
Key Points
-
Mucosal surfaces — such as the lining of the gut or the reproductive tract — are the main point of entry for viruses into the body.
-
Almost all viruses interact with epithelial cells, and make use of the normal epithelial signalling and trafficking pathways of the host cell.
-
In addition to protein receptors, carbohydrate chains of proteoglycans and epithelial membrane glycosphingolipids have emerged as a new class of receptors for viral attachment to the host cell.
-
Viruses have also developed soluble virulence factors, which interact with epithelial cells independently of the virus and participate in the development of the virally induced pathology.
Abstract
Mucosal surfaces — such as the lining of the gut or the reproductive tract — are the main point of entry for viruses into the body. As such, almost all viruses interact with epithelial cells, and make use of the normal epithelial signalling and trafficking pathways of the host cell. In addition to protein receptors, carbohydrate chains of proteoglycans and epithelial-membrane glycosphingolipids have emerged as a new class of receptors for viral attachment to the host cell.
Similar content being viewed by others


Main
Viruses enter the body through two main surfaces — the skin and the mucosal epithelia. These surfaces are covered by epithelial cells that are organized into complex structures (Box 1), and this epithelial organization often dictates the mechanisms of viral entry and translocation.
In this review, we explore the strategies that viruses have evolved to translocate across the epithelial barrier and to act as pathogens, according to the target-cell structure and the nature of the virus. To understand these mechanisms, we first map the pathways of viral entry. Then we describe, at the molecular level, the cell receptors that allow viral attachment and entry into the cell, as well as the viral proteins that interact with — and subvert — these receptors, which allows the virus to cross the cell membrane.
Our understanding of host–virus interactions at the molecular level has allowed the characterization of many viral receptors at the epithelial surface. The accepted model of viral entry that is achieved using a unique viral receptor has been challenged. Instead, viruses are now thought to use host-cell molecules — which are referred to as attachment receptors or 'co-receptors' — in addition to the protein receptor, which is often renamed the 'principal' receptor. The dynamics of these receptors in the membrane is crucial because, on binding of the virus, receptors can be recruited to or excluded from transiently organized glycosphingolipid-rich membrane microdomains, which are known as lipid RAFTS.
An important class of attachment receptors has emerged recently — namely the carbohydrate chain of PROTEOGLYCANS and epithelial-membrane GLYCOSPHINGOLIPIDS (Fig. 1). There is growing recognition of the functional importance of these biomolecules, as shown by the emergence of the field of 'glycomics', or 'the sugar code'1. Indeed, oligosaccharides transfer information to complementary effector molecules, and the virus acts as a LECTIN by binding the carbohydrate as an attachment receptor. Owing to the nature of the lectin–sugar interaction (namely charge-transfer processes that are facilitated by networks of hydrogen-bond formation), environmental factors are important. These factors relate to the characteristics of the mucosal surface, which has numerous carbohydrate groups protruding into the aqueous environment and a high ionic strength.This high ionic strength is due to the charged surface lipids and their counterions. On the viral oligomeric peptide, the lectin site is characterized by the charged amino acids and the aromatic tryptophan that surrounds the galactose ring.

a | Proteoglycans are proteins that are classified by a post-translational attachment of polysaccharide glycosaminoglycan moieties, which are each composed of repeating disaccharide units. The ubiquitously expressed glycosaminoglycan heparan sulphate (shown here) is highly polymorphic, and its sulphated structural motifs are primarily responsible for its protein binding and regulatory properties. b | Glycosphingolipids are anchored in the outer leaflet of the plasma-membrane bilayer by their common hydrophobic backbone, ceramide, which consists of a fatty-acid chain linked to the sphingosine base. The hydrophilic carbohydrate parts of neutral glycosphingolipids and gangliosides protrude into the extracellular space and partially cover the cell surface. GalNAc, (OSO3), N-acetyl galactosamine sulphate; GlcNSO3, glucosamine sulphate; GlcUA, glucuronic acid; IdUA (OSO3), iduronic acid sulphate.
Viral pathways in the epithelial barrier
Viruses have evolved several pathways to initiate entry through epithelial barriers. Viruses can enter and infect epithelial cells by accessing the cell cytosol using one of two mechanisms — direct entry at the epithelial plasma membrane, or entry through the epithelial endocytic pathway2. By contrast, some viruses do not need to infect epithelial cells to spread — they are internalized by epithelial cells and cross the epithelial barrier using TRANSCYTOSIS (Box 1), as has been described recently for the human immunodeficiency virus (HIV)3,4.
In viruses, the genome is surrounded either solely by a capsid ('naked' virus), or by both a capsid and a membrane ('enveloped' virus; Box 2). We know much about the entry mechanism of enveloped viruses, but the mechanism by which naked viruses penetrate epithelial cells is far less well understood. Nevertheless, the route by which viruses enter and then infect or cross epithelial cells is not dictated solely by whether or not the virus is enveloped2,5, and the three main modes of entry are summarized in the text below (Table 1).
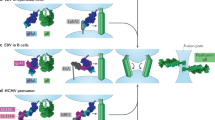
Endocytosis and transcytosis (without infection). Several enveloped and naked viruses, such as HIV-1 (Fig. 2) and poliovirus, cross simple epithelial cells by rapid transcytosis without infection. They are released — and are still infectious — at the pole of the cell that is opposite to their pole of entry, and this enables the virus to spread in the submucosa.

Cell-free human immunodeficiency virus (HIV) or infected cells encounter a mucosal surface (Box 4). Both contain viruses that use either the chemokine receptor CCR5 (R5 virus; found in most acutely infected patients, and therefore thought to be the main vector of infection) or CXCR4 (X4 virus; found later in patients, as the disease progresses) for fusion and/or infection. In the upper small intestine, galactosyl-ceramide (GalCer)+/CCR5+/CXCR4− epithelial cells endocytose cell-free R5 virus at the lumenal surface in a galactosyl ceramide/CCR5-receptor-mediated mechanism, whereas cell-free X4 viruses cannot enter these CXCR4– epithelial cells4. Alternatively, R5 (or X4)-infected MONONUCLEAR CELLS bind to the epithelial cell and induce the polarized budding of newly formed R5 (or X4) viruses, which are rapidly endocytosed through GalCer that is present in raft microdomains3,91. HIV transcytoses across epithelial cells to the serosal surface, where fusion of the transcytotic vesicles releases virus into the LAMINA PROPRIA. Here, R5 viruses infect the CCR5+/CXCR4+/CD4+ lamina propria T lymphocyte, but not the CCR5−/CXCR4−/CD4+ intestinal macrophage93. By contrast, the infection of intestinal CXCR4+/CD4+ T cells by X4 viruses that are transcytosed from X4-infected cells is blocked by stromal-cell-derived factor (SDF)-1 that is present in the mucosa94. The ability of primary human M cells to translocate R5 or X4 HIV remains to be clarified.
The Epstein–Barr virus (EBV) uses a more complicated strategy. EBV forms a complex with mucosal immunoglobulin (Ig)A that is specific for gp350, a viral surface protein that is present in latently infected people. The complex binds to the POLY-IMMUNOGLOBULIN RECEPTOR at the basal surface of epithelial cells, and is endocytosed and delivered apically without infection6. By contrast, in non-polarized cells, the entry of IgA–EBV leads to infection. Two EBV replicative proteins can stimulate the production of anti-EBV IgA7,8, thereby contributing to the development of disease in latently infected people.
Finally, Peyer's patches — specific lymphoid areas of the gastrointestinal tract — are covered by specialized epithelial cells, which are known as M CELLS. These patches might be one portal for the mucosal entry of poliovirus9, HIV-110 and reovirus11, by transcytosis across M cells in a receptor-mediated fashion.
Polarized surface entry and infection by fusion. Viruses can penetrate epithelial cells at the epithelial-cell plasma membrane directly after attachment and fusion. In polarized monostratified epithelium — where the plasma membrane is divided into two domains that have a different lipid and protein composition and different membrane dynamics — viruses usually attach and penetrate the cell cytosol preferentially at the restricted pole of the epithelial cell. Some enveloped viruses attach to, and fuse with, the epithelial-cell apical membrane12,13,14, whereas other enveloped viruses15 attach to, and fuse with, the basal membrane. Importantly, viruses from the same family do not always show the same polarity of entry14,16,17. This is due, in part, to the use of different cell receptors. However, some viruses such as poliovirus can enter at both the plasma membrane and through the endocytic pathway18,19 (see below).
Endocytosis and endosomal fusion with infection. Both enveloped and naked viruses can enter epithelial cells by endocytosis, and they usually penetrate the host-cell cytosol by fusion from an ENDOSOME. This mode of entry makes use of specific endosomal conditions such as low pH or a high concentration of calcium.
In polarized monostratified epithelium, the polarity of viral entry is a determinant for the outcome of an infection, as polarity differentially influences the processing and sorting mechanisms in apical and basolateral endosomes. However, the nature of the experimental system can change the polarity of viral entry20 (Box 3). Some enveloped viruses, such as influenza virus types A and C, bovine coronavirus21 and hepatitis A virus (HAV)22, are endocytosed at the apical pole of polarized epithelial cells, whereas vesicular stomatitis virus (VSV), which is also enveloped, is endocytosed basolaterally2. For all of these viruses, however, access to the cytosol from the endosome is pH dependent.
Other viruses can enter by endocytosis at either of the poles in epithelial cell lines23, but each route results in a specific infection outcome. Factors that dictate which entry pathway is used by the virus include differences in the interactions between cellular receptors and viral proteins (see below). For example, the infection of pigmented retinal epithelial cells by enveloped primary herpes simplex virus (HSV) and human cytomegalovirus (HCMV) occurs by endocytosis24,25,26 at either one of the poles followed by a pH-independent translocation into the cytoplasm. The outcome is either efficient viral replication followed by host-cell lysis, or, alternatively, latent infection. In the latter case, reactivation of the latent viral genome might then occur, and lead to the infection of nearby sensory neurons and the release of new virions. These virions can then re-enter epithelial cells — but now in an highly polarized manner — by fusion at the basolateral pole27. Naked adeno-associated virus (AAV)-2 enters airway epithelial cells essentially basolaterally by receptor-mediated endocytosis28. Its single-stranded (ss)DNA is then converted to a circular genome, and its polarity of entry is reinforced by a post-endocytic barrier. By contrast, AAV-2 that enters at the apical pole by a receptor-independent mechanism gives rise to persistent ssDNA that is arrested in a transcriptionally inactive form. The ubiquitin–proteasome pathway, which is involved in the regulation of endocytosis29, is instrumental in this arrest30.
In pluristratified epithelia or immature gastrointestinal cells, which lack TIGHT JUNCTIONS, viral entry is not polarized. Instead, the viruses make use of various endocytic pathways (Box 1). Filovirus, influenza virus, simian virus 40 (SV40) and measles virus are endocytosed after clustering in transient raft membrane microdomains31,32,33,34,35. Some enveloped viruses, such as the Semliki Forest virus (SFV) and Sindbis virus, as well as some naked viruses, including Jamestown Canyon (JC) polyoma virus, parvovirus, adenovirus-2, two of the picorna viruses — human echovirus-1 (E-1) and the minor group of human rhinoviruses (HRV) — and foot and mouth disease virus (FMDV), use receptor-mediated endocytosis through the CLATHRIN-COATED-VESICLE pathway36,37,38,39,40.
Once they are in endosomes, the surface proteins of viruses such as influenza, FMDV, VSV, Sindbis virus or SFV undergo a conformational change that is dependent on a mildly acidic pH, and they can then disrupt the endosomal membrane. Alternatively, other viruses, such as the poliovirus18,19 and the major group of HRV41, translocate from the endosome into the cell cytosol in a pH-insensitive manner, before gaining access to the lysosome — a compartment that is deeper in the endocytic pathway19,41. A Ca2+-dependent, but pH-independent, endocytosis and virus-uncoating model has been proposed for the rotavirus42 that would apply equally to other viruses, whether they are enveloped or naked.
Multiple viral receptors: a new model
The differences in the viral-entry pathways are due largely to the nature of the molecular interactions between the viral components and target-cell receptors. Viral pathogenesis arises from mechanisms that have been developed to block or abuse normal cell processes and, as with bacteria43, the surface proteins of enveloped or naked viruses bind to host-cell molecules that have receptor functions. So, viruses mimic the natural ligand of these receptors and interfere with their signalling to promote viral entry into the cell and the spread of infection (Box 4).
The classical concept of viral receptors has been superseded by new data, which indicate that the binding and entry of viruses is a multi-step process that involves the recognition of, and attachment to, the epithelial-cell surface. This is followed either by penetration of the host-cell cytosol, with infection of the cell, or by transcytosis. Each step involves many host-cell receptors. These receptors range from ubiquitous cell-surface-associated carbohydrate moieties of membrane glycoproteins, proteoglycans or glycolipids — which are inserted in the dynamic bilayer of the target-cell membrane and usually act as attachment receptors — to cell-specific transmembrane proteins, which can mediate numerous different steps. These many steps are not independent of each other, but their spatio-temporal sequence in the process of viral infection is difficult to assess. Indeed, at the molecular level, the kinetics of each virus–host-cell interaction is dependent on its association constant, the concentration of viral ligands and of host-cell receptors that are available to interact at any given moment, and the nature of the target cell.
To attach to the surface of the target cell, an increasing number of enveloped or naked viruses have been described as acting as lectins, by using a peptide of their envelope or capsid proteins, respectively, that has a lectin site. They compete with endogenous lectins to bind epithelial-cell-surface carbohydrates, which act as attachment receptors. Viral surface proteins are multimeric, they have several lectin sites, so they can interact with several receptor molecules at a time at the host-cell surface. Such clusters of lectin sites have a much higher affinity for oligosaccharides than their monomeric counterparts44. This multimeric interaction interferes with lipid organization and dynamics, and stabilizes raft microdomains at the epithelial-cell membrane. The lipids in these domains differ from other membrane lipids in their lateral diffusion in the membrane, and they can be separated in vitro, owing to their insolubility in detergent. The rafts are small, mobile, unstable and they probably fluctuate in their size and composition as a result of molecular interactions at the cell surface.
Attachment receptors and viral receptors can be recruited to, or excluded from, membrane microdomains such as rafts, clathrin-coated pits or CAVEOLAE5,45. This remodelling of the host-cell membrane is a determinant of the mechanism of virus entry and signal transduction in the host epithelial cell46.
Surface carbohydrates as attachment receptors
The carbohydrate moiety of host-cell glycoproteins, glycosphingolipids and proteoglycans has emerged as a widely used virus-attachment receptor47.
Sialyloligosaccharides. Influenza virus contains two major surface proteins that are involved in viral entry — haemagglutinin and neuraminidase — which bind and cleave SIALYLOLIGOSACCHARIDE, respectively. Balanced haemagglutinin and neuraminidase activities are crucial for efficient viral binding to the cell surface, and for viral replication. Influenza virus haemagglutinin is one of the best-known viral lectins. Its trimeric organization increases its affinity for sialyloligosaccharide and allows the virus to bind to the surface of the epithelial cell21. The epithelial molecules that contain this sialyloligosaccharide carbohydrate receptor remain unknown, although they could be glycoproteins or glycosphingolipids48. On endocytosis of the influenza virus, haemagglutinin is cleaved into haemagglutinin 1 and haemagglutinin 2. Whereas low endosomal pH exposes the haemagglutinin 2 fusion-peptide domain, haemagglutinin 1 mediates viral fusion with the endosomal membrane48. Similarly, human JC virus49 and Sendai50 virus, as well as sialyloligosaccharide-dependent strains of rotavirus20 and reovirus, also attach themselves to epithelial cells through sialyloligosaccharides, which are either from glycosphingolipids51 or proteoglycans52.
Glycosphingolipids. Several enveloped and naked viruses have recently been shown to interact specifically with a defined carbohydrate moiety of glycosphingolipids (Fig. 1; Table 1). Glycosphingolipids — which are characteristic components of eukaryotic plasma membranes — are a highly polymorphic class of lipids and are principal components of the apical plasma membrane of epithelial cells in the gastrointestinal and urinary tracts, myelin and neuroepithelial cells53. Glycosphingolipids are anchored in the outer leaflet of the plasma membrane bilayer by their common hydrophobic backbone, ceramide, which consists of a fatty-acid chain that is linked to the sphingosine base. The hydrophilic oligosaccharide residues of neutral glycosphingolipids and gangliosides protrude into the extracellular space and, together with the membrane glycoproteins and proteoglycans, they constitute the glycocalyx of the cell surface54.
The multimeric glycoprotein subunits gp41 (Refs 55–57) and gp120 (Refs 3,58) of the HIV-1 envelope both attach to the cell-membrane glycosphingolipid, galactosylceramide. Indeed, D-galactose is particularly eligible for stacking to the aromatic ring system of gp41 owing to the van der Waals interactions and the presence of a set of polarized C–H bonds (C–H/π-electrons) on one side of the galactose ring. Interestingly, HIV-1 and herpesvirus can enter neural cells (which are also polarized) and mucosal epithelial cells, both of which are rich in glycosphingolipids59. HIV-1 (Ref. 56), naked Ebola and Marburg viruses (through their capsid proteins) and measles virus31,32 require glycosphingolipids to be assembled in rafts for virus attachment and entry into the epithelial cell. This virus–cell interaction, which involves several species of viral surface protein, probably stabilizes raft microdomains, allowing signal transduction in the epithelial cell and endocytosis of the virus. A galactosylceramide/sphingomyelin-binding motif, which is similar to that found in HIV-1 gp120, is also found on prion protein and the amyloid-β peptide that is involved in Alzheimer's disease60.
Proteoglycans. Another class of carbohydrate attachment receptors used by viruses are found on proteoglycans. Proteoglycans are proteins that are classified by the post-translational attachment of polysaccharide GLYCOSAMINOGLYCAN moieties (Fig. 1). Glycosaminoglycan chains provide the initial docking sites for viruses to bind to eukaryotic cells. The ubiquitously expressed glycosaminoglycan HEPARAN SULPHATE is highly polymorphic, and its sulphated structural motifs are responsible primarily for its protein binding and regulatory properties, as shown recently61 for respiratory syncytial virus (RSV).
During the past decade, proteoglycans have emerged as key players in the regulatory network of the cell62. Depending on the length of the glycosaminoglycan chain, and on the sulphated structural motifs, a single glycosaminoglycan chain can bind many viral ligands on a single virion62. As the cell attachment receptors for numerous enveloped and naked viruses, the glycosyl epitopes of the epithelial-cell-surface proteoglycans mediate virus adhesion, in turn initiating signal transduction as described for the GLYCOSYNAPSE54.
HSV-1 uses glycosaminoglycan as a receptor for entry into epithelial cells as well as into primary neuronal cells. There are five HSV-1 glycoproteins — gB, gC, gD, gH and gL — and two of these (gB and gC) mediate the attachment of the virus to cellular heparan sulphate47,63. Human herpesvirus 8 (HHV-8), which is associated with Kaposi's sarcoma, has a broad cellular tropism that includes epithelial cells. This broad tropism might be due, at least in part, to interaction of the viral surface glycoproteins with heparan sulphate64. Enveloped Rous sarcoma virus (RSV)65 and adenovirus-2 and -5 (Ref. 66), as well as several naked viruses including echovirus, AAV-2, human papilloma virus (HPV)-16, HPV-33 and FMDV (Refs 30,40,67–70), interact with heparan sulphate proteoglycans, which facilitates the attachment and infectivity of the virus. The use of heparan sulphate as an alternative receptor is likely to be the result of an adaptation to growth in cell lines71.
Paradoxically, the binding of a virus to heparan sulphate might prevent the virus from reaching the cell surface. Indeed, heparan sulphates are present in proteoglycans in the extracellular matrix, and these non-cellular matrix structures can bind viruses. The basal lamina, for example, is a barrier to the spread of HSV-1 from, and back into, epithelial cells. In addition, certain bodily fluids contain heparin, heparan sulphate and heparin-binding proteins, all of which can compete with and inhibit the binding of viruses to cell-surface heparan sulphate72.
Finally, the glycosylation state of epithelial-cell-surface proteins and lipids varies with the differentiation, ageing and activation of the cell, and such modulation of surface carbohydrates has an evident effect on the susceptibility of epithelial cells to viral infection54.
Protein receptors for attachment and infection
Several classes of protein receptor, which often show a polarized distribution, are used opportunistically by viruses to attach to and infect cells. A protein receptor either mediates these sequential steps by itself, or it has to cooperate with attachment receptors. As mentioned above, few studies describe the mechanism of such cooperativity.
Integrins. Integrins are a class of surface molecules that are used by several viruses (enveloped and naked) to attach to and infect epithelial cells. One class of integrin, the cellular role of which is to maintain cell–cell contact, is expressed basolaterally in cell culture and in gut tissue adjacent to the tight-junction complex on the basolateral pole. However, other integrins, such as α2β1, are expressed apically in crypt and villus ENTEROCYTES throughout the intestine.
Integrins that are expressed at the cell surface bind to ligands that are referred to as disintegrins. These disintegrins contain motifs of several amino acids that are specific to each integrin. One of these motifs — RGD (arginine–glycine–aspartic acid) — is specific to a set of integrins that are known as RGD-sensitive integrins. Disintegrin motifs are found in viral surface proteins, which allows the virus to bind integrins, and thereby interferes with the bona fide ligand.
A restricted set of RGD-sensitive integrins often seems to be used by viruses38,73. Several viruses bind RGD-sensitive integrins in an RGD-dependent manner74. For example, the RGD motif of the HHV-8 envelope glycoprotein B interacts with integrin-α3β1 to allow attachment of HHV-8 to (and the infection of) epithelial cells and use of the integrin signalling pathway75. The RGD-containing capsid protein VP1 of FMDV attaches to the integrin-αvβ6, which is expressed on primary epithelial cells, then uses the signalling pathways that are initiated at the integrin-β6 cytoplasmic domain71. The RGD-containing capsid protein VP1 of human parechovirus 1 (HPEV-1) also interacts with integrins — αvβ3 and αvβ1 — on the epithelial cell surface, but HPEV-1 then enters the host cell through the clathrin-dependent endocytic pathway38.
As an alternative, viral proteins might use other motifs to bind RGD-sensitive integrins in an RGD-independent fashion (that is, the viral protein does not act as a physiological counter-ligand). For example, rotaviral VP4 protein binds the RGD-sensitive integrin-α2β1, which is basolaterally expressed (or, alternatively, it binds integrin-αvβ3), as a post-attachment receptor through the GDE(A) (glycine–aspartic acid–glutamic acid/alanine) amino-acid motif of the viral protein73,76,77. When both of these integrins are expressed at the epithelial-cell surface, they work together to promote viral entry78. The rotaviral VP7 protein interacts with integrin-αxβ2 through a GRP (glycine–arginine–proline) motif, and integrin-α4β1 through an LVD (leucine–valine–aspartic acid) amino-acid motif77. Direct interaction between VP4 and VP7 has been observed.
Adenoviruses use also integrin-αv as a receptor to mediate their endocytosis, but binding of the virus to the integrin activates a signalling pathway that is distinct from the physiological one79. Echoviruses bind to integrin-α2β1, and during viral entry, caveolin 1 and integrin-α2β1 co-localize with E-1 capsid proteins and migrate into the perinuclear area in the cell80.
Cell-adhesion molecules. Other adhesion molecules, such as intercellular adhesion molecule 1 (ICAM1), function as viral receptors on epithelial cells. The major group of HRV uses ICAM1 as the viral receptor81. HRVs have a cleft that encircles the fivefold axes of icosahedral symmetry, which accommodates the amino-terminal domain of ICAM1.
Cell-junction-associated proteins. Another widely used class of receptor consists of components of the epithelial-cell tight junctions. The use of such receptors to penetrate epithelial-cell cytosol at the epithelial cell–cell junction implies that the junction complex is disrupted, with an immediate effect on the integrity of the epithelial barrier. Whereas the interaction of viral components with components of the epithelial tight junction has been described biochemically69, how viruses access such receptors — which are 'hidden' in the tight junction in vivo — is difficult to clarify, despite recent studies on cell lines17.
Two cell-surface glycoproteins — the coxsackievirus and adenovirus receptor (CAR) and the junction-adhesion molecule (JAM) — were identified as transmembrane components of the tight junction in epithelial cells, as well as being entry receptors for coxsackie virus/adenovirus and reovirus, respectively69,82. Owing to the localization of CAR at the tight junction, infection by both adenovirus and coxsackievirus in vivo could require the destruction of the tight-junction complex for the virus to be able to access its receptor, as is the case in vitro82. Such destruction might be involved in regulating tissue-specific inflammatory responses to viral infection69. Coxsackievirus binds CAR through a 'canyon' at the surface of the virus. By contrast, the terminal knob portion of the fibre protein of human adenovirus (HAdV)-2 (Ref. 68) binds to CAR. This allows the viral capsid penton base protein to bind cell-surface integrin-αvβ5 and its subsequent endocytosis in clathrin-coated vesicles79.
Nectin 1 (also known as poliovirus-related protein 1 or HveC) is a Ca2+-independent cell-adhesion molecule that is localized at cadherin-based intercellular junctions. It is used by α-HSV-1 and α-HSV-2 for entry and infection83 after attachment to epithelial-cell heparan sulphate proteoglycan. In reovirus infection, human JAM — an integral membrane protein that organizes the tight junctions of epithelial cells — binds the head domain of the viral σ1 outer-capsid protein. It binds at the basolateral (serosal) pole of the epithelial layer, after reovirus has undergone transcytosis across M cells11. JAM functions as a serotype-independent receptor that can mediate virus attachment and infection69.
Other classes of polarized receptor. Several protein receptors have a polarized distribution, which allows viral entry into epithelial cells, or cell resistance to infection when a virus enters epithelial cells by the opposite pole. Apical viral receptors include the carcino-embryonic antigen family84,85, CD46 (Ref. 13; together with its co-receptor epithelial moesin), and the N-aminopeptidase CD13 (Refs 12,84), which can dimerize and probably transduce signals. The glycosylphosphatidylinositol (GPI)-anchored complement regulatory protein DAF (decay acceleration factor) is widely used by enteroviruses, including echoviruses, human enterovirus (EV)-70, coxsackievirus types B and A21 (Ref. 45), as well as other GPI-anchored proteins. These GPI-anchored proteins, which are localized in apical rafts, can be endocytosed by a Rho-GTPase-dependent mechanism (Fig. 1). So, as shown recently for E-11, they can mediate internalization of the virus in the endosome before translocation into the epithelial-cell cytosol45. Molecules that are expressed basolaterally include the transferrin receptor. By contrast, viruses can use non-polarized protein receptors27, and so they need a polarized carbohydrate co-receptor for entry.
Conclusions and perspectives
The interaction between a cellular host and a viral pathogen is an important field of research, both for unravelling polarized membrane trafficking86 and for understanding epithelial pathology (for example, see Fig. 3), with obvious significance for designing anti-viral strategies.

During the first cycle of rotavirus replication in mucosal epithelial cells, the synthesis of rotaviral proteins in the cell cytoplasm leads to an increase in the plasma-membrane permeability to Ca2+, to activation of regulatory mechanisms and to an increase in the concentration of Ca2+ in the endoplasmic reticulum (ER). The increased concentration of cytosolic Ca2+ in infected cells promotes the activation of Ca2+-dependent enzymes, which in turn induces cell lysis and the release of viral proteins and viral progeny. Non-structural protein (NSP)-4 might act as a viral enterotoxin on as-yet-uninfected cells to induce secretory diarrhoea through a | Ca2+-dependent secretion by intestinal cells, b | Ca2+-dependent secretion of peptides and amines to stimulate the enteric nervous system (ENS), and c | further activation of epithelial-cell chloride (Cl−) secretion by the ENS. In parallel, released virus infects downstream absorptive cells. This will lead to a massive cell death and, as a consequence, reduction of the absorptive surface of the intestinal epithelium and an osmotic component of diarrhoea42. Various forms of the virus along the rotavirus-maturation pathway are shown: DLP, double-layer particle; IMP, intramembrane particle; Ins(1,4,5)P3, inositol 1,4,5-trisphosphate; PLC, phospholipase C; SERCA, sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase; TLP, triple-layer particle.
It seems clear that no real correlation can be made between a family (or even a type) of virus and a defined mechanism of interaction with the epithelial cell. Viruses from families as different as naked and enveloped viruses can use the same attachment receptors83,87 (for example, carbohydrates or proteins). Conversely, closely related viruses from the same family might use completely different attachment receptors that will dictate the intracellular fate of the virus41,81. Even the same virus might use the same attachment receptor but different protein receptors depending on the type of epithelial cell, which extends its infectious potential47.
The cellular features of the epithelial target therefore seem to be essential if we are to describe the mechanisms of a viral interaction with an epithelial cell. These features are the epithelial trafficking pathways and the epithelial-specific protein and lipid composition that are associated with their signalling pathways. This description emphasizes how viruses act as pathogens — that is, by subverting the normal epithelial cell function to their own benefit.
We are just beginning to understand the molecular interactions between viral and cellular components. However, the factors that control the finely regulated specificity that is observed in vivo for each virus, for a defined target cell, remain unclear. The signalling that is induced when a virus binds its receptors should be compared with the pathways that are activated when the natural ligands bind these same receptors. Entry receptors for many viruses have now been described, and it will be important to correlate data from both cell lines and PRIMARY CELLS, despite the pitfalls inherent in each system (Box 3). In this regard, studies that use the cell type directly involved in the pathology — rather than receptor molecules transfected in standard cell-line models — could help. It will also be important to define the environmental parameters that influence binding of the virus to its receptors, and interaction with co-receptors, especially when considering lectin–sugar interactions. The use of new techniques that involve dynamic fluorescence microscopy will help, both for mapping the viral pathway inside epithelial cells and also for studying signalling in epithelial cells.
The first step of viral attachment to the cell — through either proteoglycan or glycosphingolipids — raises the question of the specific role of the glycosyl epitope, not only in viral attachment, but also in further transduction of signals. The final glycosylation state of glycoproteins and lipids varies with cell differentiation, maturation or ageing54, and it could govern the specificity of the virus–carbohydrate interaction that is involved in viral entry into epithelial cells. Lipid microdomains participate in the clustering of viral proteins on the viral membrane and cell receptors on the cell membranes, which favours virus–cell interactions. The spatio-temporal correlation of the different steps of the virus attachment to carbohydrates of the cell surface, and their further interaction with additional protein cell receptors or co-receptors, remains to be established.
Clearly, epithelia are more than just a physical barrier. They are a dynamic host defence system with sensor molecules, signalling circuits and effector molecules that coordinate and execute a graduated reaction to microbes. We are now on the verge of learning more about the molecules and pathways that are involved in these epithelial responses.


References
Gabius, H., Andre, S., Kaltner, H. & Siebert, H. The sugar code: functional lectinomics. Biochim. Biophys. Acta 19, 2–3 (2002).
Compans, R. W. Virus entry and release in polarized epithelial cells. Curr. Top. Microbiol. Immunol. 202, 209–219 (1995).
Bomsel, M. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nature Med. 3, 42–47 (1997). This reports the characterization of a novel pathway for HIV transmission at mucosal sites.
Meng, G. et al. Primary intestinal epithelial cells selectively transfer R5 HIV-1 to CCR5+ cells. Nature Med. 8, 150–156 (2002). An ex vivo system using human primary epithelial cells to show the selective filter function of an epithelial barrier during a viral infection.
Smith, G. A. & Enquist, L. W. Break ins and break outs: viral interactions with the cytoskeleton of mammalian cells. Annu. Rev. Cell Dev. Biol. 18, 135–161 (2002).
Gan, Y. J., Chodosh, J., Morgan, A. & Sixbey, J. W. Epithelial cell polarization is a determinant in the infectious outcome of immunoglobulin A-mediated entry by Epstein–Barr virus. J. Virol. 71, 519–526 (1997).
Eliopoulos, A. G. et al. Epstein–Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-κB pathway involving TNF receptor-associated factors. Oncogene 14, 2899–2916 (1997).
Desgranges, C. et al. Detection of Epstein–Barr viral DNA internal repeats in the nasopharyngeal mucosa of Chinese with IgA/EBV-specific antibodies. Int. J. Cancer 29, 87–91 (1982).
Iwasaki, A. et al. Immunofluorescence analysis of poliovirus receptor expression in Peyer's patches of humans, primates, and CD155 transgenic mice: implications for poliovirus infection. J. Infect. Dis. 186, 585–592 (2002).
Fotopoulos, G. et al. Transepithelial transport of HIV-1 by M cells is receptor-mediated. Proc. Natl Acad. Sci. USA 99, 9410–9414 (2002).
Wolf, J. L. et al. Intestinal M cells: a pathway for entry of reovirus into the host. Science 212, 471–472 (1981).
Wang, G. Human coronavirus 229E infects polarized airway epithelia from the apical surface. J. Virol. 74, 9234–9239 (2000).
Blau, D. M. & Compans, R. W. Entry and release of measles virus are polarized in epithelial cells. Virology 210, 91–99 (1995).
Ravkov, E. V., Nichol, S. T. & Compans, R. W. Polarized entry and release in epithelial cells of Black Creek Canal virus, a New World hantavirus. J. Virol. 71, 1147–1154 (1997).
Jarvis, M. A. et al. Human cytomegalovirus infection of caco-2 cells occurs at the basolateral membrane and is differentiation state dependent. J. Virol. 73, 4552–4560 (1999).
Spear, P. G. A first step toward understanding membrane fusion induced by herpes simplex virus. Mol. Cell 8, 2–4 (2001).
Yoon, M. & Spear, P. G. Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J. Virol. 76, 7203–7208 (2002).
Irurzun, A. & Carrasco, L. Entry of poliovirus into cells is blocked by valinomycin and concanamycin A. Biochemistry 40, 3589–3600 (2001).
Kronenberger, P. et al. Uptake of poliovirus into the endosomal system of HeLa cells. Arch. Virol. 143, 1417–1424 (1998).
Ciarlet, M. & Estes, M. K. Interactions between rotavirus and gastrointestinal cells. Curr. Opin. Microbiol. 4, 435–441 (2001).
Hughes, M. T., Matrosovich, M., Rodgers, M. E., McGregor, M. & Kawaoka, Y. Influenza A viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J. Virol. 74, 5206–5212 (2000).
Blank, C. A., Anderson, D. A., Beard, M. & Lemon, S. M. Infection of polarized cultures of human intestinal epithelial cells with hepatitis A virus: vectorial release of progeny virions through apical cellular membranes. J. Virol. 74, 6476–6484 (2000).
Carfi, A. et al. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8, 169–179 (2001).
Topp, K. S., Bisla, K., Saks, N. D. & Lavail, J. H. Centripetal transport of herpes simplex virus in human retinal pigment epithelial cells in vitro. Neuroscience 71, 1133–1144 (1996).
Bodaghi, B. et al. Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosis. Invest. Ophthalmol. Vis. Sci. 40, 2598–2607 (1999).
Tugizov, S., Maidji, E. & Pereira, L. Role of apical and basolateral membranes in replication of human cytomegalovirus in polarized retinal pigment epithelial cells. J. Gen. Virol. 77, 61–74 (1996).
Topp, K. S., Rothman, A. L. & Lavail, J. H. Herpes virus infection of RPE and MDCK cells: polarity of infection. Exp. Eye Res. 64, 343–354 (1997).
Duan, D., Yue, Y., Yan, Z., McCray, P. B. Jr & Engelhardt, J. F. Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum. Gene Ther. 9, 2761–2776 (1998).
Strous, G. J. & Govers, R. The ubiquitin–proteasome system and endocytosis. J. Cell Sci. 112, 1417–1423 (1999).
Duan, D., Yue, Y., Yan, Z., Yang, J. & Engelhardt, J. F. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest. 105, 1573–1587 (2000).
Bavari, S. et al. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195, 593–602 (2002).
Manie, S. N., Debreyne, S., Vincent, S. & Gerlier, D. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J. Virol. 74, 305–311 (2000).
Simons, K. & Toomre, D. Lipid rafts and signal transduction. Nature Rev. Mol. Cell Biol. 1, 31–39 (2000).
Norkin, L. C., Anderson, H. A., Wolfrom, S. A. & Oppenheim, A. Caveolar endocytosis of simian virus 40 is followed by Brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 76, 5156–5166 (2002).
Stang, E., Kartenbeck, J. & Parton, R. G. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell 8, 47–57 (1997).
Pho, M. T., Ashok, A. & Atwood, W. J. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 74, 2288–2292 (2000).
Parker, J. S., Murphy, W. J., Wang, D., O'Brien, S. J. & Parrish, C. R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 75, 3896–3902 (2001).
Joki-Korpela, P., Marjomaki, V., Krogerus, C., Heino, J. & Hyypia, T. Entry of human parechovirus 1. J. Virol. 75, 1958–1967 (2001).
DeTulleo, L. & Kirchhausen, T. The clathrin endocytic pathway in viral infection. EMBO J. 17, 4585–4593 (1998).
Fry, E. E. et al. The structure and function of a foot-and-mouth disease virus–oligosaccharide receptor complex. EMBO J. 18, 543–554 (1999).
Schober, D., Kronenberger, P., Prchla, E., Blaas, D. & Fuchs, R. Major and minor receptor group human rhinoviruses penetrate from endosomes by different mechanisms. J. Virol. 72, 1354–1364 (1998). This work tracks the entry of comparatively different viruses of the same family into cells by cell-fractionation analysis.
Ruiz, M. C., Cohen, J. & Michelangeli, F. Role of Ca2+ in the replication and pathogenesis of rotavirus and other viral infections. Cell Calcium 28, 137–149 (2000). A clear review on the role of calcium in viral infection of the epithelial cell, and the viral soluble virulence factors.
Knodler, L. A., Celli, J. & Finlay, B. B. Pathogenic trickery: deception of host cell processes. Nature Rev. Mol. Cell Biol. 2, 578–588 (2001).
Weis, W. I. & Drickamer, K. Structural basis of lectin–carbohydrate recognition. Annu. Rev. Biochem. 65, 441–473 (1996). An important review on the characteristics of lectin–carbohydrate interactions that rely on the structure and oligomerization state of the lectin peptide, as well as on the nature and conformation of the carbohydrate.
Stuart, A. D., Eustace, H. E., McKee, T. A. & Brown, T. D. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. J. Virol. 76, 9307–9322 (2002).
Dimitrov, D. S. Cell biology of virus entry. Cell 101, 697–702 (2000).
Shukla, D. & Spear, P. G. Herpesviruses and heparansulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108, 503–510 (2001). Reference 47 uses herpesviruses to emphasize the role of heparan sulphate — a ubiquitous component of cell proteoglycans — in viral entry into host cells.
Skehel, J. J. & Wiley, D. C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 (2000).
Suzuki, S. et al. Broad distribution of the JC virus receptor contrasts with a marked cellular restriction of virus replication. Virology 286, 100–112 (2001).
Markwell, M. A. & Paulson, J. C. Sendai virus utilizes specific sialyloligosaccharides as host cell receptor determinants. Proc. Natl Acad. Sci. USA 77, 5693–5697 (1980).
Delorme, C. et al. Glycosphingolipid binding specificities of rotavirus: identification of a sialic acid-binding epitope. J. Virol. 75, 2276–2287 (2001). Among the various ganglioside specificities for different rotavirus strains, a common carbohydrate minimal structural element is shown to be required for binding of these strains.
Barton, E. S., Connolly, J. L., Forrest, J. C., Chappell, J. D. & Dermody, T. S. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276, 2200–2211 (2001). Using naked reovirus as an example, this paper shows the cooperativity of carbohydrate attachment and protein receptors for virus interaction with epithelial cells.
van der Bijl, P., Lopes-Cardozo, M. & van Meer, G. Sorting of newly synthesized glycosphingolipids to the two surface domains of epithelial cells. J. Cell Biol. 132, 813–821 (1996).
Hakomori, S. I. Inaugural article: the glycosynapse. Proc. Natl Acad. Sci. USA 99, 225–232 (2002). The cell-to-cell transfer of information through the synapse, which is characteristic of neural cells, has recently been extended to other cell types such as immunological synapses. The importance of carbohydrates in such cellular functions is underlined.
Raulin, J. Human immunodeficiency virus and host cell lipids. Interesting pathways in research for a new HIV therapy. Prog. Lipid Res. 41, 27–65 (2002).
Alfsen, A., Iniguez, P., Bouguyon, E. & Bomsel, M. Secretory IgA specific for a conserved epitope on gp41 envelope glycoprotein inhibits epithelial transcytosis of HIV-1. J. Immunol. 166, 6257–6265 (2001).
Alfsen, A. & Bomsel, M. HIV-1 gp41 envelope residues 650–685 exposed on native virus act as a lectin to bind epithelial cell galactosyl ceramide. J. Biol. Chem. 277, 25649–25659 (2002).
Fantini, J. et al. Synthetic soluble analogs of galactosylceramide (GalCer) bind to the V3 domain of HIV-1 gp120 and inhibit HIV-1-induced fusion and entry. J. Biol. Chem. 272, 7245–7452 (1997).
Simons, K. A. & van Meer, G. Lipid sorting in epithelial cells. Biochemistry 27, 6197–6202 (1988).
Mahfoud, R. et al. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J. Biol. Chem. 277, 11292–11296 (2002).
Hallak, L. K., Spillmann, D., Collins, P. L. & Peeples, M. E. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J. Virol. 74, 10508–10513 (2000).
Turnbull, J., Powell, A. & Guimond, S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 11, 75–82 (2001). An informative review on the structure and function of heparan sulphate.
Immergluck, L. C., Domowicz, M. S., Schwartz, N. B. & Herold, B. C. Viral and cellular requirements for entry of herpes simplex virus type 1 into primary neuronal cells. J. Gen. Virol. 79, 549–559 (1998).
Wang, F. Z., Akula, S. M., Pramod, N. P., Zeng, L. & Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 75, 7517–7527 (2001).
Feldman, S. A., Audet, S. & Beeler, J. A. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74, 6442–6447 (2000).
Dechecchi, M. C. et al. Heparan sulfate glycosaminoglycans are receptors sufficient to mediate the initial binding of adenovirus types 2 and 5. J. Virol. 75, 8772–8780 (2001).
Goodfellow, I. G., Sioofy, A. B., Powell, R. M. & Evans, D. J. Echoviruses bind heparan sulfate at the cell surface. J. Virol. 75, 4918–4921 (2001).
Summerford, C. & Samulski, R. J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72, 1438–1445 (1998).
Barton, E. S. et al. Junction adhesion molecule is a receptor for reovirus. Cell 104, 441–451 (2001).
Giroglou, T., Florin, L., Schafer, F., Streeck, R. E. & Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75, 1565–1570 (2001).
Miller, L. C. et al. Role of the cytoplasmic domain of the β-subunit of integrin α(v)β6 in infection by foot-and-mouth disease virus. J. Virol. 75, 4158–4164 (2001).
WuDunn, D. & Spear, P. G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63, 52–58 (1989).
Guerrero, C. A. et al. Integrin α(v)β(3) mediates rotavirus cell entry. Proc. Natl Acad. Sci. USA 97, 14644–14649 (2000).
Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715 (1996).
Akula, S. M., Pramod, N. P., Wang, F. Z. & Chandran, B. Integrin lpha;3β1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108, 407–419 (2002).
Ciarlet, M. et al. VLA-2 (α2β1) integrin promotes rotavirus entry into cells but is not necessary for rotavirus attachment. J. Virol. 76, 1109–1123 (2002).
Coulson, B. S., Londrigan, S. L. & Lee, D. J. Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc. Natl Acad. Sci. USA 94, 5389–5394 (1997).
Mendez, E., Lopez, S., Cuadras, M. A., Romero, P. & Arias, C. F. Entry of rotaviruses is a multistep process. Virology 263, 450–459 (1999).
Li, E., Stupack, D., Klemke, R., Cheresh, D. A. & Nemerow, G. R. Adenovirus endocytosis via α(v) integrins requires phosphoinositide-3-OH kinase. J. Virol. 72, 2055–2061 (1998).
Marjomaki, V. et al. Internalization of echovirus 1 in caveolae. J. Virol. 76, 1856–1865 (2002).
Reischl, A. et al. Viral evolution toward change in receptor usage: adaptation of a major group of human rhinovirus to grow in ICAM-1-negative cells. J. Virol. 75, 9312–9319 (2001). The ability of a group of viruses to adapt to different cell receptors seems, in part, to be responsible for the difficulties in protecting a specific type of cell from viral entry.
Cohen, C. J. et al. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl Acad. Sci. USA 98, 15191–15196 (2001). This characterization of the cellular function of the coxsackievirus and adenovirus receptor (CAR) sheds light on the polarity of the entry of these viruses into epithelial cells.
Geraghty, R. J., Krummenacher, C., Cohen, G. H., Eisenberg, R. J. & Spear, P. G. Entry of α-herpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280, 1618–1620 (1998).
Rossen, J. W., Horzinek, M. C. & Rottier, P. J. Coronavirus infection of polarized epithelial cells. Trends Microbiol. 3, 486–490 (1995).
Lewicki, D. N. & Gallagher, T. M. Quaternary structure of coronavirus spikes in complex with carcinoembryonic antigen-related cell adhesion molecule cellular receptors. J. Biol. Chem. 277, 19727–19734 (2002).
Rodriguez-Boulan, E. & Sabatini, D. D. Assymetric budding of viruses in epithelial cells: a model system for study of epithelial polarity. Proc. Natl Acad. Sci. USA 75, 5071–5075 (1978).
Yongning, H. et al. Interaction of the poliovirus receptor with poliovirus. Proc. Natl Acad. Sci. USA 97, 79–84 (2000).
Apodaca, G. Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic 2, 149–159 (2001).
Sabharanjak, S., Sharma, P., Parton, R. G. & Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2, 411–423 (2002).
Kerneis, S., Bogdanova, A., Kraehenbuhl, J. P. & Pringault, E. Conversion by Peyer's patch lymphocytes of human enterocytes into M cells that transport bacteria. Science 277, 949–952 (1997).
Phillips, D. M. The role of cell-to-cell transmission in HIV infection. AIDS 8, 719–731 (1994).
Imai, S., Nishikawa, J. & Takada, K. Cell-to-cell contact as an efficient mode of Epstein–Barr virus infection of diverse human epithelial cells. J. Virol. 72, 4371–4378 (1998).
Smith, P. D., Meng, G., Sellers, M. T., Rogers, T. S. & Shaw, G. M. Biological parameters of HIV-1 infection in primary intestinal lymphocytes. J. Leukoc. Biol. 68, 360–365 (2000).
Agace, W. W. et al. Constitutive expression of stromal derived factor-1 by mucosal epithelia. Curr. Biol. 10, 325–328 (2000).
Elliott, G. & O'Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 88, 223–233 (1997).
Hayashi, K. Role of tight junctions of polarized epithelial MDCK cells in the replication of herpes simplex virus type 1. J. Med. Virol. 47, 323–329 (1995).
Ohara, P. T., Chin, M. S. & La Vail, J. H. The spread of herpes simplex virus type 1 from trigeminal neurons to the murine cornea: an immunoelectron microscopy study. J. Virol. 74, 4776–4786 (2000).
Miyazaki, D. et al. Neutrophil chemotaxis induced by corneal epithelial cells after herpes simplex virus type 1 infection. Curr. Eye Res. 17, 687–693 (1998).
Esclatine, A., Lemullois, M., Servin, A. L., Quero, A. M. & Geniteau-Legendre, M. Human cytomegalovirus infects Caco-2 intestinal epithelial cells basolaterally regardless of the differentiation state. J. Virol. 74, 513–517 (2000).
Chodosh, J., Gan, Y., Holder, V. P. & Sixbey, J. W. Patterned entry and egress by Epstein–Barr virus in polarized CR2-positive epithelial cells. Virology 266, 387–396 (2000).
Molesworth, S. J., Lake, C. M., Borza, C. M., Turk, S. M. & Hutt-Fletcher, L. M. Epstein–Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74, 6324–6332 (2000).
Rodriguez, D., Rodriguez, J. R., Ojakian, G. K. & Esteban, M. Vaccinia virus preferentially enters polarized epithelial cells through the basolateral surface. J. Virol. 65, 494–498 (1991).
Hammache, D. et al. Specific interaction of HIV-1 and HIV-2 surface envelope glycoproteins with monolayers of galactosylceramide and ganglioside GM3. J. Biol. Chem. 273, 7967–7971 (1998).
Saito, T. et al. Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J. Infect. Dis. 175, 497–504 (1997).
Techaarpornkul, S., Collins, P. L. & Peeples, M. E. Respiratory syncytial virus with the fusion protein as its only viral glycoprotein is less dependent on cellular glycosaminoglycans for attachment than complete virus. Virology 294, 296–304 (2002).
Fujioka, H. et al. Immunocytochemical colocalization of specific immunoglobulin A with Sendai virus protein in infected polarized epithelium. J. Exp. Med. 188, 1223–1229 (1998).
Maisner, A., Klenk, H. & Herrler, G. Polarized budding of measles virus is not determined by viral surface glycoproteins. J. Virol. 72, 5276–5278 (1998).
Naim, H. Y., Ehler, E. & Billeter, M. A. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 19, 3576–3585 (2000).
Evander, M. et al. Identification of the α6 integrin as a candidate receptor for papillomaviruses. J. Virol. 71, 2449–2456 (1997).
Zhang, J., Pekosz, A. & Lamb, R. A. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 74, 4634–4644 (2000).
Zimmer, G., Zimmer, K. P., Trotz, I. & Herrler, G. Vesicular stomatitis virus glycoprotein does not determine the site of virus release in polarized epithelial cells. J. Virol. 76, 4103–4107 (2002).
Brunet, J. P. et al. Rotavirus infection induces cytoskeleton disorganization in human intestinal epithelial cells: implication of an increase in intracellular calcium concentration. J. Virol. 74, 10801–10806 (2000).
Casola, A. et al. Rotavirus infection of cultured intestinal epithelial cells induces secretion of CXC and CC chemokines. Gastroenterology 114, 947–955 (1998).
Barton, E. S., Chappell, J. D., Connolly, J. L., Forrest, J. C. & Dermody, T. S. Reovirus receptors and apoptosis. Virology 290, 173–180 (2001).
Chappell, J. D., Duong, J. L., Wright, B. W. & Dermody, T. S. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 74, 8472–8479 (2000).
Pickles, R. J., Fahrner, J. A., Petrella, J. M., Boucher, R. C. & Bergelson, J. M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 74, 6050–6057 (2000).
Walters, R. W. et al. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem. 274, 10219–10226 (1999).
Kolatkar, P. R. et al. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18, 6249–6259 (1999).
Bayer, N., Schober, D., Huttinger, M., Blaas, D. & Fuchs, R. Inhibition of clathrin-dependent endocytosis has multiple effects on human rhinovirus serotype 2 cell entry. J. Biol. Chem. 276, 3952–3962 (2001).
Golovkina, T. V. et al. A novel membrane protein is a mouse mammary tumor virus receptor. J. Virol. 72, 3066–3071 (1998).
Rossen, J. W. et al. Coronaviruses in polarized epithelial cells. Adv. Exp. Med. Biol. 380, 135–138 (1995).
Lin, X., O'Reilly, K. L. & Storz, J. Infection of polarized epithelial cells with enteric and respiratory tract bovine coronaviruses and release of virus progeny. Am. J. Vet. Res. 58, 1120–1124 (1997).
Rossen, J. W., Strous, G. J., Horzinek, M. C. & Rottier, P. J. Mouse hepatitis virus strain A59 is released from opposite sides of different epithelial cell types. J. Gen. Virol. 78, 61–69 (1997).
Ciarlet, M., Isa, P., Conner, M. E. & Liprandi, F. Antigenic and molecular analyses reveal that the equine rotavirus strain H-1 is closely related to porcine, but not equine, rotaviruses: interspecies transmission from pigs to horses? Virus Genes 22, 5–20 (2001).
Acknowledgements
We apologize to our colleagues for omitting references in this review because of space limitations. We thank B. Wecksler and V. David for their editing of this manuscript. This work was supported by Agence Nationale de Recherche sur le Sida (ANRS) and SIDACTION/Ensemble Contre le SIDA funds to M.B.
Author information
Authors and Affiliations
Corresponding author
Related links
Related links
DATABASES
Entrez
Swiss-Prot
Glossary
- PROTEOGLYCAN
-
An acidic macromolecule that is composed of glycosaminoglycan chains attached covalently to a protein core. Proteoglycans are found in the extracellular matrix, cell surfaces, and intracellular vesicles.
- 'RAFT' MEMBRANE MICRODOMAIN
-
A dynamic assembly of cholesterol and sphingolipids in the plasma membrane that is probably involved in cell signalling.
- GLYCOSPHINGOLIPIDS
-
A highly polymorphic class of lipids with a common hydrophobic backbone — ceramide — that are composed of a fatty-acid chain linked to the sphingosine base and a hydrophilic oligosaccharide residue that protrudes into the extracellular space.
- LECTIN
-
A cell-agglutinating protein of non-immune origin, which binds carbohydrates without modifying them.
- TRANSCYTOSIS
-
A rapid and selective vesicular transcellular pathway that is characteristic of polarized epithelia. Cargo is transported from one pole of the cell to the opposite pole. The cargo remains enclosed in transcytotic vesicles, which precludes access to the cytosol and therefore viral infection of epithelial cells.
- POLY-IMMUNOGLOBULIN RECEPTOR
-
This receptor is expressed at the basolateral surface of epithelial cells, allowing specific transcytosis towards the apical pole of mucosal dimeric IgA or pentameric IgM. At the apical pole, after cleavage of the extracellular region of the receptor, which is known as secretory component (SC), the mucosal IgA or IgM is released with SC as secretory IgA or IgM, and can act as the first defence against pathogens.
- M CELL
-
'Membranous' or 'microfold' cell. This is a specialized epithelial cell covering the lymphoid Peyer's patches in the gut. M cells can internalize macromolecules and microorganisms and deliver them to the underlying lymphoid tissue.
- ENDOSOME
-
A membranous transport vesicle that is involved in endocytosis.
- TIGHT JUNCTION
-
A protein heterocomplex that connects neighbouring simple epithelial cells and controls the barrier function of the tight mucosal surface.
- CLATHRIN-COATED VESICLE/PIT
-
An invagination of the plasma membrane that is surrounded by clathrin, a cytosolic protein that is formed by a triskelion of three heavy and three light chains. Triskelions assemble into a polyhedral lattice to form the clathrin coat.
- MONONUCLEAR CELLS
-
Lymphocytes, dendritic cells and monocytes/macrophages. These are usually found in the blood, but also in tissues.
- LAMINA PROPRIA
-
(chorion). This is formed of conjunctive tissue that is traversed by blood and lymphoid vessels. It supports epithelial cells through the basal membrane.
- CAVEOLAE
-
Flask-shaped, cholesterol-rich invaginations of the plasma membrane that contain the protein caveolin. They might mediate the uptake of some extracellular material.
- SIALYLOLIGOSACCHARIDE
-
An oligosaccharide chain that is linked to a terminal sialic acid (N-acetyl neuraminic acid).
- GLYCOSAMINOGLYCAN
-
The polysaccharide moiety of proteoglycans, which is added posttranslationally and is composed of repeating disaccharide units.
- HEPARAN SULPHATE
-
One of the glycosaminoglycan parts of proteoglycans, this is a long, polyanionic carbohydrate chain that consists of a repeating disaccharide unit.
- GLYCOSYNAPSE
-
A membrane structure that provides a connection between two cells, and is involved in a glycosylation-dependent cell-adhesion/recognition processes.
- ENTEROCYTE
-
An intestinal epithelial cell that is organized in monostratified layers.
- PRIMARY CELL
-
A cell that is isolated directly from living tissues instead of transformed cells.
Rights and permissions
About this article
Cite this article
Bomsel, M., Alfsen, A. Entry of viruses through the epithelial barrier: pathogenic trickery. Nat Rev Mol Cell Biol 4, 57–68 (2003). https://doi.org/10.1038/nrm1005
Issue Date:
DOI: https://doi.org/10.1038/nrm1005
This article is cited by
-
LDLR, LRP1, and Megalin redundantly participate in the uptake of Clostridium novyi alpha-toxin
Communications Biology (2022)
-
Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells
Nature Microbiology (2019)
-
Broadly neutralizing antibodies suppress post-transcytosis HIV-1 infectivity
Mucosal Immunology (2017)
-
CD4-mimetic sulfopeptide conjugates display sub-nanomolar anti-HIV-1 activity and protect macaques against a SHIV162P3 vaginal challenge
Scientific Reports (2016)
-
Beyond RGD: virus interactions with integrins
Archives of Virology (2015)
