
Abstract
The cytokinesis-block micronucleus cytome assay is a comprehensive system for measuring DNA damage, cytostasis and cytotoxicity. DNA damage events are scored specifically in once-divided binucleated (BN) cells and include (a) micronuclei (MNi), a biomarker of chromosome breakage and/or whole chromosome loss, (b) nucleoplasmic bridges (NPBs), a biomarker of DNA misrepair and/or telomere end-fusions, and (c) nuclear buds (NBUDs), a biomarker of elimination of amplified DNA and/or DNA repair complexes. Cytostatic effects are measured via the proportion of mono-, bi- and multinucleated cells and cytotoxicity via necrotic and/or apoptotic cell ratios. Further information regarding mechanisms leading to MNi, NPBs and NBUDs formation is obtained using centromere and/or telomere probes. The assay is being applied successfully for biomonitoring of in vivo genotoxin exposure, in vitro genotoxicity testing and in diverse research fields such as nutrigenomics and pharmacogenomics as well as a predictor of normal tissue and tumor radiation sensitivity and cancer risk. The procedure can take up to 5 days to complete.
Similar content being viewed by others
Introduction
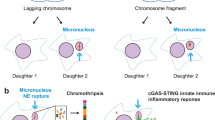
MNi originate from chromosome fragments or whole chromosomes that lag behind at anaphase during nuclear division (Fig. 1)1,2,3,4,5,6. The cytokinesis-block micronucleus (CBMN) assay is the preferred method for measuring MNi in cultured human and/or mammalian cells because scoring is specifically restricted to once-divided BN cells, which are the cells that can express MNi3,4,5,6. In the CBMN assay, once-divided cells are recognized by their BN appearance after blocking cytokinesis with cytochalasin-B (Cyt-B), an inhibitor of microfilament ring assembly required for the completion of cytokinesis3,4,5,6,7. Restricting scoring of MNi in BN cells prevents confounding effects caused by suboptimal or altered cell division kinetics, which is a major variable in micronucleus (MN) assay protocols that do not distinguish between non-dividing cells that cannot express MNi and dividing cells that can6,8,9. Because of its reliability and good reproducibility, the CBMN assay has become one of the standard cytogenetic tests for genetic toxicology testing in human and mammalian cells.

MN originate from either lagging whole chromosomes or acentric chromosome fragments. NPBs originate from dicentric chromosomes that may be caused by misrepair of double strand DNA breaks or telomere end fusions. These events can only be observed in cells completing nuclear division, which are recognized by their BN appearance after cytokinesis blocking with Cyt-B.
Over the past 17 years, the CBMN assay has evolved into a comprehensive method for measuring chromosome breakage, DNA misrepair, chromosome loss, non-disjunction, necrosis, apoptosis and cytostasis (Figs. 1,2,3,4,53,4,5,6,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22. This method is now also used to measure NPBs, a biomarker of dicentric chromosomes resulting from telomere end-fusions or DNA misrepair, and to measure NBUDs, a biomarker of gene amplification20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39. The significance of these developments and the concept of the CBMN assay as a “cytome” assay of chromosomal instability are explained in the following sections. The “cytome” concept implies that every cell in the system studied is scored cytologically for its viability status (necrosis, apoptosis), its mitotic status (mononucleated, BN, multinucleated) and its chromosomal damage or instability status (presence of MNi, NPBs, NBUDs and number of centromere probe signals among nuclei or MNi of BN cell if such molecular tools are used in combination with the assay) (Figs. 1,2,3,4,5). For these reasons, it is now appropriate to refer to this technique as the cytokinesis-block MN cytome (CBMN Cyt) assay.

Using these biomarkers within the CBMN assay, it is possible to measure the frequency of chromosome breakage (MN), chromosome loss (MN), chromosome rearrangement, for example, dicentric chromosomes (NPB), gene amplification (NBUDs), necrosis and apoptosis. In addition, cytostatic effects are readily estimated from the ratio of mono-, bi- and multinucleated cells.

(a) Mononucleated cell; (b) BN cell; (c) multinucleated cell; (d) early necrotic cell; (e) late apoptotic cell; (f) BN cell containing one or more MNi; (g) BN containing an NPB (and a MN); (h) BN cell containing NBUDs. The ratios of mononucleated, BN, multinucleated, necrotic and apoptotic cells are used to determine mitotic division rate or NDI (a measure of cytostasis) and cell death (cytotoxicity). The frequency of BN cells with MNi, NPBs or NBUDs provides a measure of genome damage and/or chromosomal instability. For a wider selection of photomicrographs of different types of cells and biomarkers scored in the CBMN Cyt assay, refer to Fenech et al.75.

The use of molecular techniques for identifying (a) an MN originating from a lagging acentric chromosome fragment, (b) a MN originating from a lagging whole chromosome and (c) non-disjunction of a specific chromosome leading to aneuploid daughter nuclei. The yellow spots in the nuclei and MNi of the BN cells on the left of each panel show the centromeric or kinetochore pattern of staining when pancentromeric probes or kinetochore antibodies are used. The light blue spots in the nuclei and MNi of the BN cells on the right of each panel show the pattern of centromeric staining when a centromeric probe specific to the chromosomes involved in MN formation or non-disjunction events is used. The example shown is for a hypothetical cell with only two pairs of chromosomes. Pancentromeric probes should be used only for distinguishing between MNi originating from chromosome breaks (centromere negative) and chromosome loss (centromere positive). Chromosome-specific centromere probes should be used only to measure malsegregation (owing to non-disjunction or chromosome loss) involving unique chromosomes. It is important to note that pancentromeric probes cannot be used to determine non-disjunction because of the difficulty in reliably counting all centromeres within nuclei.

Pancentromeric and telomeric probes can be used to distinguish (I) between MNi originating from whole chromosome loss (a) and MNi originating from acentric chromosome fragments (b) and (II) NPBs from dicentric chromosomes resulting from misrepair of DNA strand breaks (b) and dicentric chromosomes caused by telomere end fusions (c). The yellow dots represent probes that hybridize to the centromeric region of chromosomes. The light blue dots represent probes that hybridize with the telomeric sequences in chromosomes.
We have proposed that NPBs between nuclei in BN cells should be scored in the CBMN assay because they provide a measure of chromosome rearrangement, which is otherwise not achievable in this assay if only MNi are scored3,22,23,35,36,37,38,39. NPBs occur when centromeres of dicentric chromosomes are pulled to opposite poles of the cell at anaphase. Rarely is it possible to observe dicentric anaphase bridges before the nuclear membrane is formed, because cells proceed through anaphase and telophase rapidly, completing cytokinesis, which ultimately results in breakage of the NPB when the daughter cells separate26. However, in the CBMN assay, BN cells with NPBs are allowed to accumulate because cytokinesis is inhibited and the nuclear membrane is eventually formed around the chromosomes allowing an anaphase bridge to be observed as an NPB.
Various mechanisms could lead to NPB formation following DNA misrepair of strand breaks in DNA39. Typically, a dicentric chromosome and an acentric chromosome fragment are formed that result in the formation of an NPB and an MN, respectively (Figs. 1,2,3 and 5). Misrepair of DNA strand breaks could also lead to the formation of dicentric ring chromosomes and concatenated ring chromosomes which could also result in the formation of NPBs39. An alternative mechanism for dicentric chromosome and NPB formation is telomere end fusion caused by telomere shortening, loss of telomere capping proteins or defects in telomere cohesion40, but in this case the NPB is not necessarily accompanied by an acentric chromosome fragment or an MN. The formation of anaphase bridges and NPBs has been observed in models of rodent and human intestinal cancer in vivo and shown to correlate with telomere length, indicating that NPB formation may also be used as a surrogate measure of critically short telomeres25. Real-time in vitro studies showed that anaphase bridges may be broken in more than one region during late anaphase, leading to the formation of acentric chromosome fragments and eventually MNi26,27. Therefore, some MNi may also originate from broken anaphase bridges although whether this actually happens in cytokinesis-blocked cells remains unclear. The importance of scoring NPBs should not be underestimated because it provides direct evidence of genome damage resulting from misrepaired DNA breaks or telomere end fusions, which is otherwise not possible to deduce by scoring MNi only, which could originate from either acentric chromosome fragments or chromosome loss.
Over the past decade, another unique mechanism of MNi formation, known as nuclear budding, has emerged. This process has been observed in cultures grown under strong selective conditions that induce gene amplification as well as under moderate folic acid deficiency27,28,29,30,31,32,33,34,35,36,37. Shimizu et al.31,32 showed, using in vitro experiments with mammalian cells, that amplified DNA is selectively localized to specific sites at the periphery of the nucleus and eliminated via nuclear budding to form MNi during S phase of mitosis. Amplified DNA may be eliminated through recombination between homologous regions within amplified sequences forming mini-circles of acentric and atelomeric DNA (double minutes), which localize to distinct regions within the nucleus, or through the excision of amplified sequences after segregation to distinct regions of the nucleus. The process of nuclear budding occurs during S phase and the NBUDs are characterized by having the same morphology as an MN with the exception that they are linked to the nucleus by a narrow or wide stalk of nucleoplasmic material depending on the stage of the budding process (Figs. 2 and 3h). The duration of the nuclear budding process, and the extrusion of the resulting MN from the cell remain largely unknown. MNi may also be formed by a budding process following exposure to γ-irradiation33. In this process, Rad 51 recombination protein complexes are detectable throughout the entire nucleus 3 h after irradiation and then concentrate into distinct foci before being extruded from the nucleus as NBUDs.
In a series of studies on folic acid deficiency in long-term primary human lymphocyte cultures, we carefully quantified the interrelationship between MNi, NPBs and NBUDs in an attempt to validate the use of these biomarkers and to determine more comprehensively the impact of folic acid deficiency, within the physiological range, on various aspects of genomic stability35,36,37,38. Folic acid concentration correlated significantly (P<0.0001) and negatively (r = −0.63 to −0.74) with all these markers of chromosome damage, which were minimized at 60–120 nM folic acid. The strong cross-correlation between MN, NPB and NBUD frequency (r = 0.65–0.77; P<0.001) suggests a common mechanism initiated by folic acid deficiency-induced DNA breaks, the most plausible being chromosomal instability generated by breakage–fusion–bridge cycles41.
Expression of MNi could also be used as a surrogate marker of DNA hypomethylation. For example, the DNA methylation inhibitor 5-azacytidine induces undercondensation of the heterochromatin regions of chromosomes 1, 9 and 16, and the specific loss of these chromosomes as MNi in human lymphocytes in vitro42,43. The immunodeficiency, centromeric region instability, facial anomalies syndrome, which is caused by mutation in the DNA methyl transferase DNMT3B gene, is characterized by despiralization of heterochromatin of chromosomes 1, 9 and 16 and loss of this chromatin into NBUDs and MNi44. These events are likely to be relevant to the aging process because Suzuki et al.45 demonstrated that in vitro aging of normal human fibroblasts results in concomitant demethylation of satellite 2 and satellite 3 DNA (which is abundant in the juxta-centromeric DNA of chromosomes 1, 9 and 16) and the increasing frequency of MNi that specifically contained these sequences. It has been conclusively shown that in vivo aging leads to an increased MN frequency in lymphocytes and that loss of the X chromosome in females and males and loss of the Y chromosome in males are among the primary mechanisms explaining this increase6,45,46.
Although it is evident that MN expression could be increased as a result of hypomethylation of satellite DNA, it is also possible that increased genome damage may be caused by hypermethylation of CpG islands within or adjacent to the promoter regions of housekeeping genes involved in cell-cycle check points and DNA repair. For example, CpG island hypermethylation of mitotic spindle check point genes such as APC, BUBR1 and hCDC4 could reduce their expression and therefore increase the possibility of chromosome malsegregation leading to MN formation47,48. Silencing or loss of function of ATM, FANC and BRCA1 or BRCA2 genes involved in DNA repair could also result in higher baseline MN frequencies in human cells49,50,51.
To take full advantage of the CBMN Cyt assay and obtain deeper understanding of the genotoxic mechanism, it is essential to distinguish between MNi originating from whole chromosomes and those originating from acentric fragments as well as to determine whether malsegregation of chromosomes is occurring between nuclei in a BN cell that may or may not contain MNi. This is best achieved by using methods that can detect centromeric regions of chromosomes. The use of MN size as a discriminant is not recommended for human cells or other cell types in which the size of chromosomes is heterogenous because a small MN may contain either a fragment of a large chromosome or a whole small chromosome. Centromeric regions of chromosomes can be detected indirectly using antibodies to the kinetochore proteins that are assembled on centromeres52,53 but this approach does not distinguish between unique chromosomes and may not detect chromosome loss occurring due to absence of kinetochores on defective centromeres54. The preferred method is to use in situ hybridization (ISH) to identify centromeric regions using DNA or peptide nucleic acid pancentromeric probes, which allows MNi containing whole chromosomes to be reliably identified. Furthermore, use of centromeric probes that are specific for a unique chromosome can be used to detect non-disjunctional events (i.e., unequal distribution of unique homologous chromosome pairs in daughter nuclei) in BN cells12,13,14,15,16,17,18,19,55. The types of results that can be expected with the various techniques are illustrated schematically in Figure 4. It is also possible to use a combination of pantelomeric and pancentromeric probes to determine the mechanism of MN and NPB formation as explained in Figure 5. For example, an NPB originating from a telomere end fusion would be expected to have a telomere signal within the bridge, but an NPB originating from a DNA misrepair event is less likely to exhibit a telomere signal within the bridge because double strand breaks are expected to occur more frequently in the non-telomeric sequences because they are much more abundant.
Most chemical agents and different types of radiation have multiple effects at the molecular, cellular and chromosomal level, which may occur simultaneously and to varying extents depending on the dose. Interpretation of genotoxic events in the absence of data on effects on nuclear division rate and necrosis or apoptosis can be confounding because observed increases in genome damage may be due to indirect factors such as inhibition of apoptosis or defective/permissive cell-cycle checkpoints leading to shorter cell-cycle times and higher rates of chromosome malsegregation. Furthermore, determining nuclear division index (NDI) and proportion of cells undergoing necrosis and apoptosis provides important information on cytostatic and cytotoxic properties of the agent being examined that is relevant to the toxicity assessment. In human lymphocytes, the NDI also provides a measure of mitogen response, which is a useful biomarker of immune response in nutrition studies and may also be related to genotoxic exposure3,23,36,37,38,56. The cytome approach in the CBMN Cyt assay is important because it allows genotoxic (MNi, NPBs and NBUDs in BN cells), cytotoxic (proportion of necrotic and apoptotic cells) and cytostatic (proportion and ratios of mono-, bi- and multinucleated cells, NDI) events to be captured within one assay.
In conclusion, the CBMN method has evolved into an efficient “cytome” assay of DNA damage and misrepair, chromosomal instability, mitotic abnormalities, cell death and cytostasis, enabling direct and/or indirect measurement of various aspects of cellular and nuclear dysfunction such as
-
unrepaired chromosome breaks, DNA misrepair, acentric chromosome fragments and asymmetrical chromosome rearrangement (MN or NPB accompanied by MN originating from acentric chromosomal fragments),
-
telomere end fusions (NPB with telomere signals in the middle of the bridge and possibly without accompanying MN),
-
malsegregation of chromosomes due to spindle or kinetochore defects or cell-cycle checkpoint malfunction (MN containing whole chromosomes or asymmetrical distribution of chromosome-specific centromere signals in the nuclei of BN cells),
-
nuclear elimination of amplified DNA and/or DNA repair complexes (NBUDs),
-
chromosomal instability phenotype and breakage–fusion–bridge cycles (simultaneous expression of MNi, NPBs and NBUDs)
-
DNA hypomethylation (specific elevation in the frequency of MN containing whole chromosomes 1, 9 and 16),
-
altered mitotic activity and/or cytostasis (NDI) and
-
cell death by necrosis or apoptosis (ratios of necrotic and apoptotic cells)
The Micronucleus Assay Working Group of the International Workshop on Genotoxicity test procedures concluded that demonstration of cell proliferation in both control and treated cells is compulsory for the acceptance of the test because MN expression is compromised when the proportion of dividing cells is reduced by cytostatic or poor tissue culture conditions9,57,58. It is evident that the best way to eliminate any possible confounding effects of altered cell division kinetics is to use the CBMN Cyt assay regardless of the cell type used by scoring MNi in BN cells, which in addition allows measurement of NPBs as well as NBUDs, cytotoxicity and cytostatic effects. The choice not to use Cyt-B is justifiable only in those rare situations in which cytokinesis defects are being specifically investigated.
The CBMN assay has already been proven to be an effective tool for the study of cellular and nuclear dysfunction caused by in vitro or in vivo aging, micronutrient deficiency or excess, genotoxin exposure and genetic defects in genome maintenance. More recent studies indicate that this method is likely to also prove fruitful in the emerging fields of nutrigenomics and toxicogenomics and their combinations, as it becomes increasingly clear that nutrient status also impacts on sensitivity to exogenous genotoxins59. The ultimate test of the CBMN Cyt assay is its ability to predict disease outcomes prospectively. The Human Micronucleus (HUMN) project (http://www.humn.org), an international collaborative project aimed at determining the variables affecting MN frequency in humans and the pathological significance of this biomarker60, has recently completed a cohort study involving a total of 6,718 subjects from ten countries, screened for MN frequency between 1980 and 2002; a significant increase of all cancer incidence was found for subjects in the groups with medium (RR = 1.84; 95% CI: 1.28–2.66) and high MN frequency (RR = 1.53; 95% CI: 1.04–2.25) relative to those with low MN frequency61. Results from a case–control study of lung cancer in smokers showed that both spontaneous and nicotine-derived nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced MNi are associated with increased lung cancer risk (OR 2.06 and 2.32, respectively) and the association with NPB (OR 29.05 and 45.52, respectively) and NBUDs (OR 6.53 and 10.10, respectively) was even greater62. These initial results indicate the potential predictive value of the CBMN Cyt assay with respect to cancer risk and validate its use as a test for detecting nutritional, environmental and genetic factors that are potentially carcinogenic.
Use of the CBMN Cyt assay in peripheral blood lymphocytes provides the opportunity for a harmonized approach for studying genotoxicity and cytotoxicity both in vitro and ex vivo, which is important for modeling and predicting in vivo effects in humans. This provides the opportunity to test, using the same system, the extent to which the in vitro test is predictive of in vivo genotoxic events. The adoption of the CBMN Cyt assay by the pharmaceutical industry57,58,63, its application in human biomonitoring of genotoxic exposures24,60,61 and its increasing application in preventive medicine and nutrition62,64,65 and the increased investment in the automation of the CBMN Cyt assay (see http://www.imstar.fr/applications/genotoxicity/micronucleiassay/; http://www.metasystems.de/products/metafer/metafer_msearch.htm; http://www.biocompare.com/techart.asp?id=1247) are indicative of the increasing importance of this test66,67,68,69,70,71.
The standard method for the CBMN Cyt assay as applied to isolated lymphocyte cultures and its various modifications as used with other culture systems and cell types are described in detail in the next sections. Timing information can be found in Table 1.
Materials
Reagents
-
Ficoll–Paque, sterile (Amersham Pharmacia Biotech; #17-0840-03, 500 ml, #17-0840-02, 100 ml)
Caution
This product is very stable if bottle remains unopened; however, it can deteriorate if exposed to air for long periods of time. For best results, always date the bottles, use 100 ml bottles and remove from the bottle slowly using a needle and syringe without breaking the seal. Store at 4 °C. Use at room temperature (20–22 °C).
-
Hank's balanced salt solution (HBSS), sterile, with calcium and magnesium, without phenol red (Trace, #11-101-0500V). Store at 4 °C
Critical
Must be used at room temperature (20–22 °C).
-
RPMI 1640 medium, without L-glutamine, sterile liquid (Trace, #11-066-0500V, 500 ml or #11-066-100V, 100 ml). Store at 4 °C. Use at 37 °C when preparing cultures.
-
Fetal bovine serum (FBS), heat-inactivated, sterile (Trace, #15-010-0100V, 100 ml). Store frozen at −20 °C. Thaw in a 37 °C water bath before adding to the culture medium. Once thawed FBS will remain stable at 4 °C for 3–4 weeks
Caution
Avoid repeated refreezing of the thawed FBS.
-
L-Glutamine solution, 200 mM sterile solution, cell-culture tested (Sigma, #G7513, 20 ml). Store frozen at −20 °C in 1.1 ml aliquots. Thaw at room temperature before adding to the culture medium
-
Sodium pyruvate solution, sodium pyruvate 100 mM, sterile solution (Trace, #21-154-0100V, 100 ml). Store frozen at −20 °C in 1.1 ml aliquots. Thaw at room temperature before adding to the culture medium
-
Cyt-B (Sigma, #C6762)
Caution
This material is toxic and a possible teratogen. It must always be purchased in sealed vials. The preparation of this reagent must be carried out in a cytoguard cabinet and the following personal protection must be used: Tyvek gown, P2 dust mask, double nitrile gloves and safety glasses.
-
DMSO (Sigma, hybrimax, sterile–filtered, #D2650)
-
Phytohemagglutinin (PHA; Abbott Murex, #HA15 8E27)
-
Isoton II (Coulter Electronics, #8546719)
-
Zapoglobin (Coulter Electronics, #9366013)
-
DePex (or DPX) mounting medium (Ajax, 3197)
-
1% (w/v) hypochlorite solution for disinfecting cytocentrifuge cups. Change weekly
-
Diff-Quik staining set (Lab-Aids LP64851, supplied by Surgical and Medical). Consists of fixative, solution 1 (orange) and solution 2 (blue). Stains can be reused and can last several months depending on usage. Diff-Quik is a modified Wright-Giemsa stain
-
Methanol (Ajax Univar, #318) for fixing cells. This fixative can be used in place of the Diff-Quik fixative once it runs out
-
0.85% (w/v) isotonic saline, sterile, for the preparation of PHA solution: add 4.25 g sodium chloride to 500 ml Milli-Q water, autoclave at 121 °C for 30 min and then store at 4 °C
Equipment
-
Sterile vacutainer blood tubes with lithium heparin as anticoagulant (e.g., Greiner vacuette tubes 9 ml, #455084 No gel)
-
70 ml polystyrene gamma sterile containers (e.g., Sarstedt 75.9922.333)
-
10 ml graduated sterile pipettes (Falcon #7551)
-
Preferred tube for Ficoll separation—dependant on the blood volume: 50 ml polystyrene, sterile, conical (Falcon #2073), 15 ml polypropylene, sterile, conical (Falcon #2096), 10 ml polystyrene, yellow capped, sterile, V-bottom (Disposable Products #21829)
-
Sterile plugged Pasteur pipettes 9″ (22–23 cm) (Chase#93P)
-
200–1,000 μl Finnpipette + sterile 1 ml tips (Labsystems, Edward Instruments Co. #1040-800 Blue)
-
40–200 μl Finnpipette + sterile 200 μl tips (Labsystems, Edward Instrument Co. #1030-800 Yellow)
-
Biological safety cabinet (e.g., Gelman Sciences Class II BH Series)
-
Bench top centrifuge, biosafe with sealed buckets, capable of spinning at 1,000g (e.g. MSE Mistral 2000R)
-
Cell counter (e.g., Coulter Electronics model ZB1). A hemocytometer can be used instead if an electronic cell counter is not available
-
Counting vials 15 ml (Johns diluent vials #DSV002)
-
Preferred tube for culturing cells is dependant on the final culture volume. Use 6 ml polystyrene, vented cap, gamma-sterilized, round bottom culture tube (Falcon #2058), for 750 μl isolated lymphocyte culture
-
Single-use 0.22 μm filter units, syringe-driven (Millipore-GS, #SLGS0250S)
-
0.2 μm hydrophobic filter (Millipore, Dualex #SSLOVS25XS)
-
Cytocentrifuge (e.g., Shandon Cytocentrifuge from Thermo Electron Corporation, http://www.thermo.com)
-
Microscope slides, frosted end, 76 × 26 mm (3 × 1″), 1 mm thick (HD Scientific MZO2 110)—wiped with alcohol and allowed to dry before use
-
Filter cards—Shandon 3 × 1″, thick, white, boxes of 200 (from Thermo Electron Corporation)
-
Cytocentrifuge cups—supplied with instrument from Thermo Electron Corporation. Must be clean and completely dry before assembly
-
Metal slide holders—12 supplied with instrument from Thermo Electron Corporation
-
Slide holder opener—supplied with instrument from Thermo Electron Corporation
-
Staining racks and containers
-
Coverslips, no. 1, 22 × 50 mm (HD Scientific MZ LD2250)
-
Tissues or Whatman no. 1 filter paper, no. 371001240, 24 cm diameter (from Lab Supply)
-
Microscope with excellent optics for bright-field and fluorescence examination of stained slides at ×1,000 magnification (e.g., Leica DMLB, Nikon Eclipse 600)
-
Slide storage boxes (Kartell, no. 278)
Reagent setup
-
Culture medium Prepare by adding 90 ml of RPMI 1640 medium with 10 ml of 100% FBS, 1 ml of 200 mM L-glutamine and 1 ml of 100 mM sodium pyruvate solution.
Prepare the culture medium in sterile tissue-culture-grade glass or plastic bottles. Smaller volumes of culture medium may be prepared depending on the number of cultures required. Culture medium can be stored for 1 week at 4 °C before use. It is not usually necessary to sterile filter the prepared culture medium if rigorous aseptic technique is followed; however, to be absolutely certain, medium may be sterile-filtered using a 0.22 μm filter.
-
Cyt-B Dissolve 5 mg solid in 8.33 ml DMSO to give a Cyt-B solution concentration of 600 μg ml−1 as follows:
-
1
Remove the Cyt-B vial from −20 °C and allow to reach room temperature. Do not remove the seal.
-
2
Sterilize the top of the rubber seal with ethanol and allow the ethanol to evaporate.
-
3
Pipette 8.33 ml of DMSO into a 50 ml sterile Falcon tube.
-
4
Vent the vial seal with a 25 G needle and a 0.2 μm hydrophobic filter to break the vacuum.
-
5
Using a 5 ml sterile syringe and another needle, inject 4 ml of the 8.3 ml DMSO through the seal.
-
6
Mix contents gently. Cyt-B should dissolve readily in DMSO. Leave syringe and needle in place.
-
7
Remove the 4 ml from the vial into the syringe and eject into a labeled sterileTV10 tube.
-
8
Aspirate the remaining 4.3 ml of DMSO into the syringe and inject into the vial as before.
-
9
Remove this final volume from the vial and eject into the TV10 tube.
-
10
Mix and dispense 100 μl aliquots into sterile, 5 ml polystyrene yellow capped tubes labeled Cyt B with the date.
-
11
Store at −20 °C for up to 12 months. The vials of powder are guaranteed by Sigma for 2 years if stored at −20 °C.
-
1
-
PHA The preparation of this reagent has been modified slightly from the instructions in the product insert to suit this protocol. 45 mg solid is dissolved in 20 ml sterile isotonic saline to make a solution of 2.25 mg ml−1 as follows:
-
1
Remove the vial from the refrigerator and allow to reach room temperature. Do not remove the seal.
-
2
Sterilize the top of the rubber seal with ethanol and allow the ethanol to evaporate.
-
3
Pipette 20 ml of sterile isotonic saline into a 50 ml sterile Falcon tube.
-
4
Vent the seal with a 25G needle and a 0.22 μm hydrophobic filter to break the vacuum.
-
5
Using a 10 ml sterile syringe and another needle, inject 5 ml of the 20 ml isotonic saline into the vial and dissolve the solid.
-
6
Remove the 5 ml solution from the vial into the syringe and eject into another labeled sterile 50 ml Falcon tube.
-
7
Add another 5 ml of isotonic saline to the vial, using the syringe and mix gently.
-
8
Remove this volume from the vial and eject into the labeled Falcon tube.
-
9
Add the remaining 10 ml of isotonic saline to the labeled Falcon tube.
-
10
Dispense 500 μl aliquots into sterile, labeled 1.2 ml vials (cryovial type).
-
11
Store at 4 °C for up to 6 months.
Note: The 500 μl aliquots are single-use only. Discard any remaining solution in vial. Discard any vial with signs of bacterial contamination. Slight turbidity may occur but this does not affect the activity of PHA.
-
1
Procedure
Isolation and counting of leukocytes
Timing approximately 3 h
-
1
Collect fresh blood by venipuncture into vacutainer blood tubes with lithium heparin anticoagulant. Keep blood tubes at room temperature (i.e., 20–22 °C). The volume taken is dependent on the number of cells needed. (Usually one can expect to collect up to 1 × 106 leukocytes per 1 ml blood.)
Caution
All procedures until slide preparation must be carried out aseptically in a class II Biological Safety Cabinet.
-
2
Dilute whole blood 1:1 with HBSS (20–22 °C) and gently invert to mix.
-
3
Gently overlay diluted blood onto Ficoll–Paque using a ratio of 1:3 (e.g., 2 ml Ficoll–Paque:6 ml diluted blood) being very careful not to disturb the interface.
-
4
Weigh and balance the centrifuge buckets before spinning the tubes at 400g for 30 min at 18–20 °C.
-
5
Remove the leukocyte layer located at the interface of Ficoll–Paque and diluted plasma into a fresh tube, using a sterile, plugged, Pasteur pipette, taking care not to remove too much Ficoll–Paque.
-
6
Dilute the leukocyte suspension (which includes a mixture of lymphocytes and monocytes) with 3 × its volume of HBSS at room temperature (20–22 °C), mix gently and then centrifuge at 180g for 10 min.
-
7
Discard the supernatant and resuspend the cell pellet (slightly beige color) in 2× the volume removed of HBSS, using a Pasteur pipette, then centrifuge at 100g for 10 min at 18–20 °C.
-
8
Discard the supernatant and gently resuspend the cells in 1 ml culture medium at room temperature, using a Pasteur pipette.
-
9
Count the cells using the Coulter Counter.
-
10
Set instrument settings for counting human lymphocytes (e.g., Coulter Counter Model ZB1; threshold: 8, attenuation: 1, aperture: 1/4, manometer: 0.5 ml).
-
11
Dilute 15 μl cell suspension into 15 ml isoton + five drops of zapoglobin to lyse any residual red blood cells.
-
12
Perform the count in triplicate to determine the mean. If the cell count is >15,000, count 7.5 μl cells instead, then multiply by 2. Multiply the value by 2,000 (dilution factor of 1,000 and counting volume of 0.5 ml) to give the number of cells per ml.
-
13
To set the cells up at 1 × 106 per ml in 750 μl culture medium, divide 750 (μl) by the number of cells per ml and multiply by 1. This gives the volume of cells to use in “μl”. Make this volume up to 750 μl with medium.
Example: mean cell count = 12,586
12,586 × 2,000 = 25.2 × 106 cells per ml
750 μl/25.2(× 106) × 1(× 106) = 29.8 μl
29.8 μl cell suspension + 720.2 μl medium = cell suspension at 1 × 106 per ml in 750 μl.
Pause point
Cells may be left in medium at this point at room temperature in the dark for 1–2 h with the tubes sealed.
Culture of lymphocytes, addition of Cyt-B and harvesting of cells
Timing 72 h
-
14
Using the above calculation (Step 13), resuspend the cells at 1 × 106 per ml into the culture medium. Add the calculated volume of medium into labeled 6 ml round-bottomed culture tubes, then add the cells into the medium to make up a final volume of 750 μl. Set up duplicate cultures per subject and/or treatment studied (Fig. 6).
Figure 6 
An optimal sampling and scoring plan for the CBMN Cyt assay that enables an estimation of experimental variation (results for A + C versus B + D) as well as the effect of scorer bias (results for A + B versus C + D).
-
15
Stimulate mitotic division of lymphocytes by adding PHA. Add 10 μl of the PHA solution to 750 μl culture to give a final concentration of 30 μg ml−1. Discard the remaining PHA after use.
-
16
Incubate the cells at 37 °C with lids loose in a humidified atmosphere containing 5% CO2 for exactly 44 h.
Critical Step
Cyt-B must be added exactly 44 h after PHA stimulation.

Addition of Cyt-B to culture
-
17
Thaw out stock vial containing 100 μl solution of Cyt-B in DMSO at 600 μg ml−1.
Caution
Carry out all manipulations of the Cyt-B solutions in a fume hood with the base glass window down to chest level and use the following personal protection: Tyvek gown, double nitrile gloves and safety glasses. These precautions are necessary for protection against biohazardous and cytotoxic aerosols.
-
18
Aseptically add 900 μl of culture medium equilibrated to room temperature to the vial to obtain a 1,000 μl solution of 60 μg ml−1 Cyt-B.
-
19
Remove 56 μl of medium from the top of the 750 μl culture and replace with 56 μl of the 60 μg ml−1 Cyt-B solution to give a final Cyt-B concentration of 4.5 μg ml−1.
-
20
Return cultures to the incubator and incubate the cultures for a further 28 h. Discard the remaining Cyt-B solution.
-
21
At 24–28 h after addition of Cyt-B, harvest cells for slide preparation and scoring according to the procedure given below. The chosen harvest time should maximize the proportion of BN cells and minimize the frequency of mononucleated and multinucleated (three or more nuclei) cells.
Harvesting of cells using cytocentrifugation
Timing Approximately 30 min
-
22
Follow the manufacturer's instructions for assembly of the slides, filter-cards and cytocentrifuge cups within the cytocentrifuge rotor. Cytocentrifugation is performed at 18–20 °C.
-
23
Prepare a concentration of cells that is sufficient to produce a monolayer of cells on each spot. It may be necessary to spin down a culture gently and take off a proportion of the supernatant to obtain an optimal concentration of cells on the slide following cytocentrifugation. In the case of lymphocyte cultures in round-bottomed tubes, it is only necessary to take off the supernatant (approximately 300 μl from a 1 ml culture), as the cells tend to settle in the incubator.
Caution
Loading and cytocentrifugation of cell culture sample (Steps 23–28) must be carried out in a fume hood or preferably in an approved cytoguard cabinet. Appropriate safety protection including gloves must be worn.
-
24
Resuspend the cells well using a Pasteur or Gilson pipette to disaggregate the cells. It is best to suspend all the cultures to be harvested first, then return to each one to resuspend just before loading into the sample cup.
-
25
Working quickly, add 100–200 μl of cell suspension to the well of each sample cup. The typical volume for cytokinesis-blocked lymphocyte cultures is 120 μl. The required volume may need slight adjustment depending on the concentration of cells in the culture and the optimal cell density for slide scoring, which is determined by trial and error.
-
26
Replace the lid on the rotor and press down on the button in the center to lock. Place the rotor in the cytocentrifuge and click the main lid shut. Set the time and speed parameters recommended by the manufacturer.
-
27
Press START. The machine will run the program, stop and slow automatically.
-
28
Returning the rotor to the fume hood, follow the manufacturer's procedure for opening each slide holder.
Drying, fixing and staining of cells and slide preparation
Timing Approximately 30 min
-
29
Dismantle each slide holder, taking care not to smear the spots. Discard the filter cards into the biohazard bin. Place the sample cups in the 1% hypochlorite solution to soak for at least 10 min before rinsing carefully six times in pure water and drying. Do not use direct heat to dry the cups.
Caution
Appropriate protection including nitrile gloves must be worn while staining samples.
-
30
Place the slides horizontally on a slide tray and allow the cells to air-dry for exactly 10 min at room temperature.
Critical Step
Do not air-dry slides for longer than 10 min, otherwise cellular and nuclear morphology is altered making it difficult to score the slides.
-
31
Place slides vertically in a dry staining rack and place in methanol or Diff-Quik fixative for 10–15 min. The methanol can be reused.
-
32
Transfer the rack without delay to Diff-Quik solution 1 (orange) and stain for 6 s while moving the rack back and forth. Increase staining time if required.
-
33
Transfer the rack without delay to Diff-Quik solution 2 (blue) and stain for another 6 s while moving the rack back and forth. Again, staining time can be increased if required.
Critical Step
Staining time is determined by trial and error depending on how fresh the stains are. The aim is to obtain optimal contrast between nuclear and cytoplasmic staining so that various biomarkers in the CBMN Cyt assay are easily and unequivocally scored.
-
34
Keeping the slides in the rack, wash the slides very briefly and gently with tap water and rinse with pure water, making sure that the slides are not destained by residual water droplets on cells.
-
35
Immediately place the slides face-down on paper tissues (or better, Whatman no. 1 filter paper) to blot away any residual moisture. Do not place any pressure or rub on the cell spots.
-
36
Place the slides on a slide tray and allow to dry for about 10–15 min.
-
37
Examine the cells at ×100 and ×400 magnification to assess efficiency of staining and the density of the cells, remembering that for the CBMN Cyt assay, it is necessary to have at least 1,000 BN cells to score. Restaining can be carried out at this stage if the staining is too light by repeating Steps 30–37. If the cell density is too heavy or light, concentrate or dilute the cells as necessary and repeat the spinning and staining steps.
-
38
If the slides are satisfactory, seal the tubes or flasks containing the cultures and discard in the biohazard bin.
-
39
Leave the slides to dry completely for at least 30 min before putting coverslips on.
Pause point
Slides can be left overnight at room temperature to dry.
Coverslipping and storage
-
40
Place the slides to be coverslipped on tissue or paper and set out one coverslip alongside each.
Caution
Carry out coverslipping in a fume hood to avoid inhalation of organic solvent in DePex and leave slides there until completely dry.
-
41
Put two large drops of DePex (use a plastic dropper) on each of the coverslips in the approximate area where the spots correspond.
Caution
Wear nitrile gloves when applying DePex medium.
-
42
Invert the slide over the coverslip and allow the DePex to spread by capillary action. Slide the coverslip gently to and fro to expel any excess DePex and air bubbles. Ensure that the spots do not have air bubbles over them.
-
43
Wipe excess DePex from the edges of the slide and ensure that the medium or glass does not cover any of the frosted label area, as a coding label will not stick on these.
-
44
Place the slides on a tray and leave overnight in the fume hood to dry.
-
45
Store slides in slide boxes at room temperature and code with a sticky label over the frosted area before scoring.
-
46
Score the biomarkers in the CBMN Cyt assay using the criteria described in Box 1. See Box 2 for details regarding the data that should be obtained from each slide. Box 3 explains how the NDI is calculated, as well as the purpose of doing the measurement. The basic elements of a typical score sheet are listed in Table 2 and an example of a score sheet is given in Table 3. For a comprehensive photographic gallery of the various cell types scored in the CBMN Cyt assay, refer to Fenech et al.72. Figures 7,8,9,10 provide diagrammatic explanations of the criteria for selecting BN cells suitable for scoring presence of MNi, NPBs and NBUDs (Fig. 7), the other various cell types that are observed and scored in the CBMN Cyt assay (Fig. 8), typical size and appearance of MNi (Fig. 9) and other structures in BN cells that resemble MNi but in fact represent other abnormal cellular events (Fig. 10).
Table 2 Information that should be included on a score sheet for the CBMN Cyt assay. Table 3 Example of CBMN Cyt assay score sheet. Figure 7: Criteria for choosing binucleate cells in the cytokinesis-block MN assay. 
(a) Ideal binucleate cell; (b) binucleate cell with touching nuclei; (c) binucleate cell with narrow NPB between nuclei; (d) binucleate cell with relatively wide NPB. Cells with two overlapping nuclei may be considered suitable to score as BN cells if the nuclear boundaries are distinguishable. Occasionally, BN cells with more than one NPB are observed.
Figure 8: The various types of cells that may be observed in the in vitro cytokinesis-block MN assay excluding BN cells. 
These cell types shown should not be scored for MN frequency: (a) viable mono-, tri- and quadrinuclear cells; (b) mononucleated and BN cells at early stage of apoptosis when chromatin condensation has occurred but nuclear membrane has not disintegrated and late-stage apoptotic cells with intact cytoplasm, no nucleus and apoptotic chromatin bodies within the cytoplasm; (c) cells at the various stages of necrosis including early stages showing vacuolization, disintegration of cytoplasmic membrane and loss of cytoplasm with an intact nucleus and late stages in which cytoplasm is partially or completely lost and nuclear membrane is visibly damaged and nuclear material is commencing to leak from the remnant nucleus.
Figure 9: Typical appearance and relative size of MNi in BN cells. 
(a) Cell with two MNi one with 1/3 and the other 1/9 the diameter of one of the main nuclei within the cell. (b) MNi touching but not overlapping the main nuclei. (c) A BN cell with NPB between main nuclei and two MNi. (d) A BN cell with six MNi of various sizes; this type of cell is rarely seen in cells that are not exposed to high doses of genotoxins.
Figure 10: Occasionally, BN cells (or cells that resemble BN cells) may contain structures that resemble MNi but should not be scored as MNi originating from chromosome loss or chromosome breakage. 
These situations include (a) a trinucleated cell in which one of the nuclei is relatively small but has a diameter greater than 1/3 the diameter of the other nuclei; (b) dense stippling in a specific region of the cytoplasm; (c) NBUD that appears like an MN with a narrow nucleoplasmic connection to the main nucleus and (d) nuclear blebs consisting of nuclear material protruding from the nucleus but without an obvious constriction or bridge between the protruding nuclear material and nucleus.




Troubleshooting
Blood and cell storage conditions
The published evidence available suggests that storage of blood between 5 and 22 °C for up to 24 h has no significant impact on baseline or radiation-induced MN frequency73; however, these observations need to be further verified and replicated. It is possible to perform the CBMN Cyt assay using cryopreserved lymphocytes, but there are conflicting reports on whether cryopreservation alters the frequency rate of MN in BN cells74,75, which means that it is essential to optimize and verify that the freezing and thawing protocol used does not induce DNA damage.
Mitogen stimulation
The optimal PHA concentration for maximizing proportion of BN cells should be verified for each batch of PHA using the BN frequency ratio and NDI to assess mitogenesis. The success of mitogen stimulation can be determined visually 24 h following PHA stimulation. If lymphocytes have been successfully stimulated, cultures will appear clumpy and grainy. Unstimulated cultures will appear cloudy and silty.
Time of addition of Cyt-B and Cyt-B concentration
The critical aspect regarding the time of addition of Cyt-B is to ensure that it is added before the first mitotic cells start to appear so that all the observed BN cells that are captured are in fact once-divided cells only. This is important because MN tend to get lost in subsequent divisions and the MN frequency in a second division cell is likely to be less than that in a first division cell after a genotoxic insult4,5. Cyt-B may take up to 6 h before it starts to exert its cytokinesis-blocking action (unpublished observations), which means it should be added at least 6 h before cells start to enter M phase of the cell cycle. The optimal time to add Cyt-B with lymphocytes is usually 44 h after PHA stimulation; however, earlier addition of Cyt-B is acceptable if the culture conditions used cause an earlier than expected mitotic wave to occur.
To capture all once-divided cells as BN cells, it is also essential to verify that the Cyt-B concentration is optimal to maximize the ratio of cytokinesis-blocked cells by doing a dose–response across the concentration range of 2–10 μg ml−1 of Cyt-B using a 24 h cytokinesis-blocking time. The dose–response should yield a plateau in response within the optimal concentration range for cytokinesis-blocking. Choose a concentration that is at least a dose-point past the inflection point of the dose–response and on the plateau. Experience has shown that concentrations of 4.5 and 6 μg ml−1 are optimal for isolated lymphocyte and whole blood lymphocyte cultures, respectively. These concentrations are usually also optimal for mammalian cell lines but this should be checked for each cell line.
Cytokinesis-blocking time
In maximizing the number of cytokinesis-blocked BN cells by increasing exposure time to Cyt-B, there is the risk of also increasing the proportion of cytokinesis-blocked multinucleated cells, which arise from BN cells that attempt another nuclear division while cytokinesis-blocked. Ideally, the proportion of BN cells among cytokinesis-blocked cells should be in excess of 80%. The proportion of BN and multinucleated cells among all cells will depend on the proportion of dividing cells in the culture and the cell-cycle time, which in turn depends on the cell line and the culture conditions. For human cells, a cytokinesis-blocking period of 24 h is usually optimal for most conditions. In situations when mitotic delay may be expected owing to high levels of DNA damage, a longer cytokinesis-block period may be considered to capture late dividing cells as BN cells.
Controls and important variables
It is important to include a concurrent control sample from a single individual or cell line with each assay run throughout a study period to verify that there are no major variations in experimental conditions that may have impacted on the observed results. Furthermore, a positive control in which cells are challenged with an MN-inducing agent (e.g., ionizing radiation, mitomycin-C) is also recommended to verify that the system responds predictably from one assay batch to the next.
Historical control data are particularly useful in human biomonitoring studies aimed at establishing normal range values for the CBMN assay and to identify key variables affecting the observed frequency of CBMN assay biomarkers, which typically include age, gender, B vitamin status (particularly folate and vitamin B12) and smoking status. It is important in case–control studies that the groups are matched properly for these variables. For more information on the use of controls in biomonitoring studies refer to Albertini et al.76 and for important variables affecting MN frequency in the CBMN assay in human lymphocytes refer to Bonassi et al.77. The appropriate use of controls in in vitro studies is discussed in detail in the report of the In Vitro Micronucleus Assay Working Group57.
Hypotonic treatment of cells
Hypotonic treatment for slide preparation is not recommendable because it may destroy necrotic cells and apoptotic cells, making them unavailable for assay. Inclusion of necrosis and apoptosis is important for the accurate description of mechanism of action and measurement of cellular sensitivity to a chemical or radiation. Isolated lymphocyte culture assay or culture of cell lines does not require hypotonic treatment of cells for slide preparation, thus making it possible to preserve the morphology of both necrotic and apoptotic cells.
Slide preparation and staining
In our experience, most of the problems in the CBMN Cyt assay arise during slide preparation and staining. This is because the quality of the score depends on the quality of the slide. Main points to note: (i) avoid cell clumps by gently resuspending cells before harvest and transfer to slides; (ii) maintain a moderate cell density so that it is relatively easy to identify cytoplasmic boundaries; (iii) stain only one slide initially to ensure that staining is optimal before staining the whole batch.
Number of BN cells that should be scored
One of the most common questions is the number of BN cells to be scored in the CBMN assay. The accepted protocol is to score a minimum of 1,000 BN cells per treatment or time point although reports vary between 500 and 2,000 BN cells. An alternative approach is to keep on scoring BN cells until a fixed number of MNi are observed (e.g., 45 MNi). The latter has the advantage that more BN cells are scored when fewer MNi are induced, thus maintaining similar statistical power across different treatments. The main disadvantage is that more than 2,000 cells may have to be scored in cultures with low MN frequency. In our experience, scoring 1,000 BN cells from each of the duplicate cultures (total 2,000 BN cells) always yields robust results.
Slide scoring
With respect to scoring slides, it is best to first score the frequency of mononucleated, BN, multinucleated, apoptotic and necrotic cells to determine the NDI and frequency of apoptotic and necrotic cells and then focus on scoring BN cells for the presence of MNi, NPBs and NBUDs to determine the genome damage rate. Note that it is best to skip scoring a cell if one is uncertain on how to classify it.
Attention should be given to ensure that MNi, NPBs and NBUDs are scored only in BN cells and not in multinucleated cells because multinucleated cells are not once-divided cells and tend to have greatly elevated MNi frequencies relative to BN cells78, which would result in inaccurate genome damage estimates. This issue is particularly important in dense cell preparations, because it may be difficult to distinguish between the two cell types. The elevated MNi frequency in multinucleated cells is artifactual and caused by inability of the spindle to properly segregate the large number of chromosomes in multinucleated cells. For these reasons, it is important to optimize the CBMN Cyt assay protocol to maximize the frequency of BN cells and at the same time minimize the frequency of multinucleated cells.
Reproducibility
The use of duplicate cultures is critical for producing robust results because it also allows the measurement of the intraexperimental coefficient of variation (CV). Cytogenetic assays should be subject to the same rigor as analytical assays, which typically reject duplicate results with a CV greater than 10%. Owing to the visual scoring, greater latitude in the acceptable CV is understandable. However, in our experience and the results of international interlaboratory scoring comparison79, CVs greater than 40% are not acceptable for baseline data. With radiation-exposed cultures in which more than 100 MN per 1,000 BN cells are induced, CVs less than 20% are expected. Acceptable CVs for automated systems have yet to be established, but CVs of between 5.4% and 9.5% have been reported recently (see http://www.imstar.fr/applications/genotoxicity/micronucleiassay/). Scores from inexperienced personnel (e.g., students, new staff) should not be relied upon until they are able to achieve acceptable CVs (no greater than 40%) for repeat scores of standard control slides and absolute values that are within close range to those obtained by experienced personnel. Interscorer variability is one of the key sources of variation in the MN assay79. It is therefore essential that the same scorers are maintained throughout a single study and ideally two scorers are used, each providing a count from each of the duplicate cultures and their mean values calculated to take into account both experimental and scorer variation; however, it is also acceptable to use a single scorer if two experienced scorers are not available. An alternative approach is to calibrate scorers by using a common set of standard slides with “low”, “medium” and “high” MN frequencies. The scores of each scorer on the standard slides can then be used to calculate a corrected value. The latter approach is still in development but worth noting as an option because it can take account of differences in the visual capacity of scorers within the same laboratory and between laboratories. Another important source of variability between scorers and between laboratories is the quality of the microscopes and their optics. In our experience, scoring of NPBs is influenced by the quality of the microscope because fine bridges can be missed with low-quality optics. The main issue here is for scorers to avoid switching microscopes during experiments and for the laboratory manager to upgrade the optics of the microscopes to a uniform and high level whenever possible. An optimal sampling and scoring schedule for the CBMN Cyt assay is shown in Figure 6.
Is it necessary to heat-inactivate FBS in the CBMN Cyt assay?
Heat-inactivation (56 °C for exactly 30 min) is only required if it is considered necessary to destroy heat-labile complement proteins that may cause cell lysis. Triglia and Linscott80 determined that levels of complement in FBS are a fraction of adult levels and in ten samples of commercial FBS no hemolysis was detected even in undiluted serum. Furthermore prewarming of FBS to 37 °C is enough to inactivate heat-labile complement. In addition, other heat-labile components such as certain vitamins (e.g., folic acid), growth factors, amino acids, etc. may be diminished by the heat treatment. Therefore, the choice to use heat-inactivated FBS or nontreated FBS is optional and depends on whether the heat inactivation of FBS causes any significant effects on growth and genome stability of cells tested in the CBMN Cyt assay. The CBMN Cyt assay for lymphocytes can be performed using untreated FBS.
Timing
Timing information can be found in Table 1.
Anticipated results
The anticipated results with the CBMN Cyt assay depend on the culture conditions, level of exposure to genotoxic or cytotoxic agents and their potency, the nutrient composition of the medium and the genetic background, age and gender of the donor of the cells being tested.
For normal peripheral blood lymphocytes cultured under optimal conditions using the protocol described above, one can usually expect the following ranges of values
-
frequency of BN cells: 30–60%,
-
NDI: 1.3–2.2,
-
necrotic cells: 0–9%,
-
apoptotic cells: 0–7%,
-
MNi per 1,000 BN cells: 0–30,
-
NPBs per 1,000 BN cells: 0–10,
-
NBUDs per 1,000 BN cells: 0–5.
MNi frequency in human peripheral blood lymphocytes measured using the CBMN Cyt assay increases with age and tends to be higher in females relative to males by a factor of approximately 1.4 owing to the random loss of the inactive X chromosome, which may lag at anaphase as a result of defective centromere or kinetochore function6,13,65,46.
The frequency of the biomarkers in the CBMN Cyt assay may vary in their extent and relative to each other depending on the genotoxic and cytotoxic mechanism of specific chemicals, radiation type and nutrient deficiency at different doses. For example, X-rays are efficient in inducing genome damage biomarkers with relatively small effects on NDI and cell death, whereas on the other hand, exposure to hydrogen peroxide tends to induce high levels of cell death with relatively low levels of DNA damage biomarkers in human lymphocytes6,20,23,39. Cells with genetic defects in DNA repair, antioxidant response or apoptosis may exhibit abnormally high levels of MNi, NPBs, NBUDs, necrosis and apoptosis as well as reduced NDI.
Risk of false negative results if the assay is performed without cytokinesis-block
There has been an interest in exploring the possibility of performing the in vitro MN assay without Cyt-B to minimize concern of a possible confounding effect of Cyt-B while running the potential risk of obtaining a false negative result because of inadequate control of cell division kinetics because, for example, inhibition of nuclear division by the agent being tested is likely to lead to an underestimate of MN expression given that both once-divided and nondivided cells are included in the denominator of the MN frequency ratio9,81.
One of the reasons for considering performing the MN assay without Cyt-B is the concern that Cyt-B, used to accumulate BN cells, may interfere with the expression of MN63. However, studies with normal cells do not show an induction of MNi by Cyt-B or a dose–response effect of Cyt-B with MN frequency in BN cells at doses that are usually used to block cells in cytokinesis5,78,82,83,84. One study suggested that MN expression induced by spindle poisons may be less than expected in cytokinesis-blocked BN cells because of pole-to-pole distance shortening, which may increase the probability of reinclusion of lagging chromosome fragments or whole chromosomes back into a nucleus, but this did not diminish the effectiveness of the CBMN assay85.
Although the evidence of obtaining a false positive result with the CBMN assay in normal cells is lacking, there is already adequate evidence that performing the MN assay in a manner that does not account for inhibition of nuclear division can lead to false negative results or an underestimate of MN induction in human lymphocyte cultures5,6,8,81,84. An example of false negative result of MN assay without Cyt-B is shown in Figure 11, which would also have been predicted from the mathematical model that was developed to demonstrate the effect of altered cell division kinetics on MN assay outcomes9. Nevertheless, recent studies comparing the MN assay with or without Cyt-B suggest that if cell lines with good growth characteristics are used and culture and nuclear division conditions are optimal, it is possible to obtain comparable results between the CBMN assay and the MN assay without Cyt-B when strong clastogens are tested86. However, genotoxicity tended to be detected at lower concentrations with the CBMN assay because of the greater statistical power afforded by the fact that BN cells contain twice the number of MNi per cell than would be the case for their mononucleated daughter cells81,84,87,88. Furthermore, without a cytokinesis-block it becomes impossible to score NPBs, which can only be observed in BN cells and this would result in a critical genotoxic event being missed. In addition, non-disjunction events studied using chromosome-specific centromeric probes are best observed in BN cells when the actual distribution of chromosomes between daughter nuclei within one cell after anaphase can be observed directly15,16,17,18,19,55. In addition, the use of Cyt-B makes it easier to score apoptotic cells because it inhibits the disintegration of apoptotic cells into smaller apoptotic bodies because the latter process requires microfilament assembly89, which is readily inhibited by Cyt-B7.

(a) Comparison of the MN dose–response in human lymphocytes exposed in vitro in G1/S/G2 to mitomycin-C (MMC) measured either in mononucleated cells in cultures without Cyt-B (solid black bars) or in BN cells in cultures with Cyt-B (white bars). (b) The level of dividing cells assessed by measuring the percentage of BN cell in the cytokinesis-blocked cultures. It is evident that the assay without Cyt-B underestimates the extent of genetic damage induced by MMC, particularly at doses that inhibit nuclear division. The data represent the mean ±1 s.e. of three replicate cultures.
Finally, a mathematical model of MN expression predicts (i) that scoring MN in BN cells is the most reliable way of determining MN frequency and (ii) scoring MNi in mononucleated cells in cultures without cytokinesis-block is likely to generate false negative results when nuclear division is significantly inhibited by the chemical tested or the culture conditions do not allow an optimal number of dividing cells9. Consequently, results for MN frequency obtained by scoring MNi in mononucleated cells in cultures without Cyt-B cannot be considered conclusive and a negative result with this system should be confirmed using the CBMN Cyt assay. The mathematical model paper9 is an essential reading to have a thorough appreciation of the effect of nuclear and cell division kinetics on MN frequency.
When is it justifiable to also score MNi in mononucleated cells in the CBMN Cyt assay?
It is justifiable to also score MNi in mononucleated cells in the CBMN Cyt assay under the following circumstances:
(1) Information on the frequency of MNi already present within lymphocytes in vivo may be required in chronic exposure situations because this reflects the expression of MNi in lymphocyte precursor cells (in the thymus, bone marrow, spleen and lymph nodes) exposed to the genotoxic agent. This information may also be useful if it is considered necessary to correct the results of the CBMN Cyt assay for MNi already present within lymphocytes; however, this may not be justified because it remains unclear whether lymphocytes with pre-existing MNi can divide ex vivo. The best time point to measure MNi already present in lymphocytes is approximately 24 h after PHA stimulation when activated lymphocytes have a larger cytoplasmic area, which facilitates MNi scoring90,91.
(2) The agent being examined in an in vitro assay is suspected to cause mitotic slippage and thus prevent formation of BN cells following DNA replication91.
Other important aspects of the CBMN Cyt assay
In this manuscript, only the CBMN Cyt assay protocol for isolated lymphocyte cultures is described in detail; however, similar methods can be applied to whole blood lymphocyte cultures, other cell culture systems and other cell types, which are of similar importance as the isolated lymphocyte system in terms of usage. Furthermore, the CBMN Cyt assay includes other important aspects such as the use of molecular cytogenetic techniques to determine the mechanism of formation and DNA content of MNi and NPBs and abnormal distribution of chromosomes among nuclei in a BN cell. Therefore, it is the purpose of the following sections to provide further information and details on these other important aspects of the CBMN Cyt assay.
CBMN Cyt assay in other cell culture systems
Whole blood cultures for human lymphocytes
The CBMN Cyt assay in human lymphocytes can also be performed using whole blood cultures. Typically, 0.5 ml of whole blood is added to 4.5 ml of culture medium (e.g., RPMI 1640) supplemented with fetal calf serum containing L-glutamine, antibiotics (optional) and PHA. Cyt-B is added at 44 h after PHA stimulation. The recommended optimal concentration of Cyt-B for accumulating BN cells in whole blood cultures is 6 μg ml−13. The BN lymphocytes are harvested 24 h after adding Cyt-B as follows.
(1) the cells are centrifuged gently (300g) for 5 min and the supernatant culture medium is removed;
(2) the cells are hypotonically treated with 7 ml of cold (4 °C) 0.075 M KCl to lyse red blood cells and centrifuged immediately (300g) for 8 min;
(3) the supernatant is removed and replaced with 5 ml fixative consisting of methanol:acetic acid (3:1) (the fixative should be added while gently agitating the cells to prevent clumps forming);
(4) the cells are then centrifuged again at 300g for 8 min and washed with two further changes of fixative;
(5) the cells are resuspended gently and the suspension is dropped onto clean glass slides and allowed to dry.
As an alternative, it is also possible to isolate the BN lymphocytes directly from the whole blood culture using Ficoll gradient separation, followed by one wash in culture medium, and then transfer cells to slides by cytocentrifugation before fixation and staining. This approach precludes the requirement for hypotonic treatment and enables optimal preservation of the cytoplasm. The Ficoll separation method is preferable to the hypotonic treatment protocol for slide preparation because hypotonic treatment could result in loss of morphology and lysis of necrotic and apoptotic cells. Staining of cells can be carried out using either 10% Giemsa in potassium phosphate buffer (pH 7.3) or Diff-Quik for light microscopy or acridine orange (10 μg ml−1 in phosphate-buffered saline, pH 6.9) for fluorescence microscopy.
Murine lymphocyte cultures
Lymphocytes are isolated either from the spleen or peripheral blood and cultured according to the procedures described by Fenech et al.92. Because murine lymphocytes have shorter cell division cycles than human lymphocytes, it is essential to add Cyt-B no later than 18 h after stimulation by mitogen and to harvest the cells 20–30 h later. Depending on the culture conditions, it is possible to obtain good binucleate ratios at 42 h after mitogen stimulation.
Other primary cell cultures
The CBMN Cyt assay can be readily adapted to other primary cell types to assess DNA damage induced in vitro, in vivo or ex vivo. The most important points to remember are (a) to ensure that MNi, NPBs and NBUDs are scored in the first nuclear division following the genotoxic insult and (b) to perform preliminary experiments to determine the concentration of Cyt-B and incubation time at which the maximum number of dividing cells will be blocked at the binucleate stage. It is also important to remember that Cyt-B may take up to 6 h before it starts to exert its cytokinesis-blocking action (unpublished observation).
When using established or primary cell lines from dividing cell populations, it is usual to add Cyt-B shortly after exposure to genotoxin to capture all cells undergoing their first nuclear division as BN cells—this usually requires an incubation period of about 24–48 h, depending on the cell-cycle time, before harvesting the cells.
Attached cells can be trypsinized and then prepared by cytocentrifugation as described for human lymphocytes. Specific methods have been described for use with nucleated bone marrow cells93, lung fibroblasts94, skin keratinocytes95 and primary tumor cell cultures96.
It is generally more practical to assess in vivo induction of MNi by blocking cytokinesis in dividing cells after the cells have been isolated from the animal and placed in culture medium in the presence of Cyt-B; this approach has proven to be successful with a variety of cell types including fibroblasts, keratinocytes and nucleated bone marrow cells93,94,95,96.
Treatment schedules for in vitro genotoxicity testing
The cytokinesis-block MN assay has become one of the standard methods for determining the safety of chemicals and pharmaceuticals57,58,63,81,84,86. Ideally, each chemical under investigation should be tested for its genotoxic potential at the various stages of the cell cycle. Because human peripheral blood lymphocytes are in the G0 phase when collected, they can be used for assessing damage induction at this stage. However, cells are expected to be more sensitive to genotoxic effects during S, G2 and M phase, and for this purpose, it is essential to expose cell cultures when most cells are dividing. Because MN expression requires one nuclear division to be completed, the period between treatment and harvest time has to allow for this.
With human peripheral blood lymphocytes treated in G0, it is necessary to accumulate BN cells as early as possible and for as long as possible to ensure that even cells experiencing mitotic delay are examined. Typically, the standard protocol of adding Cyt-B at 44 h and harvesting cells at 72 h should suffice for this purpose; however, a second harvest of cells at 96 h may maximize the number of late-dividing BN cells available for analysis.
If treatment of cells in G1, S, G2 and M phases is required, as would be the case with cell lines, then exposure to the chemical should occur during the logarithmic growth phase of the culture, followed shortly afterwards by Cyt-B to accumulate dividing cells, and cells are then harvested 6 and 48 h later depending on the stage of the cell cycle that is being examined and whether the treatment causes substantial mitotic delay. At the very early harvest times, mainly cells exposed in G2 or late S phase are accumulated as BN cells, whereas at the later harvest time, cells exposed in all stages of the cell cycle are blocked in the binucleate stage. Thus, the harvest time relative to Cyt-B addition would affect the type of cell examined.
The most efficient way to do the MN assay in a manner that minimizes false negative results is to use the Cyt-B method and a treatment phase of 24 h duration to cover all phases of the cell cycle and harvesting cells 24 and 48 h after Cyt-B addition in the recovery phase to allow for any cell-cycle delay in damaged cells as outlined in Figure 12. In the case of lymphocytes, it would be preferable to delay the start of the treatment phase to 48 h after mitogen stimulation at which point a large proportion of the mitogen-activated cells would have progressed from G1 to S, G2 and M phases of the cell cycle. By scoring MN in both mononucleated and BN cells, it should be possible to account for MN expressed before Cyt-B block as well as cells with MN that fail to divide and cells that undergo mitotic slippage91,97. The suggested protocol also allows for harvesting of cells at the end of the treatment phase before Cyt-B addition if there is a need to test whether the chemical inhibits cytokinesis. Examples of other typical schedules for use of the CBMN Cyt assay for in vitro genotoxicity testing are summarized in Table 4. The use of a metabolic activation system such as S9 mix should be included as an option when testing new chemicals57,58,63 but this could limit the exposure period owing to the possible cytotoxicity of S9 to the target cells. A better option may be the use of metabolically competent cells such as genetically modified MCL-5 cells98.

The protocol also allows the harvesting of cells immediately before addition of Cyt-B if it was necessary to test for induction of polyploidy via multinucleation or cytokinesis-blocking action of the agent tested or if it was considered useful to score MN in cells without the Cyt-B block. It is assumed that the cell lines are already in log phase growth when tested. *The time points for lymphocytes refer to number of hours after mitogen stimulation. MNi, micronuclei; NPBs, nucleoplasmic bridges; NBUDs, nuclear buds; BN, binucleated; NDI, nuclear division index.
Molecular techniques for studying mechanisms for MN and NPB formation and non-disjunction in BN cells
To take full advantage of the CBMN assay, it is essential to distinguish between MNi originating from whole chromosomes or acentric fragments. This can be achieved by using probes that are specific for the centromeric DNA or antibodies that bind to the kinetochore proteins that are assembled at the centromeric regions of active chromosomes. The use of MN size as a discriminant is not recommended for human cells or other cell types in which the size of chromosomes is heterogenous because a small MN may contain either a fragment of a large chromosome or a whole small chromosome. The anti-kinetochore antibody method52,53 has some limitations because this approach does not distinguish between unique chromosomes and may not detect chromosome loss occurring due to absence of kinetochores on inactive centromeres as may occur in the case of the inactive X chromosome54. The use of ISH to identify centromeric regions is preferable because it does not suffer from these limitations. In addition, using centromeric probes that are specific for one or more unique chromosomes allows detection of non-disjunctional events (i.e., unequal distribution of homologous chromosomes in daughter nuclei) in BN cells, which provides further information on events causing aneuploidy and altered gene dosage12,13,14,15,16,17,18,19. For details on the use of the kinetochore detection methods, refer to the paper of Fenech and Morley53, and for methods relating to centromere detection by ISH, refer to the papers by Farooqi et al.12, Hando et al.13, Ehajouji et al.15,17, Schuler et al.18 and Wang et al.99. The types of results that can be expected with the various techniques are illustrated in Figure 4.
A combination of pancentromeric and pantelomeric probes can also be used to distinguish between (a) MN containing whole chromosomes or acentric chromosome fragments and (b) NPBs originating from misrepair of double strand breaks in non-telomeric DNA and NPB caused by telomere end fusion of chromosomes (Fig. 5). The use of molecular probes to determine the content of MNi has been extensively reviewed recently46.
Measurement of excision-repaired DNA lesions in G0/G1 human lymphocytes using the cytosine arabinoside MN assay in human lymphocytes
After assessing the MN response in human G0 lymphocytes following exposure to a variety of genotoxins, it became evident that the extent of MN formation in relation to cytotoxicity was low for chemicals and ultraviolet radiation which mainly induce base lesions and adducts on DNA rather than strand breakage or spindle damage100. We hypothesized that this was due to either efficient repair of the lesions or that such sites, if left unrepaired, do not convert to a double-stranded break in DNA following one round of DNA synthesis. Furthermore, we reasoned that inhibition of the gap-filling step in excision repair by cytosine arabinoside (ARA) would result in the conversion of such base lesions to a single-stranded break that would become a double-stranded break following DNA synthesis leading to the production of an acentric fragment, which would then be expressed as an MN within one division cycle100,101.
Using this concept (illustrated in Fig. 13), we showed that addition of ARA during the first 16 h of lymphocyte culture (i.e., before DNA synthesis) did result in a dramatic increase (tenfold or greater) in the MN dose–response following ultraviolet or N-nitroso-N-methyl urea treatment. However, the ARA-induced increase following X-ray exposure was only 1.8-fold as would be expected from the proportion of DNA adducts or base lesions relative to the induction of DNA strand breaks. This method has since been used to identify pesticides that induce excision repair and to distinguish between genotoxic agents that do or do not induce excision repair102.

DSB, double strand break; SSB, single strand break.
The ARA protocol is an important adjunct to the basic CBMN assay in lymphocytes and should be attempted particularly if strong cytotoxic effects are observed in conjunction with weak MN induction. Precise measurement of excision-repaired DNA lesions using the ARA method is only possible using the CBMN assay because (a) the conversion of excision-repaired DNA lesions to MN occurs only in cells that have completed nuclear division and (b) the addition of ARA may also result in significantly altered cell division kinetics, which could confound results in MN assays without Cyt-B.
ARA inhibition of DNA polymerase may cause DNA strand breaks in cells undergoing replicative DNA synthesis. Therefore, it is only possible to use this method in PHA-stimulated G0 lymphocytes with ARA exposure occurring during the G1 phase and before S phase, because excision repair is activated during G1. In practice, this means that lymphocytes are cultured in the presence of ARA during the first 16–20 h after PHA stimulation, following which the cells are washed to remove ARA and incubated in culture medium containing deoxycytidine to reverse ARA inhibition of DNA polymerase. After these steps, the standard CBMN protocol (described above) is followed with the exception that interleukin-2 is added to replenish this mitogenic cytokine, which is generated by activated lymphocytes but otherwise lost during the washing process. For more details on the procedure and typical results, refer to Fenech and Neville100 and Surrales et al.102.
Future developments
It is evident that the ex vivo/in vitro MN assay has evolved into a robust assay for genetic damage with applications in ecotoxicology103, nutrition104, radiation sensitivity testing both for cancer risk assessment105 and optimization of radiotherapy64,106, biomonitoring of human populations60 and importantly testing of new pharmaceuticals and other chemicals63,86. There is little doubt that there is a need for an automated scoring system for quicker and more reliable non-subjective data acquisition, which would ideally be based on the scoring of slides also prepared for visual scoring—this should enable consistent results to be obtained that are not influenced by the inter-individual and temporal variability of human scorers. For this goal to be achieved, it is essential that scoring criteria are well developed and that a robust slide preparation protocol be put in place and that slide preparations be permanent so that they can be re-examined visually if necessary. Automated image analysis systems have recently been developed for scoring of MNi in mammalian cells66,67,68,69,70,71 (see http://www.imstar.fr/applications/genotoxicity/micronucleiassay/; http://www.metasystems.de/ products/metafer/metafer_msearch.htm#Micronucleus; http://www.biocompare.com/techart.asp?id=1247) but these systems do not take into account other important events such as NPBs, NBUDs, necrosis, apoptosis and cytostasis, which are essential for the correct interpretation of the result obtained. In the future, we should expect to have an automated system that can score reliably the various end points possible with the CBMN Cyt assay outlined in this paper.
Finally, it is essential to keep abreast of more recent developments in our understanding of MN, NPB and NBUD formation and events that may alter expression of these genotoxicity end points. Furthermore, scoring criteria in the CBMN Cyt assay are continually being reviewed as part of the HUMN project activity and protocols for validation of automated systems are also currently under consideration by the HUMN project and will be developed in the near future.
Change history
22 September 2016
The version of this article initially published stated that the amount of FBS required to make up the culture medium was 10 ml of a 10% solution. The actual amount required is 10 ml of a 100% solution. The error has been corrected in the HTML and PDF versions of the article.
References
Heddle, J.A. A rapid in vivo test for chromosome damage. Mutat. Res. 18, 187–192 (1973).
Schmid, W. The micronucleus test. Mutat. Res. 31, 9–15 (1975).
Fenech, M. The in vitro micronucleus technique. Mutat. Res. 455, 81–95 (2000).
Fenech, M. & Morley, A.A. Solutions to the kinetic problem in the micronucleus assay. Cytobios 43, 233–246 (1985).
Fenech, M. & Morley, A.A. Measurement of micronuclei in lymphocytes. Mutat. Res. 147, 29–36 (1985).
Fenech, M. & Morley, A.A. Cytokinesis-block micronucleus method in human lymphocytes: effect of in vivo ageing and low-dose x-irradiation. Mutat. Res. 161, 193–198 (1986).
Carter, S.B. Effects of cytochalasins on mammalian cells. Nature 213, 261–264 (1967).
Fenech, M. The advantages and disadvantages of the cytokinesis-block micronucleus method. Mutat. Res. 392, 11–18 (1997).
Fenech, M. Mathematical model of the in vitro micronucleus assay predicts false negative results if micronuclei are not scored specifically in binucleated cells or cells that have completed one nuclear division. Mutagenesis 15, 329–336 (2000).
Degrassi, F. & Tanzarella, C. Immunofluorescent staining of kinetochores in micronuclei: a new assay for the detection of aneuploidy. Mutat. Res. 203, 339–345 (1988).
Thompson, E.J. & Perry, P. The identification of micronucleated chromosomes: a possible assay for aneuploidy. Mutagenesis 3, 415–418 (1988).
Farooqi, Z., Darroudi, F. & Natarajan, A.T. Use of fluorescence in situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes. Mutagenesis 8, 329–334 (1993).
Hando, J.C., Nath, J. & Tucker, J.D. Sex chromosomes, micronuclei and aging in women. Chromosoma 103, 186–192 (1994).
Parry, E.M., Henderson, L. & Mackay, J.M. Guidelines for testing of chemicals. Procedures for the detection of chemically induced aneuploidy: recommendations of a UK Environmental Mutagen Society working group. Mutagenesis 10, 1–14 (1995).
Elhajouji, A., Van Hummellen, P. & Kirsch-Volders, M. Indications for a threshold of chemically induced aneuploidy in vitro in human lymphocytes. Environ. Mol. Mutagen. 26, 292–304 (1995).
Zijno, A., Marcon, F., Leopardi, P. & Crebelli, R. Simultaneous detection of X-chromosome loss and non-disjunction in cytokinesis-blocked human lymphocytes by in situ hybridisation with a centromeric DNA probe; implications for the human lymphocyte in vitro micronucleus assay using cytochalasin-B. Mutagenesis 9, 225–232 (1994).
Elhajouji, A., Tibaldi, F. & Kirsch-Volders, M. Indication for thresholds of chromosome non-disjunction versus chromosome lagging induced by spindle inhibitors in vitro in human lymphocytes. Mutagenesis 12, 133–140 (1997).
Schuler, M., Rupa, D.S. & Eastmond, D.A. A critical evaluation of centromeric labelling to distinguish micronuclei induced by chromosomal loss and breakage in vitro . Mutat. Res. 392, 81–95 (1997).
Kirsch-Volders, M., Elhajouji, A., Cundari, E. & Van Hummelen, P. The in vitro micronucleus test: a multi-end-point assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss and non-disjunction. Mutat. Res. 392, 19–30 (1997).
Fenech, M., Crott, J., Turner, J. & Brown, S. Necrosis, apoptosis, cytostasis and DNA damage in human lymphocytes measured simultaneously within the cytokinesis-block micronucleus assay: description of the method and results for hydrogen peroxide. Mutagenesis 14, 605–612 (1999).
Fenech, M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discovery Today 7, 1128–1137 (2002).
Fenech, M. Cytokinesis-block micronucleus assay evolves into a “cytome” assay of chromosomal instability, mitotic dysfunction and cell death. Mutat. Res. 600, 58–66 (2006).
Umegaki, K. & Fenech, M. Cytokinesis-block micronucleus assay in WIL2-NS cells: a sensitive system to detect chromosomal damage induced by reactive oxygen species and activated human neutrophils. Mutagenesis 15 (3): 261–269 (2000).
IAEA. Biological Dosimetry: Chromosomal Aberrations Analysis for Dose Assessment, Technical Reports Series no. 260. 13–27 (International Atomic Energy Agency, Vienna, Austria, 1986).
Rudolph, K.L., Millard, M., Bosenberg, M.W. & DePinho, R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 28, 155–159 (2001).
Hoffelder, D.R. et al. Resolution of anaphase bridges in cancer cells. Chromosoma 112, 389–397 (2004).
Shimizu, N., Shingaki, K., Kaneko-Sasaguri, Y., Hashizume, T. & Kanda, T. When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 302, 233–243 (2005).
Toledo, F., Le Roscouet, D., Buttin, G. & Debatisse, M. Co-amplified markers alternate in megabase long inverted repeats and cluster independently in interphase nuclei at early steps of mammalian gene amplification. EMBO J. 11, 2665–2673 (1992).
Shimura, M. et al. Micronuclei formation with chromosome breaks and gene amplification caused by Vpr, an accessory gene of human immunodeficiency virus. Cancer Res. 59, 2259–2264 (1999).
Ma, C., Martin, S., Trask, B. & Hamlin, J.L. Sister chromatid fusion initiates amplification of the dihydrofolate reductase gene in Chinese hamster cells. Genes Dev. 7, 605–620 (1993).
Shimizu, N., Itoh, N., Utiyama, H. & Wahl, G.M. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol. 140, 1307–1320 (1998).
Shimizu, N., Shimuara, T. & Tanaka, T. Selective elimination of acentric double minutes from cancer cells through the extrusion of micronuclei. Mutat. Res. 448, 81–90 (2000).
Haaf, T. et al. Sequestration of mammalian Rad51-recombination protein into micronuclei. J. Cell Biol. 144, 11–20 (1999).
Serrano-Garcia, L. & Montero-Montaya, R. Micronuclei and chromatid buds are the result of related genotoxic events. Environ. Mol. Mutagenesis 38, 38–45 (2001).
Crott, J.W., Mashiyama, S.T., Ames, B.N. & Fenech, M. Methylenetetrahydrofolate reductase C677T polymorphism does not alter folic acid deficiency-induced uracil incorporation into primary human lymphocyte DNA in vitro . Carcinogenesis 22, 1019–1025 (2001).
Crott, J.W., Mashiyama, S.T., Ames, B.N. & Fenech, M. Folic acid deficiency increases chromosome breakage and rearrangement, gene amplification and DNA-uracil content in human lymphocytes in vitro: effect of MTHFR C677T polymorphism. Cancer Epidemiol. Biom. Prev. 10, 1089–1096 (2001).
Kimura, M., Umegaki, K., Higuchi, M., Thomas, P. & Fenech, M. MTHFR C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in cultured human lymphocytes. J. Nutrit. 134, 48–56 (2004).
Beetstra, S. et al. Lymphocytes of BRCA1 and BRCA2 germ-line mutation carriers, with or without breast cancer, are not abnormally sensitive to the chromosome damaging effect of moderate folate deficiency. Carcinogenesis 27, 517–524 (2005).
Thomas, P., Umegaki, K. & Fenech, M. Nucleoplasmic bridges are a sensitive measure of chromosome rearrangement in the cytokinesis-block micronucleus assay. Mutagenesis 18, 187–194 (2003).
Blasco, MA. Telomeres and human disease: ageing, cancer and beyond. Nat. Rev. Genet. 6, 611–622 (2005).
Fenech, M. & Crott, J.W. Micronuclei, nucleoplasmic bridges and nuclear buds induced in folic acid deficient human lymphocytes—evidence for breakage–fusion–bridge cycles in the cytokinesis-block micronucleus assay. Mutat. Res. 504, 131–136 (2002).
Guttenbach, M. & Schmid, M. Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 211, 127–132 (1994).
Satoh, T., Yamamoto, K., Miura, K.F. & Sofuni, T. Region-specific chromatin decondensation and micronucleus formation induced by 5-azacytidine in human TIG-7 cells. Cytogenet. Genome Res. 104, 289–294 (2004).
Xu, G.L. et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191 (1999).
Suzuki, T., Fujii, M. & Ayusawa, D. Demethylation of classical satellite 2 and 3 DNA with chromosomal instability in senescent human fibroblasts. Exp. Gerontol. 37, 1005–1014 (2002).
Norppa, H. & Falck, G.C. What do human micronuclei contain? Mutagenesis 18, 221–233 (2003).
Rajagopalan, H. et al. Inactivation of hCDC4 can cause chromosomal instability. Nature 428, 77–81 (2004).
Rao, C.V. et al. Colonic tumorigenesis in BubR1+/−ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc. Natl. Acad. Sci. USA 102, 4365–4370 (2005).
Gutierrez-Enriquez, S. & Hall, J. Use of the cytokinesis-block micronucleus assay to measure radiation-induced chromosome damage in lymphoblastoid cell lines. Mutat. Res. 535, 1–13 (2003).
Maluf, S.W. & Erdtmann, B. Genomic instability in Down syndrome and Fanconi anemia assessed by micronucleus analysis and single-cell gel electrophoresis. Cancer Genet. Cytogenet. 124, 71–75 (2001).
Trenz, K., Rothfuss, A., Schutz, P. & Speit, G. Mutagen sensitivity of peripheral blood from women carrying a BRCA1 or BRCA2 mutation. Mutat. Res. 500, 89–96 (2002).
Vig, B.K. & Swearngin, S.E. Sequence of centromere separation: kinetochore formation in induced laggards and micronuclei. Mutagenesis 1, 464–465 (1986).
Fenech, M. & Morley, A.A. Kinetochore detection in micronuclei: an alternative method for measuring chromosome loss. Mutagenesis 4, 98–104 (1989).
Earnshaw, W.C. & Migeon, B.R. Three related centromere proteins are absent from the inactive centromere of a stable dicentric chromosome. Chromosoma 92, 290–296 (1985).
Wang, X., Thomas, P., Xue, J. & Fenech, M. Folate deficiency induces aneuploidy in human lymphocytes in vitro—evidence using cytokinesis-blocked cells and probes specific for chromosomes 17 and 21. Mutat. Res. 551, 167–180 (2004).
Wu, D., Han, S.N., Meydani, M. & Meydani, S.N. Effect of concomitant consumption of fish oil and vitamin E on T cell mediated function in the elderly: a randomized double-blind trial. J. Am. Coll. Nutr. 25, 300–306 (2006).
Kirsch-Volders, M. et al. Report from the in vitro micronucleus assay working group. Mutat. Res. 540, 153–163 (2003).
Kirsch-Volders, M. et al. Report from the In Vitro Micronucleus Assay Working Group. Environ. Mol. Mutagenesis 35, 167–172 (2000).
Beetstra, S., Thomas, P., Salisbury, C., Turner, J. & Fenech, M. Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. Mutat. Res. 78, 317–26 (2005).
Fenech, M., Holland, N., Chang, W.P., Zeiger, E. & Bonassi, S. The Human MicroNucleus Project—an international collaborative study on the use of the micronucleus technique for measuring DNA damage in humans. Mutat. Res. 428, 271–283 (1999).
Bonassi, S. et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 28(3), 625–631 (2006).
El-Zein, R.A. et al. Cytokinesis-blocked micronucleus assay as a novel biomarker for lung cancer risk. Cancer Res. 66, 6449–6456 (2006).
Lorge, E. et al. SFTG international collaborative study on in vitro micronucleus test I. General conditions and overall conclusions of the study. Mutat. Res. 60 (2006).
Lee, T.K. et al. Lymphocyte radiosensitivity correlated with pelvic radiotherapy morbidity. Int. J. Radiat. Oncol. Biol. Phys. 57, 222–229 (2003).
Fenech, M. et al. Low intake of calcium, folate, nicotinic acid, vitamin E, retinol, beta-carotene and high intake of pantothenic acid, biotin and riboflavin are significantly associated with increased genome instability—results from a dietary intake and micronucleus index survey in South Australia. Carcinogenesis 26, 991–999 (2005).
Tates, A.N., van Welie, M.T. & Ploem, J.S. The present state of the automated micronucleus test for lymphocytes. Int. J. Radiat. Biol. 58, 813–825 (1990).
Castelain, P., Van Hummelen, P., Deleneer, A. & Kirsch-Volders, M. Automated detection of cytochalasin-B blocked binucleated lymphocytes for scoring micronuclei. Mutagenesis 8, 285–293 (1993).
Bocker, W., Muller, W.U. & Streffer, C. Image processing algorithms for the automated micronucleus assay in binucleated human lymphocytes. Cytometry 19, 283–294 (1995).
Frieauff, W., Potterlocher, F., Cordier, A. & Suter, W. Automatic analysis of the in vitro micronucleus test on V79 cells. Mutat. Res. 413, 57–68 (1998).
Verhaegen, F. et al. Scoring of radiation-induced micronuclei in cytokinesis-blocked human lymphocytes by automated image analysis. Cytometry 17, 119–127 (1994).
Varga, D. et al. An automated scoring procedure for the micronucleus test by image analysis. Mutagenesis 19, 391–397 (2004).
Fenech, M. et al. HUMN Project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat. Res. 534, 65–75 (2003).
Lee, T.K., O'Brien, K., Eaves, G.S., Christie, K.I. & Varga, L. Effect of blood storage on radiation-induced micronuclei in human lymphocytes. Mutat. Res. 444, 201–206 (1999).
Burrill, W., Levine, E.L., Hindocha, P., Roberts, S.A. & Scott, D. The use of cryopreserved lymphocytes in assessing inter-individual radiosensitivity with the micronucleus assay. Int. J. Radiat. Biol. 76, 375–382 (2000).
O'Donovan, M.R. et al. Extended-term cultures of human T-lymphocytes: a practical alternative to primary human lymphocytes for use in genotoxicity testing. Mutagenesis 10, 189–201 (1995).
Albertini, R.J. et al. IPCS guidelines for the monitoring of genotoxic effects of carcinogens in humans. International Programme on Chemical Safety. Mutat. Res. 463, 111–172 (2000).
Bonassi, S. et al. Human MicroNucleus project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes: I. effect of laboratory protocol, scoring criteria, and host factors on the frequency of micronuclei. Environ. Mol. Mutagen. 37, 31–45 (2001).
Lindholm, C., Norrpa, H., Hayashi, M. & Sorsa, M. Induction of micronuclei and anaphase aberrations by cytochalasin-B in human lymphocyte cultures. Mutat. Res. 260, 369–375 (1991).
Fenech, M. et al. Intra- and inter-laboratory variation in the scoring of micronuclei and nucleoplasmic bridges in binucleated human lymphocytes. Results of an international slide-scoring exercise by the HUMN project. Mutat. Res. 534, 45–64 (2003).
Triglia, R.P. & Linscott, W.D. Titers of nine complement components, conglutinin and C3b inactivator in adult and fetal bovine sera. Mol. Immunol. 17, 741–748 (1980).
Fenech, M. Commentary on the SFTG international collaborative study on the in vitro micronucleus test: to Cyt-B or not to Cyt-B? Mutat. Res. 60, 9–12 (2006).
Wakata, A. & Sasaki, M.S. Measurement of micronuclei by cytokinesis-block method in cultured Chinese hamster cells: comparison with types and rates of chromosome aberrations. Mutat. Res. 190, 51–57 (1987).
Prosser, J.S., Moquet, J.E., Lloyd, D.C. & Edwards, A.A. Radiation induction of micronuclei in human lymphocytes. Mutat. Res. 199, 37–45 (1988).
Curren, R.D., Mun, G.C., Gibson, D.P. & Aardema, M.J. Development of a method for assessing micronucleus induction in a 3D human skin model (EpiDerm). Mutat. Res. 607, 192–204 (2006).
Minissi, S., Gustavino, B., Degrassi, F., Tanzarella, C. & Rizzoni, M. Effect of cytochalasin-B on the induction of chromosome missegregation by colchicine at low concentrations in human lymphocytes. Mutagenesis 14, 43–49 (1999).
Lorge, E. et al. SFTG international collaborative study on in vitro micronucleus test I. General conditions and overall conclusions of the study. Mutat. Res. 60, 13–36 (2006).
Kalweit, S., Utesch, D., von der Hude, W. & Madle, S. Chemically induced micronucleus formation in V79 cells—comparison of three different test procedures. Mutat. Res. 439, 183–190 (1999).
Matsushima, T. et al. Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU). Mutagenesis 14, 569–580 (1999).
Atencia, R. et al. A structural analysis of cytoskeletal components during the execution phase of apoptosis. Protoplasma 198, 163–169 (1997).
Fenech, M., Perepetskaya, G. & Mikhalevich, L. A more comprehensive application of the micronucleus technique for biomonitoring of genetic damage rates in human populations—experiences from the Chernobyl catastrophe. Environ. Mol. Mutagen. 30, 112–118 (1997).
Kirsch-Volders, M. & Fenech, M. Inclusion of micronuclei in non-divided mononuclear lymphocytes and necrosis/apoptosis may provide a more comprehensive cytokinesis block micronucleus assay for biomonitoring purposes. Mutagenesis 16, 51–58 2001.
Fenech, M.F., Dunaiski, V., Osborne, Y. & Morley, A.A. The cytokinesis-block micronucleus assay as a biological dosimeter in spleen and peripheral blood lymphocytes in the mouse following acute whole body irradiation. Mutat. Res. 263, 119–126 (1991).
Odagiri, Y., Takemoto, K. & Fenech, M. Micronucleus induction in cytokinesis-blocked mouse bone-marrow cells in vitro following in vivo exposure to X-irradiation and cyclophosphamide. Environ. Mol. Mutagen. 24, 61–67 (1994).
Heddle, J.A., Bouch, A., Khan, M.A. & Gingerich, J.D. (1990) Concurrent detection of gene mutations and chromosomal aberrations induced in vivo in somatic cells. Mutagenesis 5, 179–184.
He, S. & Baker, R.S.U. Initiating carcinogen, triethylenemelamine, induces micronuclei in skin target cells. Environ. Mol. Mutagen. 14, 1–5 (1989).
Masunaga, S., Ono, K. & Abe, M. A method for the selective measurement of the radiosensitivity of quiescent cells in solid tumours—combination of immunofluorescence staining to BrdU and micronucleus assay. Radiat. Res. 125, 243–247 (1991).
Rosefort, C., Fauth, E. & Zankl, H. Micronuclei induced by aneugens and clastogens in mononucleate and binucleate cells using the cytokinesis block assay. Mutagenesis 19, 277–284 (2004).
White, N.H. et al. Genotoxic potential of tamoxifen and analogues in female Fischer F344/n rats, DBA/2 and C57BL/6 mice and in human MCL-5 cells. Carcinogenesis 13, 2197–2203 (1992).
Wang, X., Thomas, P., Xue, J. & Fenech, M. Folate deficiency induces aneuploidy in human lymphocytes in vitro-evidence using cytokinesis-blocked cells and probes specific for chromosomes 17 and 21. Mutat. Res. 551, 167–180 (2004).
Fenech, M. & Neville, S. Conversion of excision-repairable DNA lesions to micronuclei within one cell cycle in human lymphocytes. Environ. Mol. Mutagen. 19, 27–36 (1992).
Fenech, M., Rinaldi, J. & Surrales, J. The origin of micronuclei induced by cytosine arabinoside and its synergistic interaction with hydroxyurea in human lymphocytes. Mutagenesis 9, 273–277 (1994).
Surrales, J., Xamena, N., Creus, A. & Marcos, R. The suitability of the micronucleus assay in human lymphocytes as a new biomarker of excision repair. Mutat. Res. 341, 43–59 (1995).
Gauthier, J.M., Dubeau, H., Rassart, E., Jarman, W.M. & Wells, R.S. Biomarkers of DNA damage in marine mammals. Mutat. Res. 444, 427–439 (1999).
Fenech, M. & Rinaldi, J. A comparison of lymphocyte micronuclei and plasma micronutrients in vegetarians and non-vegetarians. Carcinogenesis 16, 223–230 (1995).
Scott, D., Barber, J.P.B., Levine, E.L., Burrill, W. & Roberts, S.A. Radiation-induced micronucleus induction in lymphocytes identifies a high frequency of radiosensitive cases among breast cancer patients: a test for predisposition? 77, 614–620 (1998).
Shibamoto, Y., Streffer, C., Fuhrmann, C. & Budach, V. Tumour radiosensitivity prediction by the cytokinesis-block micronucleus assay. Radiat. Res. 128, 293–300 (1991).
Eastmond, D.A. & Tucker, J.D. Identification of aneuploidy–inducing agents using cytokinesis–blocked human lymphocytes and an antikinetochore antibody. Environ. Mol. Mutgen. 13, 34–43 (1989).
Acknowledgements
The development of the cytokinesis-block micronucleus assay was the result of research I initially performed at the Medical School of the Flinders University of South Australia in Professor Alec Morley's laboratory with the support of the Cancer Council of South Australia and later at CSIRO Human Nutrition. I greatly appreciate Alec Morley's support and inspiration during my formative years in his laboratory. I acknowledge Kevin Trainor, a good friend and colleague, for teaching me tissue culture and a humorous down-to-earth approach to life in a laboratory. I acknowledge Ms J. Rinaldi, Ms C. Aitken, Ms S. Neville, Ms J. Turner, Ms F. Bulman, Ms C. Salisbury, Mr P. Thomas, Dr J. Crott, Dr S. Beetstra, Dr S. Brown, Mr W. Greenrod, Dr Michiyo Kimura, Dr Keizo Umegaki, Dr Jordi Surralles and Professor Wang Xu for their significant contribution to the development and validation of the various biomarkers in the CBMN cytome assay carried out at CSIRO Human Nutrition. The HUMN project coordinating group and participants are also recognized for their contribution to help standardize the application of the CBMN assay, to identify important variables affecting the measured indices and to investigate the prospective association of MN frequency in lymphocytes with cancer risk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat Protoc 2, 1084–1104 (2007). https://doi.org/10.1038/nprot.2007.77
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.77
This article is cited by
-
Evaluating the role of cytokinesis-block micronucleus assay as a biomarker for oxidative stress-inducing DNA damage in type 2 diabetes mellitus patients
Beni-Suef University Journal of Basic and Applied Sciences (2023)
-
In vitro hyperspectral biomarkers of human chondrosarcoma cells in nanoparticle-mediated radiosensitization using carbon ions
Scientific Reports (2023)
-
Realistic scenarios of pesticide exposure alters multiple biomarkers in BOANA PULCHELLA (ANURA) Adult Frogs
Ecotoxicology (2023)
-
Flounder fish (Paralichthys sp.) collagen a new tissue regeneration: genotoxicity, cytotoxicity and physical–chemistry characterization
Bioprocess and Biosystems Engineering (2023)
-
Fate of micronuclei and micronucleated cells after treatment of HeLa cells with different genotoxic agents
Archives of Toxicology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
