
Abstract
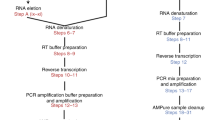
Single-cell analysis of gene expression is increasingly important for the analysis of complex tissues, including cancer, developing organs and adult stem cell niches. Here we present a detailed protocol for quantitative gene expression analysis in single cells, by the sequencing of mRNA 5′ ends. In all, 96 cells are lysed, and their mRNA is converted to cDNA. By using a template-switching mechanism, a bar code and an upstream primer-binding sequence are introduced simultaneously with reverse transcription. All cDNA is pooled and then prepared for 5′ end sequencing, including fragmentation, adapter ligation and PCR amplification. The chief advantage of this approach is the great reduction in cost and time, afforded by the early bar-coding strategy. Compared with previous methods, it is more suitable for large-scale quantitative analysis, as well as for the characterization of transcription start sites, but it is unsuitable for the detection of alternatively spliced transcripts. Sample preparation takes 3 d, and two sets of 96 cells can be prepared in parallel. Finally, the sequencing and data analysis can take an additional 4 d altogether.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Huang, S. Non-genetic heterogeneity of cells in development: more than just noise. Development 136, 3853–3862 (2009).
Elowitz, M.B., Levine, A.J., Siggia, E.D. & Swain, P.S. Stochastic gene expression in a single cell. Science 297, 1183–1186 (2002).
Li, H.H. et al. Amplification and analysis of DNA sequences in single human sperm and diploid cells. Nature 335, 414–417 (1988).
Kurimoto, K. et al. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 34, e42 (2006).
Esumi, S. et al. Method for single-cell microarray analysis and application to gene-expression profiling of GABAergic neuron progenitors. Neurosci. Res. 60, 439–451 (2008).
Taniguchi, K., Kajiyama, T. & Kambara, H. Quantitative analysis of gene expression in a single cell by qPCR. Nat. Methods 6, 503–506 (2009).
Guo, G. et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell 18, 675–685 (2010).
Tang, F. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Islam, S. et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21, 1160–1167 (2011).
Salimullah, M., Sakai, M., Plessy, C. & Carninci, P. NanoCAGE: a high-resolution technique to discover and interrogate cell transcriptomes. Cold Spring Harb. Protoc. published online doi:10.1101/pdb.prot5559 (2011).
Raj, A., Peskin, C.S., Tranchina, D., Vargas, D.Y. & Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309 (2006).
Chubb, J.R., Trcek, T., Shenoy, S.M. & Singer, R.H. Transcriptional pulsing of a developmental gene. Curr. Biol. 16, 1018–1025 (2006).
Jiang, L. et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 21, 1543–1551 (2011).
Kurimoto, K., Yabuta, Y., Ohinata, Y. & Saitou, M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat. Protoc. 2, 739–752 (2007).
Morris, J., Singh, J.M. & Eberwine, J.H. Transcriptome analysis of single cells. J. Vis. Exp. published online doi:10.3791/2634 (2011).
Zhu, Y.Y., Machleder, E.M., Chenchik, A., Li, R. & Siebert, P.D. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30, 892–897 (2001).
Levin, J.Z. et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010).
Kivioja, T. et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74 (2012).
Maheshri, N. & O'Shea, E.K. Living with noisy genes: how cells function reliably with inherent variability in gene expression. Annu. Rev. Biophys. Biomol. Struct. 36, 413–434 (2007).
Tang, F. et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 6, 468–478 (2010).
Tang, F. et al. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE 6, e21208 (2011).
Dalerba, P. et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 29, 1120–1127 (2011).
Lundin, S., Stranneheim, H., Pettersson, E., Klevebring, D. & Lundeberg, J. Increased throughput by parallelization of library preparation for massive sequencing. PLoS ONE 5, e10029 (2010).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Fujita, P.A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 39, D876–D882 (2011).
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5, 621–628 (2008).
Acknowledgements
We thank P. Sekyrova (Karolinska Institutet) for providing cultured cells. This work was supported by grants from the Swedish Foundation for Strategic Research (MDB09-0052) and from the European Research Council (261063).
Author information
Authors and Affiliations
Contributions
S.I. developed the method and performed optimization experiments, analyzed data and co-wrote the manuscript; U.K. performed initial experiments; A.M. performed cell culture and FACS sorting; P.Z. performed experiments to optimize the method; J.-B.F. performed sequencing experiments and advised on method development; P.L. developed the bioinformatics pipeline and analyzed data; S.L. conceived of the method, supervised the development, developed the bioinformatics pipeline and co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.L. has applied for patents covering parts of the method.
Supplementary information
Supplementary Tables 1 and 2
Supplementary table 1: List of Template Switching Oligos (DOCX 134 kb)
Supplementary table 2: Orientation of the template switching oligo in 96 well plate
Rights and permissions
About this article
Cite this article
Islam, S., Kjällquist, U., Moliner, A. et al. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat Protoc 7, 813–828 (2012). https://doi.org/10.1038/nprot.2012.022
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2012.022
This article is cited by
-
Molecular profiling of human non-small cell lung cancer by single-cell RNA-seq
Genome Medicine (2022)
-
Live-seq enables temporal transcriptomic recording of single cells
Nature (2022)
-
High-depth spatial transcriptome analysis by photo-isolation chemistry
Nature Communications (2021)
-
Microdroplet-based one-step RT-PCR for ultrahigh throughput single-cell multiplex gene expression analysis and rare cell detection
Scientific Reports (2021)
-
Dyslexia Candidate Gene and Ciliary Gene Expression Dynamics During Human Neuronal Differentiation
Molecular Neurobiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
