBO J. 12, 4885–4895 (1993).
Teysset, L., Dang, V. D., Kim, M. K. & Levin, H. L. A long terminal repeat-containing retrotransposon of Schizosaccharomyces pombe expresses a Gag-like protein that assembles into virus-like particles which mediate reverse transcription. J. Virol. 77, 5451–5463 (2003).
Ganser, B. K., Cheng, A., Sundquist, W. I. & Yeager, M. Three-dimensional structure of the M-MuLV CA protein on a lipid monolayer: a general model for retroviral capsid assembly. EMBO J. 22, 2886–2892 (2003).
Westaway, E., Mackenzie, J., Kenney, M., Jones, M. & Khromykh, A. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 71, 6650–6661 (1997).
Schwartz, M., Chen, J., Lee, W. M., Janda, M. & Ahlquist, P. Alternate, virus-induced membrane rearrangements support positive-strand RNA virus genome replication. Proc. Natl Acad. Sci. USA 101, 11263–11268 (2004). Shows that outwardly distinct membrane rearrangements, similar to those associated with RNA replication by different viruses, can be induced by the same viral replication proteins.
Aizaki, H., Lee, K. J., Sung, V. M., Ishiko, H. & Lai, M. M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324, 450–461 (2004).
Gosert, R., Kanjanahaluethai, A., Egger, D., Bienz, K. & Baker, S. C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76, 3697–3708 (2002).
Schlegel, A., Giddings, J., T., Ladinsky, M. & Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during polivirus infection. J. Virol. 70, 6576–6588 (1996).
Teterina, N. L., Bienz, K., Egger, D., Gorbalenya, A. E. & Ehrenfeld, E. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 237, 66–77 (1997).
Egger, D., Pasamontes, L., Bolten, R., Boyko, V. & Bienz, K. Reversible dissociation of the poliovirus replication complex: functions and interactions of its components in viral RNA synthesis. J. Virol. 70, 8675–8683 (1996).
Nibert, M. L. & Schiff, L. A. in Fields Virology (eds Knipe, D. M. & Howley, P. M.) 1679–1728 (Lippincott, Williams & Wilkins, Philadelphia, 2001).
Patton, J. T. & Spencer, E. Genome replication and packaging of segmented double-stranded RNA viruses. Virology 277, 217–225 (2000).
Koonin, E. V., Gorbalenya, A. E. & Chumakov, K. M. Tentative identification of RNA-dependent RNA polymerases of dsRNA viruses and their relationship to positive strand RNA viral polymerases. FEBS Lett. 252, 42–46 (1989).
Koonin, E. V., Choi, G. H., Nuss, D. L., Shapira, R. & Carrington, J. C. Evidence for common ancestry of a chestnut blight hypovirulence-associated double-stranded RNA and a group of positive-strand RNA plant viruses. Proc. Natl Acad. Sci. USA 88, 10647–10651 (1991).
Reinisch, K. M., Nibert, M. L. & Harrison, S. C. Structure of the reovirus core at 3.6 Å resolution. Nature 404, 960–967 (2000).
Zhang, X., Walker, S. B., Chipman, P. R., Nibert, M. L. & Baker, T. S. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nature Struct. Biol. 10, 1011–1018 (2003). References 94 and 95 define the structure of transcriptionally active cores of dsRNA reovirus.
Gomez de Cedron, M., Ehsani, N., Mikkola, M. L., Garcia, J. A. & Kaariainen, L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 448, 19–22 (1999).
Vasiljeva, L., Merits, A., Auvinen, P. & Kaariainen, L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of Nsp2. J. Biol. Chem. 275, 17281–17287 (2000).
Li, Y. I. et al. The helicase-like domain of plant potexvirus replicase participates in formation of RNA 5′ cap structure by exhibiting RNA 5′-triphosphatase activity. J. Virol. 75, 12114–12120 (2001).
Wang, X. et al. Brome mosaic virus 1a nucleoside triphosphatase/helicase domain plays crucial roles in recruiting RNA replication templates. J. Virol. 79, 13747–13758 (2005).
Bisaillon, M., Bergeron, J. & Lemay, G. Characterization of the nucleoside triphosphate phosphohydrolase and helicase activities of the reovirus λ1 protein. J. Biol. Chem. 272, 18298–18303 (1997).
Bisaillon, M. & Lemay, G. Characterization of the reovirus λ1 protein RNA 5′-triphosphatase activity. J. Biol. Chem. 272, 29954–29957 (1997).
Kao, C. C., Quadt, R., Hershberger, R. P. & Ahlquist, P. Brome mosaic virus RNA replication proteins 1a and 2a form a complex in vitro. J. Virol. 66, 6322–6329 (1992).
Luongo, C. L., Contreras, C. M., Farsetta, D. L. & Nibert, M. L. Binding site for S-adenosyl-L-methionine in a central region of mammalian reovirus λ2 protein. Evidence for activities in mRNA cap methylation. J. Biol. Chem. 273, 23773–23780 (1998).
Luongo, C. L., Reinisch, K. M., Harrison, S. C. & Nibert, M. L. Identification of the guanylyltransferase region and active site in reovirus mRNA capping protein λ2. J. Biol. Chem. 275, 2804–2810 (2000).
Li, Y. I., Chen, Y. J., Hsu, Y. H. & Meng, M. Characterization of the AdoMet-dependent guanylyltransferase activity that is associated with the N terminus of bamboo mosaic virus replicase. J. Virol. 75, 782–788 (2001).
Pirttimaa, M. J., Paatero, A. O., Frilander, M. J. & Bamford, D. H. Nonspecific nucleoside triphosphatase P4 of double-stranded RNA bacteriophage φ6 is required for single-stranded RNA packaging and transcription. J. Virol. 76, 10122–10127 (2002).
Kainov, D. E. et al. RNA packaging device of double-stranded RNA bacteriophages, possibly as simple as hexamer of P4 protein. J. Biol. Chem. 278, 48084–48091 (2003).
Goregaoker, S. P. & Culver, J. N. Oligomerization and activity of the helicase domain of the tobacco mosaic virus 126- and 183-kilodalton replicase proteins. J. Virol. 77, 3549–3556 (2003).
Jayaram, H., Taraporewala, Z., Patton, J. T. & Prasad, B. V. Rotavirus protein involved in genome replication and packaging exhibits a HIT-like fold. Nature 417, 311–315 (2002).
Taraporewala, Z. F. & Patton, J. T. Nonstructural proteins involved in genome packaging and replication of rotaviruses and other members of the Reoviridae. Virus Res. 101, 57–66 (2004).
Kim, J., Parker, J. S., Murray, K. E. & Nibert, M. L. Nucleoside and RNA triphosphatase activities of orthoreovirus transcriptase cofactor μ2. J. Biol. Chem. 279, 4394–4403 (2004).
Kroner, P. A., Young, B. M. & Ahlquist, P. Analysis of the role of brome mosaic virus 1a protein domains in RNA replication, using linker insertion mutagenesis. J. Virol. 64, 6110–6120 (1990).
Ishikawa, M., Kroner, P., Ahlquist, P. & Meshi, T. Biological activities of hybrid RNAs generated by 3′-end exchanges between tobacco mosaic and brome mosaic viruses. J. Virol. 65, 3451–3459 (1991).
Ahlquist, P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 296, 1270–1273 (2002).
Silvestri, L. S., Taraporewala, Z. F. & Patton, J. T. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J. Virol. 78, 7763–7774 (2004).
Gitlin, L., Stone, J. K. & Andino, R. Poliovirus escape from RNA interference: short interfering RNA-target recognition and implications for therapeutic approaches. J. Virol. 79, 1027–1035 (2005).
Ishikawa, M., Meshi, T., Ohno, T. & Okada, Y. Specific cessation of minus-strand RNA accumulation at an early stage of tobacco mosaic virus infection. J. Virol. 65, 861–868 (1991).
Strauss, J. H. & Strauss, E. G. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58, 491–562 (1994).
Wengler, G., Wengler, G. & Gross, H. J. Terminal sequences of Sindbis virus-specific nucleic acids: identity in molecules synthesized in vertebrate and insect cells and characteristic properties of the replicative form RNA. Virology 123, 273–283 (1982).
Collmer, C. W. & Kaper, J. M. Double-stranded RNAs of cucumber mosaic virus and its satellite contain an unpaired terminal guanosine: implications for replication. Virology 145, 249–259 (1985).
Diprose, J. M. et al. Translocation portals for the substrates and products of a viral transcription complex: the bluetongue virus core. EMBO J. 20, 7229–7239 (2001).
Hansen, J., Long, A. & Schultz, S. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 5, 1109–1122 (1997).
Lesburg, C. A. et al. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nature Struct. Biol. 6, 937–943 (1999).
Siegel, R. W., Bellon, L., Beigelman, L. & Kao, C. C. Use of DNA, RNA, and chimeric templates by a viral RNA-dependent RNA polymerase: evolutionary implications for the transition from the RNA to the DNA world. J. Virol. 73, 6424–6429 (1999). Shows that a (+)RNA-virus RNA-dependent RNA polymerase has substantial activity on DNA templates.
Gao, G., Orlova, M., Georgiadis, M. M., Hendrickson, W. A. & Goff, S. P. Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl Acad. Sci. USA 94, 407–411 (1997).
Rolls, M. M., Webster, P., Balba, N. H. & Rose, J. K. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell 79, 497–506 (1994).
Lai, M. M. RNA recombination in animal and plant viruses. Microbiol. Rev. 56, 61–79 (1992).
Nagy, P. D. & Simon, A. E. New insights into the mechanisms of RNA recombination. Virology 235, 1–9 (1997).
Worobey, M. & Holmes, E. C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 80, 2535–2543 (1999).
Ahlquist, P. et al. Viral and host determinants of RNA virus vector replication and expression. Vaccine 23, 1784–1787 (2005).
Estes, M. K. in Fields Virology (eds Knipe, D. M. & Howley, P. M.) 1747–1785 (Lippincott, Williams & Wilkins, Philadelphia, 2001).
Lopez, T. et al. Silencing the morphogenesis of rotavirus. J. Virol. 79, 184–192 (2005).
Silvestri, L. S., Tortorici, M. A., Vasquez- Del Carpio, R. & Patton, J. T. Rotavirus glycoprotein NSP4 is a modulator of viral transcription in the infected cell. J. Virol. 79, 15165–15174 (2005). References 132 and 133 reveal links between rotavirus transmembrane protein nsP4 and formation of viroplasms in which dsRNA virion-core assembly occurs.
Laurinavicius, S., Kakela, R., Bamford, D. H. & Somerharju, P. The origin of phospholipids of the enveloped bacteriophage φ6. Virology 326, 182–190 (2004).
Lee, W. M., Ishikawa, M. & Ahlquist, P. Mutation of host Δ9 fatty acid desaturase inhibits brome mosaic virus RNA replication between template recognition and RNA synthesis. J. Virol. 75, 2097–2106 (2001).
Lee, W. M. & Ahlquist, P. Membrane synthesis, specific lipid requirements, and localized lipid composition changes associated with a positive-strand RNA virus RNA replication protein. J. Virol. 77, 12819–12828 (2003).
Ono, A. & Freed, E. O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl Acad. Sci. USA 98, 13925–13930 (2001).
Kushner, D. B. et al. Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc. Natl Acad. Sci. USA 100, 15764–15769 (2003).
Hu, J., Toft, D. O. & Seeger, C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16, 59–68 (1997).
Tomita, Y. et al. Mutation of host dnaJ homolog inhibits brome mosaic virus negative-strand RNA synthesis. J. Virol. 77, 2990–2997 (2003).
Held, D. M., Kissel, J. D., Patterson, J. T., Nickens, D. G. & Burke, D. H. HIV-1 inactivation by nucleic acid aptamers. Front. Biosci. 11, 89–112 (2006).
Resh, M. D. Intracellular trafficking of HIV-1 Gag: how Gag interacts with cell membranes and makes viral particles. AIDS Rev. 7, 84–91 (2005).
Khromykh, A. A., Kondratieva, N., Sgro, J. Y., Palmenberg, A. & Westaway, E. G. Significance in replication of the terminal nucleotides of the flavivirus genome. J. Virol. 77, 10623–10629 (2003).
Yang, H., Makeyev, E. V., Butcher, S. J., Gaidelyte, A. & Bamford, D. H. Two distinct mechanisms ensure transcriptional polarity in double-stranded RNA bacteriophages. J. Virol. 77, 1195–1203 (2003).
Wickner, R. B. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 60, 250–265 (1996).
Acknowledgements
We thank all members of our laboratory for helpful discussions. This work was supported by the National Institutes of Health.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Related links
Related links
DATABASES
Entrez Genome
FURTHER INFORMATION
Glossary
- Positive-strand RNA virus
-
A virus, the infectious virions of which contain the genome in a single-stranded, messenger-sense RNA form.
- Negative-strand RNA virus
-
A virus, the infectious virions of which contain the genome in a single-stranded, anti-messenger-sense RNA form.
- Retroplasmid
-
A DNA plasmid that replicates by transcription and reverse transcription of an RNA intermediate.
- BrUTP labelling
-
Labelling RNA by replacing the substrate UTP with 5-bromo-UTP (BrUTP). The labelled RNA can be localized using electron microscopy following immunogold labelling with an antibody directed against BrU.
- Translational frameshift
-
A site-specific, programmed shift of some translating ribosomes from one reading frame to another, allowing a fraction of translation products to be extended in the new frame.
- Translational readthrough
-
Programmed translation of some ribosomes through a termination codon, allowing a fraction of translation products to be extended beyond the normal stop site.
Rights and permissions
About this article
Cite this article
Ahlquist, P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol 4, 371–382 (2006). https://doi.org/10.1038/nrmicro1389
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro1389
This article is cited by
-
Multifaceted role of natural sources for COVID-19 pandemic as marine drugs
Environmental Science and Pollution Research (2022)
-
Cypovirus capsid protein VP5 has nucleoside triphosphatase activity
Virologica Sinica (2017)
-
A capsidless ssRNA virus hosted by an unrelated dsRNA virus
Nature Microbiology (2016)
-
Physiological and pathophysiological role of nonsense-mediated mRNA decay
Pflügers Archiv - European Journal of Physiology (2016)
-
Nuclear import of Maize chlorotic mottle virus capsid protein is mediated by importin-α
European Journal of Plant Pathology (2016)
