
Key Points
-
Chemical predictive modelling encompasses empirical computational methods based on observed patterns in data that guide the design of future compounds.
-
Simple physicochemical property-based guidelines and structure-based chemical filters, such as AstraZeneca's AZFilters, are used to identify poor-quality compounds in screening set selection and compound design.
-
Despite their limitations, quantitative structure–activity relationship (QSAR) models of ADMET (absorption, distribution, metabolism, excretion and toxicity) properties are widely used in compound design; advice and guidance on the judicious use of QSAR methods has been published.
-
A key problem with QSAR methods is estimating confidence in the predictions, which is linked to the definition of the model's domain of applicability.
-
Project- or chemical series-specific QSAR models are one approach to solve the 'domain of applicability' problem but this approach requires automated model building to be practical for a large organization with multiple projects.
-
Interpretable models and inverse QSAR methods provide additional information to inform the design of compounds with improved properties.
-
Matched molecular pair analysis is complementary to standard QSAR, is interpretable and can be used to propose new compounds.
-
Despite the progress in chemical predictive modelling techniques, their impact on improving compound quality is difficult to assess and is limited by cultural factors.
-
These include continued debate over the application of compound quality guidelines and the diversity of opinions among medicinal chemists on attractive versus unattractive structures.
-
Current techniques are most successful in modelling ADMET properties, whereas prediction of potency or efficacy is more challenging.
-
Areas of active research include descriptors to incorporate chirality, multi-objective optimization and expert systems for compound optimization.
Abstract
The 'quality' of small-molecule drug candidates, encompassing aspects including their potency, selectivity and ADMET (absorption, distribution, metabolism, excretion and toxicity) characteristics, is a key factor influencing the chances of success in clinical trials. Importantly, such characteristics are under the control of chemists during the identification and optimization of lead compounds. Here, we discuss the application of computational methods, particularly quantitative structure–activity relationships (QSARs), in guiding the selection of higher-quality drug candidates, as well as cultural factors that may have affected their use and impact.
Similar content being viewed by others

Main
Reducing attrition rates remains a major challenge in drug development. Recent estimates indicate that the chances of a drug candidate successfully reaching the start of Phase II trials, where the role of the target biology in the disease can be tested, are only ∼37%1. Moreover, the probability of success in Phase II trials is only ∼34%. Although a substantial proportion of failures in Phase II trials may be due to flaws in the underlying biological hypothesis, the physicochemical properties of small-molecule drug candidates also have an important impact, through their influence on ADMET (absorption, distribution, metabolism, excretion and toxicity) characteristics and on the effectiveness of the compounds at selectively engaging their targets in humans2.
Given such issues, there has long been interest in the use of computational approaches to help guide the selection and optimization of compounds for synthesis and testing in order to reduce the risks of failure related to their physicochemical properties3. Many papers have been published that discuss the properties of a 'quality compound' — a compound that is more likely to robustly test the biological hypothesis in the clinic.
Such computational approaches can be broadly divided into physics-based and empirically based methods. Physics-based methods encompass, for example, molecular dynamics and the prediction of binding affinity by methods such as free energy perturbation and quantum chemical calculations. Empirical methods are based on observed patterns in existing data, which are used to guide the design of future compounds; examples of such methods include quantitative structure–activity relationships (QSARs), rule-based systems and expert systems. They do not rely on any understanding of the physics of the system, although they can indicate what the controlling physical properties might be. QSAR methods use statistical regression and classification-based approaches to identify quantitative patterns that are present within the existing data. The rules in rule-based methods may be either manually or automatically generated. Physics-based approaches are often used in conjunction with QSAR methods, but the large scale of data sets available can limit the degree to which a physics-based approach may be rigorously applied owing to limitations in computational resources. Empirical methods, however, are particularly suited for the analysis of the large volumes of data that are now available from the routine use of high- and medium-throughput in vitro biological and ADMET assays in drug discovery. We term the suite of available empirically based methods 'chemical predictive modelling' (Table 1).
In this Review, we describe the development of some of the most important chemical predictive modelling tools that are currently used in the industry and discuss some of their limitations as well as the cultural aspects that may prevent these approaches from realizing their potential.
What defines compound quality?
A universally accepted definition of compound quality has not been established. However, multiple landmark publications over the past 15 years or so have indicated the importance of various physicochemical properties, particularly lipophilicity.
The pioneering 'rule of five' guidelines published by Lipinski et al. in 1997 proposed simple physicochemical property-based guidelines for drug permeability4 (Box 1). By analysing a set of drugs that had entered clinical trials, it was found that the following rules pertained to a large proportion of the compounds: molecular mass ≤500 Da; calculated LogP (cLogP) ≤5; number of hydrogen-bond donors ≤5; and number of hydrogen-bond acceptors ≤10. It was suggested that compounds that violate any two of the 'rule of five' conditions are unlikely to be oral drugs4.
The recognition that lead optimization often resulted in increased lipophilicity and molecular size prompted the definition of the 'lead-like' concept5,6. This suggested that screening libraries should be preferentially populated with smaller and less lipophilic compounds than those described by Lipinski's 'drug-like' definitions. Leads that are smaller and less lipophilic than drug-like compounds would provide 'headroom' for lead optimization. These publications had a huge impact on how medicinal chemists defined compound quality and led to an increase in the use of in silico approaches for drug design; for example, medicinal chemists would computationally filter compound collections and compounds proposed for synthesis to only include those with calculated physiochemical properties that were sufficiently lead- or drug-like.
Over the past 15 years, many developments on these guidelines have been published7,8 to supplement and fine-tune recommended molecular property ranges for fragments9, target classes, disease areas10 and ADMET characteristics11,12. Some of the guidelines have been challenged and new ones proposed. Based on studies of the temporal invariance of physicochemical properties, it has been questioned whether the focus on molecular mass is justified, as lipophilicity is the more fundamental controlling property13. The importance of lipophilicity has been reaffirmed in multiple studies, such as a study of the toxicological outcomes of 245 compounds in development at Pfizer, which found that compounds with cLogP >3 and total polar surface area <75 Å2 were six times more likely to show an adverse event in a rat or dog in vivo safety study than a compound with cLogP <3 and total polar surface area >75 Å2 (Ref. 12). Flexibility, molecular complexity and shape are additional properties that have received attention from some in the field14,15.
Where potency information is available, the most recent evolution of these guidelines is the proposal of various ligand efficiency concepts that that build on the concepts of lead-likeness and of discriminating between optimal and non-optimal binders, as first suggested by Andrews16. Ligand efficiency17, ligand lipophilic efficiency (LLE)18 and LogP divided by ligand efficiency (LELP)19 are size-, lipophilicity- and size-plus-lipophilicity-corrected measures of potency that help to identify compounds that are maximizing the use of their chemical structure in desirable binding and are therefore likely to be better leads.
For example, scientists at Astex have reported that companies focusing on leads with ligand efficiency and lipophilic efficiency find more robust SARs and produce candidate drugs with a more acceptable compound property profile20. Similar findings were reported by Leeson and St Gallay21; in their target-by-target comparison, companies that applied rigorous ligand efficiency and LLE optimization produced candidate drugs that were smaller and less lipophilic. Finally, Keserü and colleagues analysed data on the ADMET properties of compounds published by Pfizer and showed that LELP can discriminate between compounds with acceptable ADMET profiles and those with significant ADMET liabilities22,23.

Structure-based chemical filters
Computational structural filters were developed to assist drug design teams in identifying compounds with undesirable chemical features24. They rely on knowledge of medicinal chemistry and retrospective analysis of high-throughput screening (HTS) outputs or analysis of highly annotated compound subsets (such as hits, leads, clinical candidates and marketed drugs). Such filters usually include unattractive chemical features, known toxicophores, metabolically labile compounds and functionalities that could potentially generate false positives in HTS or interfere with biochemical assays (for example, fluorescent and coloured compounds or aggregate-forming compounds)25 (Box 1).
For example, AZFilters (see Box 2 and Supplementary information S1 (box)) include both physicochemical properties and chemical filters26. The chemical filters are largely exclusion filters for 'ugly' functionality, but they are also complemented by inclusion filters; for example, compounds should have at least one polar atom and at least one single bond.
One drawback of such compound quality rules is that most of them use hard cut-offs for molecular properties and pass/fail assignments for the chemistry filters, and so they cannot be used for compound ranking; all compounds that comply with the rules are treated equally, as are all that violate them. Consequently, various scoring models for lead- and drug-likeness have been suggested, which are often derived by machine learning methods27. These have proved to be useful for compound prioritization but they lack the intuitiveness, transparency and ease of implementation associated with simple filters. To address this problem, Hopkins et al.28 used the concept of desirability to provide a quantitative metric for assessing drug-likeness, known as the quantitative estimate of drug-likeness (QED); values can range from zero (all properties unfavourable) to one (all properties favourable). This metric combines the simplicity of rule-based methods with the ranking output of scoring models.
QSAR models
The next step from simple structure-based filters is the application of QSAR models, which offer more quantitative predictions. These can be used in large-scale library filtering but are particularly suitable for lead optimization, where more precise prediction of properties is required.
QSAR models are empirical models in which a quantitative description of a chemical structure is related to biological activity through an algorithm to guide future drug design. The emergence of the QSAR field was driven by the work of Hansch, Fujita and colleagues in the early 1960s, who — in a series of landmark papers — developed predictive models for potency and ADMET end points based on physicochemical properties, often in congeneric series29.
In recent years, the growth in the study of ADMET properties has resulted in data sets that span chemical series, as compounds that are designed for many drug targets are screened through a limited number of assays, providing the opportunity to build cross-project 'global' ADMET models. However, the term 'global' is misleading in this context; although the aspiration is that if the model is built on enough compounds then it can effectively predict the properties of any future compound, owing to the nature of QSAR models they are unlikely to ever fulfil this aspiration. These models are not based on an understanding of the underlying physics of the system; rather, they are simply attempts to describe the SARs observed in the data set used to train the model. It is unlikely that a few thousand compounds — or even tens of thousands of compounds — in a training data set will confidently represent the entire pharmaceutical chemistry space. Nevertheless, based on either in-house data or literature data, these 'global' models have been almost universally adopted in industry as a method for guiding compound property design.
Experience indicates that the application of QSAR models in drug discovery is fraught with difficulties, complications, confusion and failure, not least owing to limitations in the data, problems in combining data from multiple sources, limitations of the molecular descriptors, inappropriate use of machine learning models and the inherent limitation of empirical models to extrapolate beyond their domain of applicability (for further discussion, see Refs 30,31,32,33). Even more fundamentally, the structures need to be correct34, and a call has recently been made for the accurate representation of chemical structures in publicly available SAR databases35.
Regulatory authorities and the other international bodies such as the Organization for Economic Co-operation and Development (OECD) have also stepped in to provide guidance and tools to stimulate good QSAR modelling practice (see the OECD Quantitative Structure–Activity Relationships Project [(Q)SARs] for further information). To facilitate the consideration of a QSAR model for regulatory purposes, the OECD recommends that the model should be associated with the following properties: a defined end point; an unambiguous algorithm; a defined domain of applicability; appropriate measures of goodness of fit, robustness and predictivity; and a mechanistic interpretation, if possible. Some research journals have also imposed more rigorous acceptance criteria on QSAR papers to raise the quality of submitted articles as well as the transparency of the models in publication36. However, the requirement to publish all the data and molecular structures used to carry out the study can be problematic for pharmaceutical companies.
QSAR models will always have limitations, as noted above, but these papers and guidance documents give good advice on how to avoid common problems. When judiciously used, QSAR models can be the most accurate and precise prediction tools available, often exceeding the capability of physics-based models.
Domain of applicability of QSAR models. At present, a key problem that needs to be addressed in the application of QSAR models is estimating confidence in their predictions. Root mean squared errors of a chosen test set are the simplest estimate of the model's likely ability to predict the properties of an average set of compounds external to the model. It is a widely held belief that compounds that are 'close' to the model space (in terms of similarity to the training set) are likely to have their properties more accurately and precisely predicted than compounds that are more 'distant' from the model space. So, the problem then becomes quantification of the domain of applicability of the model, the distance of the new compound from it, and the relationship between that distance and error in prediction.
In its simplest sense, the applicability domain can be described by a Euclidean box defined by the descriptor properties of the training set, and a future compound can be within or outside that box. The distance of future compounds can be measured in the Euclidean space or, better, by probability-based distances that include information on the co-linearity of the descriptor set. Distance measures can be based on property-based distances or on structural descriptors such as molecular fingerprints. Descriptor-based distances can either be weighted according to the contribution of each descriptor in the QSAR model or given equal weight. It has been suggested that descriptor-based distances that are weighted according to their contribution to the model provide higher-quality applicability domain assessments than those obtained using the equally weighted descriptors of the training set molecules37. In situations where the QSAR model is an ensemble of models, the standard deviation of predictions of the model ensemble also outperformed descriptor-based distance measures as a measure of confidence in prediction38.
In a recent review of many different definitions of applicability domains that were applied to bioconcentration factor models, developed according to the OECD guidelines under the EU project CAESAR (Computer Assisted Evaluation of Industrial Chemical Substances According to Regulations) with two test sets, it was found that the different approaches each had strengths and limitations. Although excluding compounds from prediction that were 'outside' the model's domain of applicability improved model statistics, applicability domain methods that excluded many compounds also limited the utility of the model39. There appears to be no universally successful method for describing the applicability domain of a QSAR model, nor a universal measure of the distance from the model space, and this topic remains a focus for QSAR scientists.
Some QSAR models, such as those used in the field of environmental toxicology, attempt to cover the chemical space of likely interest; that is, it is anticipated that the compounds being predicted will be either within the applicability domain of the model or not far from it. However, in drug discovery, the evolution of a compound series involves using prior data to predict the next compound to be synthesized, and hence compound optimization usually drives chemistry away from the domain of applicability of the QSAR model.
Within the global models used at AstraZeneca, we have observed that predictions for different chemical series have differing degrees of accuracy and precision. This may be due to: deficiencies in our descriptor set in identifying discriminating molecular features across chemical series or subseries; or the balance that the machine learning method needs to strike between different and perhaps conflicting SARs to minimize the unexplained error in prediction averaged across all chemical series in the training set; and/or the weight of representation of different chemical series in the training set itself. Maggiora described the concept “lack of invariance of chemical space” for instances where neighbourhood relationships may be significantly altered across chemical series or subseries; compounds that are nearest neighbours in one chemical space representation may not be nearest neighbours in another40.
One approach to circumvent the local series description problem within a global QSAR model would be to build project-specific or chemical-series-specific models. For each project, a decision could be made on which model is most appropriate for future predictions: the global model, the project model or even a chemical series-based model. This would maximize our ability to make accurate and precise predictions for all current structure optimizations. However, for a large pharmaceutical company with hundreds of ongoing projects, this might involve building thousands of project- or chemical-series-specific QSAR models and managing their comparisons with global models on a regular cycle. To minimize the distance between the current chemistry and the applicability domain of the model, QSAR scientists can manually update the global models, but this is a time-consuming activity. If possible, it would be ideal to automatically keep global QSAR models up to date.
Automated QSAR models. Although informatics technology has been capable of automatically building and maintaining QSAR models — as described above — for several years, the applications of such systems in drug discovery have only recently become apparent. The hurdle towards adoption is more psychological than technological; we need to become more confident that machines can build models of similar or superior quality to those built by computational chemistry specialists, and that the models will be stable and robust as the system evolves with time. Other fields have been more courageous in using machine learning and pattern recognition models in automated systems, particularly in automated online monitoring for fields as diverse as manufacturing, the food industry and in monitoring the reprocessing of nuclear fuel waste41.
Nevertheless, there are some reported examples of automated QSAR model building for drug discovery. Oprea and colleagues42 used an automated partial least squares (PLS) engine to build 1,632 QSAR models based on the WOMBAT (World of Molecular Bioactivity) database. The OCHEM database contains models and data sets, and offers automated generation of QSARs43. Leahy and coworkers44 pioneered automated QSAR modelling with the development of the Discovery Bus technology. Discovery Bus is an automated machine learning environment based on “the competitive workflow”, where new models are compared with old ones for their predictive ability on a common test set. Different machine learning agents and descriptor sets can compete to find the best model for a given data set, and a QSAR specialist can compete with the machine learning agents, which could help to build confidence in the automated system. ChemModLab provides a similar framework; it is a web-based automated QSAR platform that allows users to upload data sets, descriptor sets or modelling methods, which can then be compared with other data sets, descriptors or methods45.
A few other groups have also reported investigations of automated QSAR modelling, including a study by Segall and colleagues46 on ADME properties. Additionally, in an interesting development, automated modelling has become the basis for a published patent47. Wood, Rodgers and colleagues at AstraZeneca have attempted to answer some of the concerns over the automation of QSAR model development using real-world data to demonstrate the benefits of updating global and QSAR models48,49. Over a 2-year period, using in-house data for solubility, logD7.4 and protein binding, they showed that static models lose their predictive power over time, that different machine learning methods can be considered best as the criteria for decision-making change from a static model to an updating model, and that project- or series-specific models outperform global static models and even global updating models (Fig. 1).

Results of a 2-year performance comparison between models of logD7.4 (panel a) and solubility (panel b) across ten active AstraZeneca projects are shown. The models compared were a static global model, an updating global quantitative structure–activity relationship (QSAR) model, and an automated QSAR (AutoQSAR) hierarchy model. The best model (static global, updating global or local project model) is selected on a project-by-project basis according to the latest month's test set performance. The graphs show that, as the models mature, month by month the AutoQSAR hierarchy outperforms the static or updating global model48,49.
One area in which automation of QSAR model building may not help is where only a weak model can be built in the first place, which is often the case for potency end points in situations in which there is a high degree of molecular recognition between small molecules and the receptor. Maggiora described the concept of an “activity cliff”, where molecules that are structurally highly similar can produce very different biological responses owing to subtle structural differences affecting receptor fit or lack of it40. In an attempt to define a universal confidence metric as well as one that is robust enough to potential problems due to activity cliffs, a group at Pfizer has included the 'activity landscape' of structural near neighbours within a QSAR confidence metric50. At the heart of the method is a weighted root mean square error estimation that combines the predicted value, the experimental values of the nearest neighbours and the relative distance of those neighbours within the model space. A calibration procedure based on a test set allows a method-independent confidence metric to be defined. It has been reported that this new approach has had a substantial impact on drug discovery efforts at Pfizer50, which suggests it has been accepted by their medicinal chemists.
Automation may allow modellers to search through the model, descriptor and machine learning space to find where good models exist. The Discovery Bus methodology described above allows such exploration, which may be necessary to find the right descriptor data set combination for end points that are difficult to model. However, care must be exercised as such a tool could easily lead to another old problem: when multiple tests are carried out on the same data set, the likelihood of finding a model by chance alone increases. Demanding increased confidence in the robustness of the model before it is accepted is one approach for addressing this problem. Livingstone and Salt used an adjusted F-statistic to counter the misuse of the standard multiple regression algorithm to select important variables from a larger pool of available variables51,52. However, although adjusting the confidence level at which to accept or reject the model (when multiple comparisons are made) protects against false positives, it comes with the cost of increasing the chance of rejecting all models when a real one is present (false negatives).
Permutation tests can also provide confidence in the robustness of the model53 but, depending on how they are executed, these tests can themselves lead to a biased estimate of the model's robustness. The full model generation procedure must be repeated, including variable selection, rather than just permuting the y-variables of the final model54.
Further work is required to understand and quantify the risk of chance correlations. The iterative nature of drug discovery offers a further opportunity to test model robustness, as last week's predictions become next week's measurements, and so confidence in the model can be strengthened by real-world experience.
Interpretable QSAR models and inverse QSAR. An important limitation of the standard QSAR approach is that the medicinal chemistry designer must first generate an idea for a new compound before QSAR models are used to predict its properties. The designer is always looking for an answer to the question: “Which compound do I make next?” One way to address this issue is to improve the interpretability of QSAR models by helping the designer to understand the SAR that is encoded by the model55. The ability to do this depends on the nature of the descriptors and the modelling method used. Linear models that are built on familiar physicochemical descriptors are the most readily interpreted, whereas nonlinear models are generally viewed as opaque or 'black box' (that is, not amenable to interpretation). As nonlinear methods often lead to more accurate predictions, there is frequently a trade-off between interpretability and prediction accuracy.
Johansson et al.56 compared the prediction accuracy of three ADMET and 13 potency classification end points using both interpretable and opaque modelling methods; they found that although the interpretable models — such as the decision list algorithm Chipper — performed less effectively than state-of-the-art ensemble methods, the loss of accuracy was relatively small (lower than 5%).
Carlsson et al.57 have described a general method for the interpretation of nonlinear QSAR models. The method provides the most important model attributes in the context of a particular prediction molecule rather than the globally most important attributes. This helps the medicinal chemists to identify possible changes to their molecule that, according to the model, would be expected to have the greatest impact on the predicted property. The approach was illustrated using support vector machine and random forest models and applied to mutagenicity data. Signature descriptors58 were used, although the authors say that their method can be used with other descriptors. In an extension of this work, a system was developed that colours the atoms of a predicted molecule according to whether they contribute positively or negatively to the property being predicted: in this case, mutagenicity, carcinogenicity and aryl hydrocarbon receptor activation59. A similar visual interpretation of ADMET QSAR models is implemented in the Glowing Molecule tool within the StarDrop software package60.
Another way of tackling the interpretability issue is to use information embedded in the QSAR model to direct the transformation of a lead compound in an approach known as 'inverse-QSAR'. A general approach for automated, iterative, QSAR-driven compound optimization was described by Lewis61. In an extension of the work using the signature descriptors mentioned above, Helgee et al.62 have described a method for automated optimization. Substructures that are identified by a QSAR model as significantly contributing to the prediction are systematically replaced, leading to the generation of new structures to improve the property being modelled. The approach was demonstrated using the Ames mutagenicity test but can be applied to any end point and in combination with other end points in a multi-objective optimization.
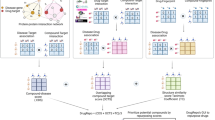
A chemical predictive modelling approach that is inherently interpretable is the automated matched molecular pair analysis (MMPA) of structure–property databases63,64. MMPA reveals the change in a measured property resulting from a specific small change in the structure (Fig. 2; Tables 2, 3). Hence, by applying the technique to local (project- or series-specific) or global (cross-project) sets of SAR data, rules can be derived that may be used to predict the properties of new analogues and to generate new compound suggestions in an inverse-QSAR fashion65. MMPA can be viewed as complementary to QSAR (Table 4). It is appealing to medicinal chemists because of its straightforward interpretability, but it is limited to molecular transformations that have previously been explored in a strict pairwise manner. The two approaches can be combined whereby MMPA-derived transformation rules are used to make prospective suggestions, and QSAR models are used to predict the properties of the proposed virtual compounds.

Matched molecular pair (MMPA) analyses can be divided into two types: supervised and unsupervised. In supervised MMPA (Table 2), the chemical transformations are predefined, then the corresponding matched pair compounds are found within the data set and the change in end point computed for each transformation. With unsupervised (or automated) MMPAs (Table 3), an algorithm finds all possible matched pairs in a data set according to defined rules. This results in much larger numbers of matched pairs and unique transformations, which are typically filtered within the algorithm to identify those transformations that correspond to statistically significant changes in a property with a reasonable number of matched pairs. pSol, –Log10 (molar aqueous solubility).
As an alternative to standard QSAR models, MMPA-derived rules can be used to predict the properties of the compounds that are proposed by these rules by applying the average change in property to a measured value — a strategy that is termed 'QSAR-by-MMPA'66. The authors used MMPA on a structurally diverse set of 322 inhibitors of the KCNQ1–KCNE1 potassium voltage-gated ion channel complex to predict prospectively the inhibitory potencies of 36 additional compounds. Comparison with predictions from a nearest-neighbour approach and a random forest QSAR model showed that the MMPA-derived predictions were superior66. These results suggest that QSAR-by-MMPA may be a successful approach for data sets for which useful QSAR models cannot be derived, because it identifies specific structural changes that control activity rather than attempting to fit a model to the whole data set.
The application of MMPA across large SAR databases for the optimization of ligand potency is problematic because the same structural transformation may increase potency against some targets, leave some targets unaffected and decrease potency against other targets67. How does one select those transformations from a global SAR data set that are most likely to increase (or maintain) potency against a given target? Mills et al.68 have described one potential approach to address this bioisostere identification problem; they used pairwise analysis of chemical series to identify those with correlated SAR patterns and then applied MMPA to generate relevant transformation rules. The approach was successfully applied to the design of more potent antagonists of transient receptor potential cation channel, subfamily A, member 1 (TRPA1)68.
Chemical predictive modelling in practice
Chemical predictive modelling is now a core part of drug discovery. For example, AstraZeneca's C-Lab platform69 has been used to make over 2 billion calculations in the past 12 years. An internal analysis of the newly synthesized compounds registered in the AstraZeneca corporate database during the 2011–2012 period showed that for 55% of these compounds one or more properties had been predicted by C-Lab before synthesis. AstraZeneca's global HERG (a potassium voltage-gated channel; also known as KCNH2) QSAR model70 has also contributed to the reduction in the synthesis of 'red flag' compounds (compounds that are measured to have an HERG potency of <1μM), from 25.8% of all compounds tested in 2003 to only 6% in 2010.
The true negative prediction rate of potentially genotoxic impurities by in silico models was recently surveyed across eight companies. The methods for prediction were given and the approaches used across the companies were very similar. The true negative prediction rate was found to be 94%, and this increased to 99% when expert evaluation of the results was included in the decision71. The results of this analysis are currently being written into guideline M7 of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) on genotoxic impurities.
There are many published examples of the successful application of QSAR models, so we have just selected one illustrative example here. A QSAR model predicting the functional duration of a series of dopamine receptor D2 and β2-adrenergic receptor agonists led to optimization of the in vivo duration of sibenadet and various other follow-on developmental compounds72. It also led to a more detailed understanding of the way these drugs interact with phospholipid bilayers and was cited in the development of long-acting β2-adrenergic receptor agonists by Pfizer73, as well as in the design of indacaterol — the recently approved long-acting β2-adrenergic receptor agonist from Novartis74. Below, we discuss some important general issues in the application of QSAR models.
Commercial and public models versus in-house models. Commercial and public-domain predictive ADMET models are available, and one important issue for companies is the performance of these models compared with in-house models, given that the predictive ability of QSAR models is limited by the domain of applicability (see above). In an assessment of the success in genotoxic predictions, models that were based on data sets in the public domain were adequate in predicting compounds in the public domain but did not perform as well on proprietary active compound data sets from pharmaceutical companies, which tend to be more complex and generally do not contain obvious reactive functional groups71.
Similarly, in an assessment of QSAR models of solubility, Bruneau found that a solubility model based on literature data was successful at predicting literature compounds, and an in-house solubility model was successful at predicting in-house compounds, but the cross-prediction of each model was markedly poorer75. Stouch, in his assessment of Bristol Myers Squibbs' search for useful global ADMET models, identified the same problem of the domain of applicability of even large literature data sets32. A model of Caco-2 permeability based on 800 literature compounds proved to have very little predictive ability on Bristol Myers Squibbs' compounds, and subsequent analysis found very little structural similarity between the literature compounds defining the Caco-2 model and the in-house compounds. A further weakness of literature data sets highlighted by Stouch is the diverse experimental methods that are used to collect the measured data32.
In our view, models in the public domain should be used with caution and, wherever possible, they should be based on structures as close as possible to those for which predictions are desired, with experimental data obtained from a consistent, relevant assay. Even models that are based on data sets in the public domain and that contain many thousands of compounds may not be as useful as a model containing fewer compounds of relevant structural similarity.
Cultural aspects of chemical predictive modelling. Notwithstanding the explosion in available SAR data and the enormous progress in predictive modelling techniques, it is difficult to assess the real impact of these advances on the practice of drug design and on improvements in compound quality76. Although we can point to illustrative case studies of improvements in key compound quality indicators on a project-by-project basis, or even within a research site or company, the impact is less convincing across the industry overall.
In 2007, Leeson and Springthorpe reported that in the 10 years since the publication of the 'rule of five' guidelines, the drug-likeness concept had apparently not greatly influenced the design decisions of chemists in some major companies, as judged by the physicochemical properties of their patented compounds18. In a follow-up article in 2011, Leeson and St-Gallay discussed the influence of organizational factors on compound quality, revealing some striking differences in the drug-likeness of synthesized compounds among organizations pursuing the same drug targets21. They proposed various cultural and organizational factors that could contribute to such differences, including varying tolerance of the companies to (or lack of awareness of) compound-related risks in clinical development; the lack of uptake or impact of computational tools; pressures on medicinal chemistry from proscribed project timelines and meeting corporate objectives; and a lack of innovation in the use of chemical templates and chemical synthesis. An example of the influence of organizational factors from AstraZeneca's experience is highlighted in Fig. 3.

a | The chart shows the distributions of the measured aqueous solubility of ∼2,000 compounds synthesized at AstraZeneca's Södertälje site in the early 2000s (good compounds have a solubility higher than 100 μM; medium compounds have a solubility of 10–100 μM; poor compounds have a solubility lower than 10 μM). Managers at AstraZeneca's Södertälje site were tracking the solubility of synthesized compounds over time. Even though good computational models of aqueous solubility had been available from before 2001 (Ref. 101), this did not result in an improvement in the solubility of the compounds synthesized. b | A marked improvement in the properties of compounds only occurred when management expectations on quality at the design stage were enforced in 2004; the chart shows the distributions for ∼14,000 compounds synthesized between 2004 and 2012.
All of the statistical studies on compound quality (many of which are cited in this Review) point to one key lesson: increasing potency without controlling lipophilicity (LogP) is detrimental to the chances of further progression for a medicinal chemistry project. So, it may be surprising that there is still continuing debate about whether drug-like concepts have improved compound quality or overly restricted compound design. However, Kenny and Montanari77 have highlighted that the importance — or strength — of many of the reported relationships has been overstated because the approaches that are used to analyse and visually represent the data exaggerate trends in data. For example, one analysis highlighted the apparent overlooked importance of the fraction of sp3 carbons78. In this analysis, binning of the fraction of sp3 carbons as a function of Log(solubility) showed a high correlation (r = 0.97), which is indicative of a very strong relationship. As these data were publicly available, Kenny and Montanari were able to re-analyse them without the use of data binning, and found a correlation coefficient of only r = 0.25. The correlation of molecular weight with Log(solubility) for this series of molecules was actually much higher (r = −0.62), thus questioning the importance that the authors of the original analysis placed on the fraction of sp3 carbons, which only described 6% of the variance in Log(solubility). Kenny and Montanari make some suggestions for good practice in data analysis77, and in our view some of the most highly cited compound quality papers would benefit from reassessment following these recommendations.
A recent analysis of 150 of AstraZeneca's compounds in development showed that Pfizer's '3/75' rule and the fraction of sp3 carbons did not discriminate between compounds that successfully reached Phase II trials and those that did not progress owing to toxicity79. The authors caution against using these simple guidelines as hard cut-offs, as many successful drugs would not pass them. Some chemists are even making a call to arms because more difficult targets may require us to step outside the drug-like space80. Nevertheless, despite challenges and even apparent opposing conclusions based on differing analyses of the same data sets, the weight of evidence indicates that compound quality guidelines have some value, and it would be foolish to ignore the potential of drug-likeness concepts harnessed from the successes and failures of hundreds of previous compounds in development. This is not to say that the definitions of drug-like space preclude discoveries in areas at the extremities, as some recent drug registrations have demonstrated81,82. The guidelines are based on statistical analyses (assuming that there is statistical validity) and therefore should be interpreted in a probabilistic manner. Projects working in the non-drug-like space should be prepared for a longer, higher-risk and more expensive journey. Some projects may be prepared for that risk, and for some it may be worth it, but it may not be wise to base a whole portfolio on the extremes of a probability distribution.
The organizational or cultural factor is also apparent in the definition of the chemical filters described above. Various validation exercises have shown that there can be little consensus among chemists on what constitutes a chemically attractive or unattractive structure. For example, Pharmacia evaluated how chemists selected and rejected compounds in lists of 2,000 compounds, seeded with 250 compounds that were previously rejected by a very senior medicinal chemist83. The average pairwise agreement among the 13 chemists in the study was only 28%. Nine of the chemists reviewed two lists of 2,000 compounds containing the same set of 250 probe compounds. The average consistency in rejection was only 51%, with the most consistent chemist only achieving a value of 71%. Based on an analysis of the full 2,000 compound sets, the average pairwise agreement was only 23%. The chemists who had been selected had experience ranging from 3 to 25 years, but it appeared that experience was not related to consistency of opinion; two of the reviewers had over 25 years of experience but they still showed very low consistency in their rejections.
In another example, Novartis gave 19 chemists 4,000 structures and asked them to identify desirable or undesirable fragments; only 8% of fragments were identified by more than 75% of chemists84. The consensus was uneven, with the agreement on good fragments being only 1%, whereas the consensus on bad fragments was 7%. Although still low, the higher consensus with 'bad' fragments suggests that chemists do a better job at carrying and sharing their bad experiences than they do with good ones.
The lack of consistency among the same group of chemists, as highlighted by the Pharmacia research, supports the use of computational filtering methods, which can at least objectively apply defined rules (but only if we can agree on what they may be) with 100% accuracy. A lack of agreement among experts may demonstrate a lack of shared expertise. As highlighted by the Novartis group, chemists show higher consensus in assessing synthetic accessibility, where the correlation coefficient for the consensus was as high as 0.73–0.84 (Ref. 84). It may be that although the underlying rules of chemical synthesis are mature and are the cornerstone of education in chemistry, the rules of medicinal chemistry are much less clear. This was further exemplified in an evaluation of 65 chemical probes identified from the US National Institutes of Health (NIH) molecular libraries programme, in which 11 experts were asked to rank the suitability of the identified probes as research tools for the elucidation of biological pathways (and not necessarily as lead compounds). The expertise of the panel was not in doubt; each expert was identifiable to many medicinal chemists by only their surname and reputation. The lack of general agreement among these 11 experts is obvious across the heatmap85 shown in Fig. 4.

As part of an evaluation of the US National Institutes of Health (NIH) molecular libraries initiative, 11 experts were asked to rank the suitability of 65 chemical probe compounds as research tools for the elucidation of biological pathways (and not necessarily as lead compounds). The heatmap illustrates the confidence scores in the chemical probes (on the y-axis) for each of the 11 experts (listed on the x-axis). Red and orange indicate high dubiosity, whereas shades of blue indicate low dubiosity (that is, a good probe), with yellow indicating a median value. The probes are sorted by the median score on the y-axis. The x-axis is sorted by the research area of the voting experts: the two on the far left are pharmacokinetics and toxicology experts, the middle five are experts in chemoinformatics, whereas the last four are high-throughput screening and chemical experts. Image reproduced, with permission, from Ref. 85 © (2009) Macmillan Publishers Ltd. All rights reserved.
One conclusion is that chemists are all victims of their own experiences in medicinal chemistry projects. That experience is gained through several chemical series; most chemists experience a small number of projects, and many of the 'rules' derived may be specific to those chemical series explored. Those 'rules' then become their guiding principles in forthcoming projects. This is supported by Leeson's observations of organizational differences in drug optimization. It may be, as Leeson implies, that some companies are simply working in a less fruitful space, but from the chemist's point of view they are applying those optimization approaches that — based on their experiences — are more likely to succeed. Past success guiding behaviours that could lead to perceived future success is thought to be a strong driver for organizational culture86. Few cross-target rules have so far emerged or at least been accepted. This provides an opportunity for empirical predictive modelling to define the rules if we can overcome prejudices about using them.
From a statistical perspective, when trying to identify a weak signal it could be beneficial to increase the sample size, and this could be achieved with a crowdsourcing evaluation. AstraZeneca's AZFilters were initially defined in 2001 by a small group of chemists who were experienced in HTS hit evaluation. In 2003, AstraZeneca took a crowdsourcing approach to validate and refine AZFilters. Over 100 chemists from 9 sites were asked to vote on groups of 1,000 compounds taken randomly from more than 65,000 representatives from the internal AstraZeneca and external vendors' compound collections (including discontinued drugs) to assess for medicinal chemistry acceptability (whether the chemist would buy the compounds and would consider chemically modifying them). Statistical analysis led to 21 new chemical filters in addition to the original 150, a refinement of the existing filters and a tightening of the LogP window for the 'core' screening set (Box 2).
In practice, medicinal chemistry experience and knowledge of the research area domain play an important part in the general assessment of compound quality and influence series prioritization for further development. Hence, instead of using structural filters as 'rules', a different approach is to present them as 'alerts' and rely on the medicinal chemistry design team's combined expertise to apply them appropriately. This is especially the case when evaluating the risk of reactive metabolites and undertaking safety or toxicity assessments, where the presence of certain functional groups should not automatically lead to the dismissal of compounds87. Alerts can also catalyse the 'frontloading' of a test for a liability to quantify the risk at an early stage.
Automated QSAR systems also impose good modelling practice on all models built and remove much of the subjectivity involved in QSAR model building, which is crucial if QSAR models are to achieve their potential utility. However, the introduction of automated QSAR methods to AstraZeneca generated some cultural challenges. The validation papers published by the AstraZeneca group were written as much to convince our own organization of the value, performance and safety of these procedures as to inform the wider research community of the value of automated modelling48,49. Internally, it required adjustments in the expectations of medicinal chemists, who are wary of predictions that potentially constantly change as the model is updated. Although completely automated model building is possible, including the definition of a project and chemical series, the majority of those decisions were left to the project team. This made the system simpler and also meant that the models belonged to the project rather than being imposed on the team by an automated system. Automation also changes the role of computational chemists, as model building by the chemist is replaced. However, to ensure that automated methods succeed, computational chemists are still required to act as model evaluators and as model interpreters, which arguably demands more of their expertise. Furthermore, it is crucial that computational chemists provide medicinal chemists with confidence in the model in order for it to be applied88.
Automated MMPA and automated QSAR were introduced to AstraZeneca at around the same time, and in some senses this generated a conflict in their application. Which approach should medicinal chemists use to make predictions? As discussed above, MMPA appeals to medicinal chemists as the results are readily comprehensible structural features, whereas QSAR models are the domain of computational chemists and use descriptors that are often difficult to interpret. It is likely that the two approaches are complementary, as shown in Table 4, but the full evaluation of when and where to apply these methods is still being defined. Objective assessments of where particular tools fit into the 'design–make–test–analyse' cycle are critical, and medicinal chemists should consider multiple approaches when deciding what to make next.
Outlook
In general, there are relatively good models available for the prediction of in vitro ADMET end points owing to large data sets, sometimes with over 100,000 data points that have been collected in large screening centres and with high consistency in the assays. The predictivity is often sufficient for distinguishing among good, medium or bad compound quality profiles and can provide a solid basis for selecting which molecules to progress with. However, predictions for potency and efficacy are more challenging. Efficacy is distinct from potency and usually refers to a functional response in a more complex model; it can be as simple as an agonist response in a functional cellular assay or a change in the course of disease in an in vivo situation, and therefore embodies both potency and the pharmacokinetic/pharmacodynamic (PK/PD) relationship. In this case, there are far fewer data points available and the models need to perform well in predictions that are extrapolations beyond the chemical space of the compounds used to train the models.
QSAR models are effective when the property being modelled changes smoothly as the descriptors change. Many enzymes, transporters and receptors involved in ADMET are designed to recognize a broad range of substrates and so ADMET end points are largely controlled by bulk properties. For this reason, QSAR models based on physicochemical descriptors — such as LogP, molecular volume, hydrogen bond counts, and so on — have proved to be successful in modelling many ADMET end points.
However, to model potency end points embodying a substantial degree of molecular recognition, we would need to have descriptors that are able to capture subtle structural changes within chemical series that are relevant to the SAR. It is not so surprising, therefore, that in a recent study it was found that for six potency end points, described by two descriptor sets and modelled with three machine learning methods, the descriptor choice was much more important than the machine learning method89. In instances where a model could be built, results from the different machine learning methods were generally not substantially different from each other89. Fragment-based descriptors and molecular fingerprints have potential in modelling potency end points. As molecular recognition involves both bulk property control and specific molecular recognition, it likely that methods that are based on combining multiple types of descriptors will be required in the future if QSAR is to be valuable in modelling potency.
Many of the properties that medicinal chemists need to optimize are dependent on the configuration of chiral centres in the molecule90. However, QSAR models typically use only achiral molecular descriptors and therefore cannot model these stereochemical effects. Three-dimensional QSAR methods such as comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) are available, and they have shown some promise in modelling potency end points91. They are dependent on the initial three-dimensional alignment, and results would be relative to this alignment of the molecular structures.
CoMFA appears to have maintained its popularity as a QSAR method. In our experience, three-dimensional QSAR methods are useful for understanding potential SAR patterns within the data set, but less useful in prediction. For example, a CoMFA analysis of the duration of action of dual dopamine D2 receptor and β2-adrenergic receptor agonists suggested that the three-dimensional positioning of hydrogen-bond acceptors near a basic amine was important for the duration of action. This was indeed the case, but the correct positioning of hydrogen bond acceptors alone did not result in a long duration of action. The real role of those groups that positioned hydrogen bonds was their through-bond electronic effect on the pKa of the basic amine, and the overall contribution to lipophilicity72. The CoMFA model was reporting SARs only indirectly. If we hope to build chirality into automated QSAR models for potency, we need models that require less manual intervention. There is a need for an approach to describe chirality in a way that can cross chemical series and be incorporated into global automated QSAR models. Carbonell et al.92 have recently described a method for incorporating stereochemistry into the algorithm that generates the signature descriptors discussed above, and this method was applied to QSAR predictions.
The last step in any design workflow is to decide which compounds to actually synthesize. Having applied the available predictive modelling approaches, the medicinal chemist is subsequently faced with a large data set of more or less accurate predictions of all the individual properties of the candidate molecules. A common way to deal with this problem is to colour the different properties green, amber and red, and to select the optimal compounds manually. However, this approach is not practical for the selection of compounds from huge virtual libraries. There is also a need to take into account the uncertainties in the predictions and deal effectively with error propagation from the multitude of models applied to each virtual compound60. Simply applying the predictions as 'hard' filters is likely to remove potentially good compounds from consideration, or it may just eliminate every idea. Seeking a compromise in the potency, selectivity, pharmacokinetic and toxicological profiles to discover a safe and efficacious drug is a complex task and although several methods have been described for molecular multi-objective optimzation93, this is a field that still merits further research. A more precise quantification of the uncertainty in any given prediction will also reap considerable benefits.
QSAR models of both potency and ADMET properties are increasingly becoming integrated in expert systems that aim to optimize an input compound against a given set of parameters in an iterative process. In addition to the inverse-QSAR systems discussed above, they are being applied as constraints alongside physics-based approaches in de novo drug design algorithms94,95 and in approaches using general molecular transformations to generate new compounds96. In a recent article, Hopkins and colleagues described a successful proof of concept for using such algorithms to design ligands with different polypharmacological profiles97. First, using Bayesian probabilistic activity models built on data from the CHEMBL database, they identified donepezil — an acetylcholinesterase inhibitor — as a moderately potent inverse agonist of the D4 receptor with minimal D2 receptor activity. The activity was further improved using a multi-objective optimization approach. Using a set of acceptable medicinal chemistry transformations, guided by the QSAR models, they were able to optimize donepezil into a dual D2 receptor and D4 receptor agonist with blood–brain barrier permeability. They were also able to optimize donepezil into a brain-penetrant D4 receptor agonist, increase D4 receptor activity by 69-fold (with 95-fold selectivity over the D2 receptor) and retain high blood–brain barrier permeability97. Again, further advances in prediction accuracy, estimates of uncertainty and a description of the domain of applicability will be essential to improving such expert systems.
Various commentators have speculated on how far chemical predictive modelling may go in the future. Will it ever be possible to design a drug completely on a computer in the way that modern aeroplanes are designed?98 In our view, this will probably not be possible until physics-based methods evolve to the same level as the mathematical equations of fluid dynamics and materials science; even then, the vast complexity and unpredictability of biological systems will always present a formidable challenge99. A more achievable goal in the short term is for the predictions of each of the assays in the first wave of a screening cascade to become sufficiently accurate and reliable that they can be used as a 'wave zero' virtual screening assay. This means that the predicted parameters are solely used as a basis for the next round of design and the method can be used to refine the ideas of molecules to be synthesized, resulting in a high probability that the project will synthesize a new 'best compound' in each round of optimization.
The advances in chemical predictive modelling over the past few years have provided an increased understanding of the relationship between chemical structure and compound quality. Automated approaches enable the extraction of information from huge compound property databases and its application to compound selection as well as the optimization of lead compounds to high-quality candidate drugs. Although scientific and cultural challenges remain, chemical predictive modelling approaches are leading to considerable improvements in both the quality of all compounds synthesized during each phase of the drug discovery process and in the efficiency of that process, which will have a beneficial impact on the productivity of the pharmaceutical industry.
References
Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature Rev. Drug Discov. 9, 203–214 (2010). This is a useful source of data on timelines, the probability of technical success and the costs associated with running drug discovery and development projects.
Morgan, P. et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov. Today 17, 419–424 (2012). This paper describes Pfizer's drug development experience, and introduces the concept of target engagement as a key confidence builder in projects.
van de Waterbeemd, H. & Gifford, E. ADMET in silico modelling: towards prediction paradise? Nature Rev. Drug Discov. 2, 192–204 (2003).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 (1997). This seminal paper introduced the 'rule of five' guidelines for oral bioavailability; these are the original compound quality guidelines based on simple calculated physicochemical properties.
Teague, S. J., Davis, A. M., Leeson, P. D. & Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed Engl. 38, 3743–3748 (1999). This paper introduces the lead-like concept, which has been highly influential on the lead generation activities of many companies.
Hann, M. M. & Oprea, T. I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 8, 255–263 (2004).
Lipinski, C. A. in Annual Reports in Computational Chemistry (ed. David, C. S.) 155–168 (Elsevier, 2005).
Walters, W. P. Going further than Lipinski's rule in drug design. Expert Opin. Drug Discov. 7, 99–107 (2012).
Congreve, M., Carr, R., Murray, C. & Jhoti, H. A 'rule of three' for fragment-based lead discovery? Drug Discov. Today 8, 876–877 (2003).
Wager, T. T. et al. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 1, 420–434 (2010).
Gleeson, M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 51, 817–834 (2008).
Hughes, J. D. et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 18, 4872–4875 (2008).
Leeson, P. D. & Davis, A. M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 47, 6338–6348 (2004).
Hann, M. M., Leach, A. R. & Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41, 856–864 (2001).
Vistoli, G., Pedretti, A. & Testa, B. Assessing drug-likeness — what are we missing? Drug Discov. Today 13, 285–294 (2008).
Andrews, P. R., Craik, D. J. & Martin, J. L. Functional group contributions to drug-receptor interactions. J. Med. Chem. 27, 1648–1657 (1984).
Kuntz, I. D., Chen, K., Sharp, K. A. & Kollman, P. A. The maximal affinity of ligands. Proc. Natl Acad. Sci. USA 96, 9997–10002 (1999).
Leeson, P. D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Rev. Drug Discov. 6, 881–890 (2007). This is a provocative publication that challenges medicinal chemists' decision-making practices.
Keseru, G. M. & Makara, G. M. The influence of lead discovery strategies on the properties of drug candidates. Nature Rev. Drug Discov. 8, 203–212 (2009).
Murray, C. W., Verdonk, M. L. & Rees, D. C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 33, 224–232 (2012).
Leeson, P. D. & St-Gallay, S. The influence of the 'organizational factor' on compound quality in drug discovery. Nature Rev. Drug Discov. 10, 749–765 (2011).
Tarcsay, A., Nyiri, K. & Keseru, G. M. Impact of lipophilic efficiency on compound quality. J. Med. Chem. 55, 1252–1260 (2012).
Tarcsay, A., Nyiri, K. & Keseru, G. M. Correction to impact of lipophilic efficiency on compound quality. J. Med. Chem. 56, 3120 (2013).
Gilbert, M. R. Reactive compounds and in vitro false positives in HTS. Drug Discov. Today 2, 382–384 (1997).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 (2010).
Davis, A. M., Keeling, D. J., Steele, J., Tomkinson, N. P. & Tinker, A. C. Components of successful lead generation. Curr. Top. Med. Chem. 5, 421–439 (2005).
Ursu, O., Rayan, A., Goldblum, A. & Oprea, T. I. Understanding drug-likeness. Wiley Interdiscip. Rev. Comput. Mol. Sci. 1, 760–781 (2011).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nature Chem. 4, 90–98 (2012).
Hansch, C. in QSAR and Molecular Modelling in Rational Design of Bioactive Molecules: Programs and Abstracts (eds Aki-Sener, E. & Yalcin, I.) 3–22 (Proceedings of the 15th European Symposium on Structure-Activity Relationships (QSAR) and Molecular Modelling, 2006).
Huang, J. & Fan, X. Why QSAR fails: an empirical evaluation using conventional computational approach. Mol. Pharm. 8, 600–608 (2011).
Doweyko, A. M. QSAR: dead or alive? J. Comput. Aided Mol. Des. 22, 81–89 (2008).
Stouch, T. R. et al. In silico ADME/Tox: why models fail. J. Comput. Aided Mol. Des. 17, 83–92 (2003). This is a textbook case study on how not to build QSARs.
Cronin, M. T. D. & Schultz, T. W. Pitfalls in QSAR. J. Mol. Struct. 622, 39–51 (2003).
Young, D., Martin, T., Venkatapathy, R. & Harten, P. Are the chemical structures in your QSAR correct? QSAR Combinatorial Sci. 27, 1337–1345 (2008).
Williams, A. J., Ekins, S. & Tkachenko, V. Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov. Today 17, 685–701 (2012).
Jorgensen, W. L. QSAR/QSPR and proprietary data. J. Chem. Inf. Model. 46, 937 (2006).
Tetko, I. V., Bruneau, P., Mewes, H., Rohrer, D. C. & Poda, G. I. Can we estimate the accuracy of ADME–Tox predictions? Drug Discov. Today 11, 700–707 (2006).
Tetko, I. V. et al. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 48, 1733–1746 (2008).
Sahigara, F. et al. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 17, 4791–4810 (2012).
Maggiora, G. M. On outliers and activity cliffs — why QSAR often disappoints. J. Chem. Inf. Model. 46, 1535 (2006).
Schwantes, J. M., Orton, C. R., Fraga, C. G., Douglas, M. & Christensen, R. N. The multi-isotope process (MIP) monitor: a near-real-time, non-destructive, indicator of spent nuclear fuel reprocessing conditions. Proceedings of the 50th Annual Meeting of the Institute of Nuclear Materials [online], (2009).
Olah, M., Bologa, C. & Oprea, T. I. An automated PLS search for biologically relevant QSAR descriptors. J. Comput. Aided Mol. Des. 18, 437–449 (2004).
Sushko, I. et al. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 25, 533–554 (2011).
Cartmell, J., Krstajic, D. & Leahy, D. E. Competitive workflow: novel software architecture for automating drug design. Curr. Opin. Drug Discov. Devel. 10, 347–352 (2007).
Hughes-Oliver, J. M. et al. ChemModLab: a web-based cheminformatics modeling laboratory. In Silico Biol. 11, 61–81 (2011).
Obrezanova, O., Gola, J. M., Champness, E. J. & Segall, M. D. Automatic QSAR modeling of ADME properties: blood–brain barrier penetration and aqueous solubility. J. Comput. Aided Mol. Des. 22, 431–440 (2008).
Fischer, H. & Kansy, M. Automated generation of multi-dimensional structure activity and structure property relationships. US Patent 7400982 (2008).
Rodgers, S. L., Davis, A. M., Tomkinson, N. P. & van de Waterbeemd, H. Predictivity of simulated ADME AutoQSAR models over time. Mol. Inform. 30, 256–266 (2011).
Wood, D. J. et al. Automated QSAR with a hierarchy of global and local models. Mol. Inform. 30, 960–972 (2011).
Keefer, C. E., Kauffman, G. W. & Gupta, R. R. Interpretable, probability-based confidence metric for continuous quantitative structure–activity relationship models. J. Chem. Inf. Model. 53, 368–383 (2013).
Kramer, C. et al. Sharpening the toolbox of computational chemistry: a new approximation of critical f-values for multiple linear regression. J. Chem. Inf. Model. 49, 28–34 (2009).
Livingstone, D. J. & Salt, D. W. Judging the significance of multiple linear regression models. J. Med. Chem. 48, 661–663 (2005).
Kubinyi, H. in Handbook of Chemoinformatics: From Data to Knowledge in 4 Volumes (ed. Gasteiger, J.) 1532–1554 (Wiley-VCH Weinheim, 2003).
Rucker, C., Rucker, G. & Meringer, M. y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 47, 2345–2357 (2007).
Guha, R. On the interpretation and interpretability of quantitative structure–activity relationship models. J. Computer-Aided Mol. Design 22, 857–871 (2008).
Johansson, U., Sonstrod, C., Norinder, U. & Bostrom, H. Trade-off between accuracy and interpretability for predictive in silico modeling. Future Med. Chem. 3, 647–663 (2011).
Carlsson, L., Helgee, E. A. & Boyer, S. Interpretation of nonlinear QSAR models applied to Ames mutagenicity data. J. Chem. Inf. Model. 49, 2551–2558 (2009).
Faulon, J. L., Visco, D. P. Jr & Pophale, R. S. The signature molecular descriptor. 1. Using extended valence sequences in QSAR and QSPR studies. J. Chem. Inf. Comput. Sci. 43, 707–720 (2003).
Spjuth, O., Eklund, M., Ahlberg Helgee, E., Boyer, S. & Carlsson, L. Integrated decision support for assessing chemical liabilities. J. Chem. Inf. Model. 51, 1840–1847 (2011).
Segall, M., Champness, E., Obrezanova, O. & Leeding, C. Beyond profiling: using ADMET models to guide decisions. Chem. Biodivers. 6, 2144–2151 (2009).
Lewis, R. A. A general method for exploiting, QSAR models in lead optimization. J. Med. Chem. 48, 1638–1648 (2005).
Helgee, E. A., Carlsson, L. & Boyer, S. A. Method for automated molecular optimization applied to Ames mutagenicity data. J. Chem. Inform. Model. 49, 2559–2563 (2009).
Griffen, E., Leach, A. G., Robb, G. R. & Warner, D. J. Matched molecular pairs as a medicinal chemistry tool. J. Med. Chem. 54, 7739–7750 (2011).
Dossetter, A. G., Griffen, E. J. & Leach, A. G. Matched molecular pair analysis in drug discovery. Drug Discov. Today 18, 724–731 (2013).
Griffen, E. The rise of the intelligent machines in drug hunting? Future Med. Chem. 1, 405–408 (2009).
Warner, D. J., Bridgland-Taylor, M. H., Sefton, C. E. & Wood, D. J. Prospective prediction of antitarget activity by matched molecular pairs analysis. Mol. Inform. 31, 365–368 (2012).
Hajduk, P. J. & Sauer, D. R. Statistical analysis of the effects of common chemical substituents on ligand potency. J. Med. Chem. 51, 553–564 (2008).
Mills, J. E. J. et al. SAR mining and its application to the design of TRPA1 antagonists. Med. Chem. Commun. 3, 174–178 (2012).
Dalke, A., Bache, E., Van De Waterbeemd, H. & Boyer, S. C-Lab: a web tool for physical property and model calculations. Dalke Scientific [online], (2008).
Gavaghan, C., Arnby, C., Blomberg, N., Strandlund, G. & Boyer, S. Development, interpretation and temporal evaluation of a global QSAR of hERG electrophysiology screening data. J. Comput. Aided Mol. Des. 21, 189–206 (2007).
Dobo, K. L. et al. In silico methods combined with expert knowledge rule out mutagenic potential of pharmaceutical impurities: an industry survey. Regul. Toxicol. Pharmacol. 62, 449–455 (2012).
Austin, R. P. et al. QSAR and the rational design of long-acting dual D2-receptor/β2-adrenoceptor agonists. J. Med. Chem. 46, 3210–3220 (2003).
Brown, A. D. et al. The discovery of indole-derived long acting β2-adrenoceptor agonists for the treatment of asthma and COPD. Bioorg. Med. Chem. Lett. 17, 6188–6191 (2007).
Baur, F. et al. The identification of indacaterol as an ultralong-acting inhaled β2-adrenoceptor agonist. J. Med. Chem. 53, 3675–3684 (2010).
Bruneau, P. Search for predictive generic model of aqueous solubility using Bayesian neural nets. J. Chem. Inf. Comput. Sci. 41, 1605–1616 (2001).
Loughney, D., Claus, B. L. & Johnson, S. R. To measure is to know: an approach to CADD performance metrics. Drug Discov. Today 16, 548–554 (2011).
Kenny, P. W. & Montanari, C. A. Inflation of correlation in the pursuit of drug-likeness. J. Comput. Aided Mol. Des. 27, 1–13 (2013). This study challenges various highly cited papers on the robustness of their conclusions and provides good statistical guidance on studying drug-likeness through database analysis.
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Muthas, D., Boyer, S. & Hasselgren, C. A critical assessment of modeling safety-related drug attrition Med. Chem. Commun. 4, 1058–1065 (2013).
Bennani, Y. L. Drug discovery in the next decade: innovation needed ASAP. Drug Discov. Today 16, 779–792 (2011).
Vaidyanathan, S., Jarugula, V., Dieterich, H. A., Howard, D. & Dole, W. P. Clinical pharmacokinetics and pharmacodynamics of aliskiren. Clin. Pharmacokinet. 47, 515–531 (2008).
Springthorpe, B. et al. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg. Med. Chem. Lett. 17, 6013–6018 (2007).
Lajiness, M. S., Maggiora, G. M. & Shanmugasundaram, V. Assessment of the consistency of medicinal chemists in reviewing sets of compounds. J. Med. Chem. 47, 4891–4896 (2004).
Kutchukian, P. S. et al. Inside the mind of a medicinal chemist: the role of human bias in compound prioritization during drug discovery. PLoS ONE 7, e48476 (2012). This is an investigation into the role of cognitive biases in medicinal chemistry decision-making.
Oprea, T. I. et al. A crowdsourcing evaluation of the NIH chemical probes. Nature Chem. Biol. 5, 441–447 (2009).
Schein, E. H. The Corporate Culture Survival Guide (Wiley, 2009).
Stepan, A. F. et al. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 24, 1345–1410 (2011).
Martin, Y. C. What works and what does not: lessons from experience in a pharmaceutical company. QSAR Comb. Sci. 25, 1192–1200 (2006).
Young, S. S., Yuan, F. & Zhu, M. Chemical descriptors are more important than learning algorithms for modelling. Mol. Inform. 31, 707–710 (2012).
Leach, A. G. et al. Enantiomeric pairs reveal that key medicinal chemistry parameters vary more than simple physical property based models can explain. Med. Chem. Commun. 3, 528–540 (2012).
Hillebrecht, A. & Klebe, G. Use of 3D QSAR models for database screening: a feasibility study. J. Chem. Inf. Model. 48, 384–396 (2008).
Carbonell, P., Carlsson, L. & Faulon, J. Stereo signature molecular descriptor. J. Chem. Inf. Model. 53, 887–897 (2013).
Segall, M. D. Multi-parameter optimization: identifying high quality compounds with a balance of properties. Curr. Pharm. Des. 18, 1292–1310 (2012).
Schneider, G. & Fechner, U. Computer-based de novo design of drug-like molecules. Nature Rev. Drug Discov. 4, 649–663 (2005).
Kutchukian, P. S. & Shakhnovich, E. I. De novo design: balancing novelty and confined chemical space. Expert Opin. Drug Discov. 5, 789–812 (2010).
Segall, M. et al. Applying medicinal chemistry transformations and multiparameter optimization to guide the search for high-quality leads and candidates. J. Chem. Inf. Model. 51, 2967–2976 (2011).
Besnard, J. et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 (2012). This paper demonstrates the value of predictive modelling in developing an expert system for drug design.
Segall, M. Why is it still drug discovery? BioFocus [online], (2008).
Hann, M. M. Molecular obesity, potency and other addictions in drug discovery. Med. Chem. Commun. 2, 349–355 (2011).
Ashby, J. Fundamental structural alerts to potential carcinogenicity or noncarcinogenicity. Environ. Mutagen. 7, 919–921 (1985).
Bergstrom, C. A., Norinder, U., Luthman, K. & Artursson, P. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm. Res. 19, 182–188 (2002).
Steinbeck, C. et al. The Chemistry Development Kit (CDK): an open-source java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 43, 493–500 (2003).
Tetko, I. V. et al. Virtual computational chemistry laboratory — design and description. J. Comput. Aided Mol. Des. 19, 453–463 (2005).
Berthold, M. R. et al. in Data Analysis, Machine Learning and Applications 319–326 (Springer, 2008).
Leach, A. G. et al. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J. Med. Chem. 49, 6672–6682 (2006). This paper introduced the MMPA technique.
Gleeson, P., Bravi, G., Modi, S. & Lowe, D. ADMET rules of thumb II: a comparison of the effects of common substituents on a range of ADMET parameters. Bioorg. Med. Chem. 17, 5906–5919 (2009).
Lewis, M. L. & Cucurull-Sanchez, L. Structural pairwise comparisons of HLM stability of phenyl derivatives: introduction of the Pfizer metabolism index (PMI) and metabolism-lipophilicity efficiency (MLE). J. Comput. Aided Mol. Des. 23, 97–103 (2009).
Dossetter, A. G. A statistical analysis of in vitro human microsomal metabolic stability of small phenyl group substituents, leading to improved design sets for parallel SAR exploration of a chemical series. Bioorg. Med. Chem. 18, 4405–4414 (2010).
Dossetter, A. G., Douglas, A. & O'Donnell, C. A matched molecular pair analysis of in vitro human microsomal metabolic stability measurements for heterocyclic replacements of di-substituted benzene containing compounds — identification of those isosteres more likely to have beneficial effects. Med. Chem. Commun. 3, 1164–1169 (2012).
Dossetter, A. G. A matched molecular pair analysis of in vitro human microsomal metabolic stability measurements for methylene substitution or replacements — identification of those transforms more likely to have beneficial effects. Med. Chem. Commun. 3, 1518–1525 (2012).
Papadatos, G. et al. Lead optimization using matched molecular pairs: inclusion of contextual information for enhanced prediction of hERG inhibition, solubility, and lipophilicity. J. Chem. Inform. Model. 50, 1872–1886 (2010).
Keefer, C. E., Chang, G. & Kauffman, G. W. Extraction of tacit knowledge from large ADME data sets via pairwise analysis. Bioorg. Med. Chem. 19, 3739–3749 (2011).
Warner, D. J., Griffen, E. J. & St-Gallay, S. WizePairZ: a novel algorithm to identify, encode, and exploit matched molecular pairs with unspecified cores in medicinal chemistry. J. Chem. Inform. Model. 50, 1350–1357 (2010).
Acknowledgements
We thank H. Van de Waterbeemd and N. Blomberg for their input to the shaping of this Review, and E. Griffen for providing input on Table 4. We also thank P. Kocis and J. Li for their contributions to AZFilters, and the chemistry community of AstraZeneca for participating in the AZFilters crowdsourcing exercise. Finally we thank the reviewers for their helpful suggestions for improving the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
FURTHER INFORMATION
Supplementary information
Supplementary information S1 (box)
AZFilters (PDF 300 kb)
Glossary
- cLogP
-
The calculated logarithm of the 1-octanol–water partition coefficient of the non-ionized molecule.
- Congeneric series
-
A set of molecules belonging to the same class, usually with chemical changes limited to changes in substituents on a fixed chemical core.
- LogD7.4
-
Log10 of the octanol–water partition coefficient of a molecule (for example, a drug) at pH 7.4.
- Support vector machine
-
A machine learning method that uses kernel functions to map input data into high-dimensional feature space. Support vector machines can be used for classification or regression.
- Random forest
-
A machine learning method that constructs a multitude of decision trees with a random selection of features to split each node. Random forests can be used for classification or regression.
- Ames mutagenicity test
-
A biological assay that uses Salmonella bacteria to test the mutagenic potential of compounds and thereby assess their potential to cause cancer.
- pKa
-
The pH at which a group would be protonated in 50% of molecules. More molecules will become protonated with decreasing pH, and vice versa.
Rights and permissions
About this article
Cite this article
Cumming, J., Davis, A., Muresan, S. et al. Chemical predictive modelling to improve compound quality. Nat Rev Drug Discov 12, 948–962 (2013). https://doi.org/10.1038/nrd4128
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd4128
This article is cited by
-
Integrating synthetic accessibility with AI-based generative drug design
Journal of Cheminformatics (2023)
-
Exploring the Potential of Compounds Isolated from Laranthus micranthus for the Treatment of Benign Prostatic Hyperplasia: Comprehensive Studies on Spectroscopic, Reactivity, and Biological Activity
Chemistry Africa (2023)
-
Tandem Synthesis of Novel thiazole-substituted pyrrolo[1,2-d][1,2,4]triazin-4(3H)-one Derivatives and their Theoretical Pharmacokinetic Profiles
Chemistry of Heterocyclic Compounds (2023)
-
Investigation of Novel Imidazole Analogues with Terminal Sulphonamides as Potential V600E-BRAF Inhibitors Through Computational Approaches
Chemistry Africa (2023)
-
Transformer-based molecular optimization beyond matched molecular pairs
Journal of Cheminformatics (2022)
