
Key Points
-
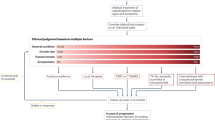
Over half of all phaeochromocytoma and/or paraganglioma (PPGL) can be attributed to genetic alterations
-
80% of inherited PPGLs are caused by a germline mutation in VHL or the SDHx group of genes
-
Genetic testing is indicated for all patients with PPGLs, as identification of the underlying mutation guides patient management and genetic counselling
-
Gene expression profiling can be used to classify PPGLs, assigning them to either an angiogenic cluster or a kinase signalling cluster, each of which has specific treatment targets
-
DNA methylation profiling has revealed a hypermethylated cluster that is specific to tumours related to the SDHx genes and FH that accounts for the malignant and noradrenergic phenotype of tumours related to mutations in these genes
-
Genomic studies of PPGLs are generating important findings that should pave the way for personalized medicine for affected patients
Abstract
Paragangliomas and phaeochromocytomas are neuroendocrine tumours whose pathogenesis and progression are very strongly influenced by genetics. A germline mutation in one of the susceptibility genes identified so far explains ∼40% of all cases; the remaining 60% are thought to be sporadic cases. At least one-third of these sporadic tumours contain a somatic mutation in a predisposing gene. Genetic testing, which is indicated in every patient, is guided by the clinical presentation as well as by the secretory phenotype and the immunohistochemical characterization of the tumours. The diagnosis of an inherited form drives clinical management and tumour surveillance. Different 'omics' profiling methods have provided a neat classification of these tumours in accordance with their genetic background. Transcriptomic studies have identified two main molecular pathways that underlie development of these tumours, one in which the hypoxic pathway is activated (cluster 1) and another in which the MAPK and mTOR (mammalian target of rapamycin) signalling pathways are activated (cluster 2). DNA methylation profiling has uncovered a hypermethylator phenotype in tumours related to SDHx genes (a group of genes comprising SDHA, SDHB, SDHC, SDHD and SDHAF2) and revealed that succinate acts as an oncometabolite, inhibiting 2-oxoglutarate-dependent dioxygenases, such as hypoxia-inducible factor prolyl-hydroxylases and histone and DNA demethylases. 'Omics' data have suggested new therapeutic targets for patients with a malignant tumour. In the near future, new 'omics'-based tests are likely to be transferred into clinical practice with the goal of establishing personalized medical management for affected patients.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Baysal, B. E. et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000).
Astuti, D. et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet 357, 1181–1182 (2001).
Dluhy, R. G. Pheochromocytoma—death of an axiom. N. Engl. J. Med. 346, 1486–1488 (2002).
Pacak, K. et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat. Clin. Pract. Endocrinol. Metab. 3, 92–102 (2007).
Dahia, P. L. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat. Rev. Cancer 14, 108–119 (2014).
Buffet, A. et al. A decade (2001–2010) of genetic testing for pheochromocytoma and paraganglioma. Horm. Metab. Res. 44, 359–366 (2012).
Viskochil, D. et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62, 187–192 (1990).
Gutmann, D. H. et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278, 51–57 (1997).
Walther, M. M., Herring, J., Enquist, E., Keiser, H. R. & Linehan, W. M. von Recklinghausen's disease and pheochromocytomas. J. Urol. 162, 1582–1586 (1999).
Bausch, B. et al. Comprehensive mutation scanning of NF1 in apparently sporadic cases of pheochromocytoma. J. Clin. Endocrinol. Metab. 91, 3478–3481 (2006).
Bausch, B. et al. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N. Engl. J. Med. 354, 2729–2731 (2006).
Hersh, J. H. et al. Health supervision for children with neurofibromatosis. Pediatrics 121, 633–642 (2008).
Burnichon, N. et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum. Mol. Genet. 21, 5397–5405 (2012).
Mulligan, L. M. et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458–460 (1993).
Wagner, A. J. et al. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod. Pathol. 26, 289–294 (2013).
Brandi, M. L. et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab. 86, 5658–5671 (2001).
American Thyroid Association Guidelines Task Force. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19, 565–612 (2009).
Latif, F. et al. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science 260, 1317–1320 (1993).
Maher, E. R., Neumann, H. P. & Richard, S. von Hippel–Lindau disease: a clinical and scientific review. Eur. J. Hum. Genet. 19, 617–623 (2011).
Castro-Vega, L. J. et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 23, 2440–2446 (2014).
Amar, L. et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 23, 8812–8818 (2005).
Lenders, J. W. et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 99, 1915–1942 (2014).
Niemann, S. & Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 26, 268–270 (2000).
Burnichon, N. et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 19, 3011–3020 (2010).
Astuti, D. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 49–54 (2001).
Hao, H. X. et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 (2009).
Pollard, P. J. et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 14, 2231–2239 (2005).
Briere, J. J. et al. Mitochondrial succinate is instrumental for HIF1α nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum. Mol. Genet. 14, 3263–3269 (2005).
Letouze, E. et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 739–752 (2013).
Baysal, B. E. Mitochondrial complex II and genomic imprinting in inheritance of paraganglioma tumors. Biochim. Biophys. Acta 1827, 573–577 (2013).
Pasini, B. & Stratakis, C. A. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma–paraganglioma syndromes. J. Intern. Med. 266, 19–42 (2009).
Gimenez-Roqueplo, A. P. et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 63, 5615–5621 (2003).
Amar, L. et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 92, 3822–3828 (2007).
Else, T. et al. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J. Clin. Endocrinol. Metab. 99, E1482–E1486 (2014).
Ricketts, C. et al. Germline SDHB mutations and familial renal cell carcinoma. J. Natl Cancer Inst. 100, 1260–1262 (2008).
Pasini, B. et al. Clinical and molecular genetics of patients with the Carney–Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 16, 79–88 (2008).
Qin, Y. et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat. Genet. 42, 229–233 (2010).
Comino-Mendez, I. et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 43, 663–667 (2011).
Qin, Y. et al. The tumor susceptibility gene TMEM127 is mutated in renal cell carcinomas and modulates endolysosomal function. Hum. Mol. Genet. 23, 2428–2439 (2014).
Cascon, A. & Robledo, M. MAX and MYC: a heritable breakup. Cancer Res. 72, 3119–3124 (2012).
Abermil, N. et al. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J. Clin. Endocrinol. Metab. 97, E805–E809 (2012).
Burnichon, N. et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 18, 2828–2837 (2012).
Zhuang, Z. et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 367, 922–30 (2012).
Buffet, A. et al. Mosaicism in HIF2A-related polycythemia–paraganglioma syndrome. J. Clin. Endocrinol. Metab. 99, E369–E373 (2014).
Schlisio, S. et al. The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 22, 884–93 (2008).
Ladroue, C. et al. Distinct deregulation of the hypoxia inducible factor by PHD2 mutants identified in germline DNA of patients with polycythemia. Haematologica 97, 9–14 (2012).
Gaal, J. et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 95, 1274–1278 (2010).
Crona, J. et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J. Clin. Endocrinol. Metab. 98, E1266–E1271 (2013).
Oudijk, L. et al. H-RAS mutations are restricted to sporadic pheochromocytomas lacking specific clinical or pathological features: data from a multi-institutional series. J. Clin. Endocrinol. Metab. 99, E1376–E1380 (2014).
van Hulsteijn, L. T., Dekkers, O. M., Hes, F. J., Smit, J. W. & Corssmit, E. P. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J. Med. Genet. 49, 768–776 (2012).
King, K. S. et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J. Clin. Oncol. 29, 4137–4142 (2011).
Lenders, J. W. et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287, 1427–1434 (2002).
Peitzsch, M. et al. Analysis of plasma 3-methoxytyramine, normetanephrine and metanephrine by ultraperformance liquid chromatography-tandem mass spectrometry: utility for diagnosis of dopamine-producing metastatic phaeochromocytoma. Ann. Clin. Biochem. 50, 147–155 (2013).
van Nederveen, F. H. et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 10, 764–771 (2009).
Korpershoek, E. et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 96, E1472–E1476 (2011).
Bardella, C. et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 225, 4–11 (2011).
Crona, J. et al. Next-generation sequencing in the clinical genetic screening of patients with pheochromocytoma and paraganglioma. Endocr. Connect. 2, 104–111 (2013).
Welander, J. et al. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 99, E1352–E1360 (2014).
McInerney-Leo, A. M. et al. Whole exome sequencing is an efficient and sensitive method for detection of germline mutations in patients with phaeochromcytomas and paragangliomas. Clin. Endocrinol. (Oxf.) 80, 25–33 (2014).
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014).
Canu, L. et al. Pitfalls in genetic analysis of pheochromocytomas/paragangliomas—case report. J. Clin. Endocrinol. Metab. 99, 2321–2326 (2014).
Gimenez-Roqueplo, A. P. et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA Investigators. J. Clin. Endocrinol. Metab. 98, E162–E173 (2013).
Kaji, P. et al. The role of 6-18F-fluorodopamine positron emission tomography in the localization of adrenal pheochromocytoma associated with von Hippel–Lindau syndrome. Eur. J. Endocrinol. 156, 483–487 (2007).
Taieb, D. et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 39, 1977–1995 (2012).
Timmers, H. J. et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 94, 4757–4767 (2009).
Walz, M. K. Adrenalectomy for preservation of adrenocortical function. Indication and results [German]. Chirurg. 80, 99–104 (2009).
Grubbs, E. G. et al. Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J. Am. Coll. Surg. 216, 280–289 (2013).
Alesina, P. F. et al. Minimally invasive cortical-sparing surgery for bilateral pheochromocytomas. Langenbecks Arch. Surg. 397, 233–238 (2012).
Rohmer, V. et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Francais d'Etude des Tumeurs Endocrines. J. Clin. Endocrinol. Metab. 96, E509–E518 (2011).
Thosani, S. et al. The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 98, E1813–E1819 (2013).
Bickmann, J. K. et al. Phenotypic variability and risk of malignancy in SDHC-linked paragangliomas: lessons from 3 unrelated cases with an identical germline mutation (p.Arg133*). J. Clin. Endocrinol. Metab. 99, E489–E496 (2014).
Jasperson, K. W. et al. Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Fam. Cancer 13, 257–265 (2014).
Timmers, H. J. et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J. Natl Cancer Inst. 104, 700–708 (2012).
Eisenhofer, G. et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel–Lindau syndrome. Endocr. Relat. Cancer 11, 897–911 (2004).
Dahia, P. L. et al. A HIF1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 1, 72–80 (2005).
Favier, J. et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE 4, e7094 (2009).
Lopez-Jimenez, E. et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol. Endocrinol. 24, 2382–2391 (2010).
Favier, J., Buffet, A. & Gimenez-Roqueplo, A. P. HIF2A mutations in paraganglioma with polycythemia. N. Engl. J. Med. 367, 2161 (2012).
Welander, J. et al. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum. Mol. Genet. 21, 5406–5416 (2012).
Joshua, A. M. et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J. Clin. Endocrinol. Metab. 94, 5–9 (2009).
Jimenez, C. et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel–Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel–Lindau disease-related tumors. J. Clin. Endocrinol. Metab. 94, 386–391 (2009).
Hahn, N. M. et al. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J. Clin. Oncol. 27, 460–463 (2009).
Ayala-Ramirez, M. et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 97, 4040–4050 (2012).
US National Library of Medicine. ClinicalTrials.gov[online], (2012).
US National Library of Medicine. ClinicalTrials.gov[online], (2013).
US National Library of Medicine. ClinicalTrials.gov[online], (2014).
Bergers, G. & Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 (2008).
Druce, M. R., Kaltsas, G. A., Fraenkel, M., Gross, D. J. & Grossman, A. B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 41, 697–702 (2009).
Oh, D. Y. et al. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 118, 6162–6170 (2012).
Giubellino, A. et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology 154, 646–655 (2013).
Killian, J. K. et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 3, 648–657 (2013).
Loriot, C. et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J. Clin. Endocrinol. Metab. 97, E954–E962 (2012).
Hadoux, J. et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 135, 2711–2720 (2014).
Cameron, E. & Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl Acad. Sci. USA 75, 4538–4542 (1978).
Acknowledgements
The authors thank Mrs Julie Sappa for English-language assistance (service provided by Alex Edelman & Associates). The research led by A.-P.G.-R. and J.F. is supported by the Programme Hospitalier de Recherche Clinique grant COMETE 3 (AOM 06 179), by grants from the Agence Nationale de la Recherche (ANR 08 GENOPATH 029 MitOxy & ANR 2011-JCJC-00701 MODEOMAPP) and from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 259735.
Author information
Authors and Affiliations
Contributions
J.F. researched data for the article, contributed to discussion of the content, and reviewed and edited the manuscript before submission. L.A. researched data for the article and contributed to discussion of the content. A.-P.G.-R. researched data for the article, contributed to discussion of the content, wrote the article and reviewed and edited the article before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Favier, J., Amar, L. & Gimenez-Roqueplo, AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 11, 101–111 (2015). https://doi.org/10.1038/nrendo.2014.188
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrendo.2014.188
This article is cited by
-
Paraganglioma in pregnancy, a mimic of preeclampsia: a case report
Journal of Medical Case Reports (2023)
-
Genotype–Phenotype Correlations and Clinical Outcomes in 155 Cases of Pheochromocytoma and Paraganglioma
World Journal of Surgery (2023)
-
Research in the genetics of pheochromocytoma and paraganglioma: a bibliometric analysis from 2002 to 2022
Clinical and Experimental Medicine (2023)
-
Pheochromocytoma with Takotsubo Syndrome and acute heart failure: a case report
World Journal of Surgical Oncology (2022)
-
Increased expression of Nrf2 and elevated glucose uptake in pheochromocytoma and paraganglioma with SDHB gene mutation
BMC Cancer (2022)
