
Abstract
This protocol describes the syntheses and applications of two metallointercalators, Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+, that target single base mismatches in DNA. The complexes bind mismatched DNA sites specifically and, upon photoactivation, promote strand scission neighboring the mismatch. Owing to their high specificity and sequence context independence, targeting mismatches with these complexes offers an attractive alternative to current mismatch- and SNP-detection methodologies. This protocol also describes the synthesis of these complexes and their use in marking mismatched sites. Irradiation of 32P-labeled duplex DNA with either intercalator followed by denaturing PAGE allows the detection of mismatches in oligonucleotides. The protocol also outlines a method for efficient detection of single nucleotide polymorphisms (SNPs) in larger genes or plasmids. Pooled genes are denatured and re-annealed to form heteroduplexes; they are then incubated with either complex, irradiated and analyzed using capillary electrophoresis to probe for mismatches (SNP sites). The synthesis of the metallointercalators requires approximately 5–7 d. The mismatch- and SNP-detection experiments each require approximately 3 d.
Similar content being viewed by others
Introduction
The synthesis and study of octahedral metal complexes that bind DNA have long been pursuits of our laboratory1. The unique modularity of metal complexes has allowed us to investigate systematically the factors that contribute to DNA site recognition. For example, the site selectivity of rhodium complexes bearing the 5,6-phenanthrenequinone diimine (phi) intercalating ligand varies dramatically with the identity of the ancillary ligands: Rh(bpy)2(phi)3+ shows little site selectivity, whereas Rh(R,R-dimethyltrien)(phi)3+ binds specifically to 5′-TGCA-3′ sites1,2.
In recent years, we have applied our understanding of molecular recognition elements to the development of complexes that selectively bind mispaired sites in DNA3,4,5. DNA mismatches occur in the cell as a result of polymerase errors or DNA damage6,7. To preserve the fidelity of its genome, the cell has developed a complex mismatch repair (MMR) machinery to find and correct these mismatches, thus preventing consequent mutations8,9. Abnormalities in this machinery, however, can lead to the accumulation of mismatches, and thus mutations, in the genome, with a likelihood of cancerous transformation. Indeed, mutations in MMR genes have been identified in 80% of hereditary non-polyposis colon cancers, and 15–20% of biopsied solid tumors have shown evidence of somatic mutations associated with MMR10,11,12. Mismatch detection may provide an early indicator of these cancerous transformations13.
Given the wealth of sequence information available through the Human Genome Project, much attention has turned to the discovery of single nucleotide polymorphisms (SNPs), the single base differences that lead to variations in disposition to disease or response to pharmaceuticals14. Significantly, DNA mismatches can also serve as important intermediates in the search for SNPs. When a test gene fragment is mixed with the wild-type fragment and the DNA is heated and re-annealed, 50% of the re-annealed duplexes will contain single base mismatches if the test DNA contains an SNP. These mismatches thus provide the target for SNP detection15. Figure 1 schematically illustrates how mismatch targeting can be usefully applied in both situations: the discovery of SNPs and the detection of deficiencies in MMR.

Schematic of the use of mismatch-selective metallointercalators in the detection of both single nucleotide polymorphisms (SNPs) (top) and single base mismatches (bottom).
In our laboratory, two families of mismatch-specific metallointercalators have been developed based on a pair of bulky intercalating ligands, 5,6-chrysenequinone diimine (chrysi) and 3,4-benzo[a]phenazine quinone diimne (phzi) (Fig. 2)3,13. The preparation of the simplest complexes in each family, Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+, is described here. In both cases, the sterically expansive ligand is too large to intercalate easily into the base stack of regular B-form DNA. However, each binds with high affinity to the thermodynamically destabilized mismatched sites. Binding affinities are in the order of 106 M−1 for Rh(bpy)2(chrysi)3+ and 108 M−1 for Rh(bpy)2(phzi)3+10,13. Above all, affinities correlate with the destabilization associated with a mismatch. Our structural model is shown in Figure 3. The correlation between affinity and mismatch stabilization can be understood on the basis of the ease of extruding the mismatched bases when the metal complex is inserted into the base pair stack16. The most destabilized sites are most easily bound by the metal complexes. In all, the compounds bind more than 80% of mismatch sites in all possible sequence contexts5.

In some cases (e.g., ref. 15), these complexes are referred to as Rh(chrysi) and Rh(phzi).

The bulky metal complex (red) inserts into the DNA base stack (gray) and the mismatched bases (blue) are extruded.
Despite their differences in binding strength, the complexes exhibit 1,000-fold or higher selectivity for mismatched DNA sites over Watson–Crick base-paired DNA sites. Site selectivity in solution can be readily discerned through DNA photocleavage experiments. In addition to binding mismatches tightly and selectively, the complexes promote direct strand scission adjacent to the mismatch site with photoactivation3,13. The selectivity of the complexes is a tremendous asset; Rh(bpy)2(chrysi)3+, for example, is capable of binding and cleaving a single CC mismatch in a 2,725 bp plasmid4. As the complexes target only those sites that are thermodynamically destabilized in the base stack, however, more stable mismatches, for example those containing Gua, are not readily detected based upon photocleavage. Interestingly, the Δ-enantiomers of the complexes bind far better than the Λ-enantiomers. Thus, although racemic complexes may be used for all the experiments described here, only the Δ-enantiomer is required.
This protocol first describes the synthesis of Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+ and the enantiomeric separation of Δ- and Λ-Rh(bpy)2(chrysi)3+ (ref. 17). First, it explains the synthesis of the two intercalating ligands (Figs. 4, 5). The two compounds are synthesized identically from RhCl3 until the last step, in which the ligand diones are condensed onto Rh(bpy)2(NH3)23+ under basic conditions (Fig. 6). It should be pointed out that other methods for the formation of the diimine complexes are known. All these others, however, require the cumbersome anaerobic coordination of the ligand diamine followed by the subsequent oxidation of the diamine to the diimine18; this alternative methodology is further limited by low product yields and the necessity of using the ligand diamine precursor. In contrast, the condensation method we employ is far more synthetically facile and produces the desired products in high yield. The UV-visible spectra for the complexes are given in Figure 7. The enantiomeric separation of Δ- and Λ-Rh(bpy)2(chrysi)3+ is achieved through the somewhat unorthodox use of 0.15 M (+)-potassium antimonyl tartrate [(+)-KSb-tartrate] as a chiral eluant for a cation exchange column (Fig. 8). Other methods, including HPLC using a chiral column, have been attempted but have yielded poorer enantiomeric separation. The circular dichrosim spectra are provided in Figure 9.

Synthesis of 3,4-benzo[a]phenazine quinone.

Synthesis of 5,6-chrysene quinone.

Synthetic route to rac-Rh(bpy)2(chrysi)3+ and rac-Rh(bpy)2(phzi)3+.

Extinction coefficients for Rh(bpy)2(chrysi)3+: 303 nm (ε=57,000 M−1), 315 nm (ε=52,200 M−1), 391 nm (ε=10,600 M−1). Extinction coefficients for Rh(bpy)2(phzi)3+: 304 nm (ε=65,800 M−1), 314 nm (ε=67,300 M−1), 343 nm (ε=39,300 M−1).

Column setup for enantiomeric separation of Rh(bpy)2(chrysi)3+.

Δε values for Δ-[Rh(bpy)2(chrysi)](Cl)3: 233 (34), 264 (26), 286 (−12), 308 (−42), 318 (−100), 341 (6). Δε values for Λ-[Rh(bpy)2(chrysi)](Cl)3: 233 (−34), 264 (−26), 286 (12), 308 (42), 318 (100), 341 (−6).
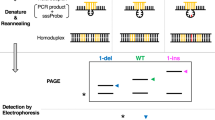
In addition to the syntheses and enantiomeric separation, two experimental applications of the mismatch-selective complexes are described. In the first application, a PAGE sequencing gel can be used to visualize the presence and location of mismatched photocleavage sites in oligonucleotide DNA sequences. In this method, samples of duplex DNA in which a very small amount of one of the strands has been 5′-32P-labeled are irradiated in the presence of Rh(bpy)2(chrysi)3+ or Rh(bpy)2(phzi)3+. The radioactive samples are then eluted using a denaturing polyacrylamide gel and visualized via phosphorimagery. If the original duplex strand contains mismatches, bands corresponding to shorter fragments created by photocleavage at the mismatched site are evident in addition to the full-length parent band (see Fig. 10 for a sample PAGE experiment). Although this methodology may be somewhat limited by the DNA length, it provides information on both the presence (or absence) of a mismatched site and, with the help of parallel sequencing ladders, the exact location of the mismatched site in the sequence5. Of course, other procedures have been developed to search DNA for mismatches using enzymatic or chemical methods, such as RNAase cleavage and chemical cleavage with osmium tetroxide or hydroxylamine19. However, none of these combines the accuracy, selectivity, robustness and ease desired in a clinical testing procedure.

Conditions employed: 1 μM duplex DNA, 50 mM NaCl per 10 mM NaPi pH 7.1 buffer, 1 μM Rh complex when applicable; irradiations performed on an Oriel Instruments solar simulator (320–450 nm) for 15 min. Lanes 1 and 2: matched DNA, Maxam Gilbert sequencing reactions. Lane 3: matched DNA, sample irradiated without rhodium. Lane 4: matched DNA, sample with rhodium but no irradiation. Lane 5: matched DNA irradiated with Δ-Rh(bpy)2(chrysi)3+. Lane 6: mismatched DNA, sample irradiated without rhodium. Lane 7: mismatched DNA, sample with rhodium but no irradiation. Lane 8: mismatched DNA irradiated with Δ-Rh(bpy)2(chrysi)3+. Lanes 9 and 10: mismatched DNA, Maxam Gilbert sequencing reactions.
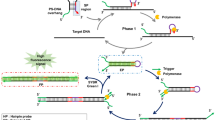
The mismatch specificity of our complexes can also be used to detect SNPs19. In this second application, a region of the genome, perhaps containing an SNP, is amplified, denatured and re-annealed in the presence of a pooled sample to create heteroduplexes that may contain a mismatch at the site of the original SNP. After annealing, the samples are incubated with Rh(bpy)2(chrysi)3+ or Rh(bpy)2(phzi)3+, irradiated to promote photocleavage, end-labeled with a fluorophore and analyzed using capillary gel electrophoresis. If an SNP is present in one of the amplified original segments, the capillary electrophoresis trace will display both a parent peak for the full, uncleaved strand and a peak that corresponds to a fragment resulting from photocleavage at the mismatched site (see Fig. 11, a schematic of the procedure). Above all, this technique allows for the detection of SNPs with allele frequencies as low as 5% from a single pooled sample. This method greatly reduces the cost of discovering new SNPs. Perhaps the greatest advantage of our method is the ease of use; specifically, fewer PCRs, and no cycle sequencing are required.

Adapted with permission from ref. 15.
An alternative method for SNP detection, re-sequencing, is known20,21,22,23. However, re-sequencing is expensive in terms of materials, labor and data processing. Further, although the current technique must scan a particular region of the genome many times to detect an SNP, the false positive rate is high24. Re-sequencing does, however, hold certain advantages over our method, namely, that it can provide a clear picture of both the sequence identity of the SNP and its frequency. We recommend using our SNP protocol first for discovery followed by re-sequencing of the specific region marked by the metal complex for characterization. Perhaps, then, in an ideal situation, metallointercalators and re-sequencing could be used in concert to discover new SNPs and create extensive genomic SNP maps.
The possible uses of our mismatch-specific metallointercalators extend well beyond the two applications outlined here. For example, trisheteroleptic complexes bearing both a mismatch-binding intercalating ligand and a linker-modified bipyridine ligand have been synthesized for the development of mismatch-directed bifunctional conjugates. To date, this strategy has been employed to make bifunctional conjugates for platination25 and alkylation26 near mismatched sites as well as conjugates for fluorescence-based mismatch detection27, and to improve nuclear uptake of the mismatch-specific complexes28. Moreover, recent experiments have revealed that Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+ may have chemotherapeutic value. Both complexes have been shown to inhibit selectively the cell proliferation of MMR-deficient cells over MMR-proficient cells29.
A general note of caution should be sounded for the syntheses: 5,6-chrysene quinone, 3,2-benzo[a]phenazine quinone (precursors of the complexes) and their metal complexes are DNA intercalators. Although their exact toxicity profile is unknown, they are almost certainly highly toxic and most likely carcinogenic. These compounds should be handled with extreme care.
Note that for the sake of simplicity, the section of the protocol relating to the detection of SNPs is specific to the synthetic plasmid described earlier15. The procedure, however, can be easily adapted to any biologically derived gene. For example, ref. 15 also discusses the application of the procedure to SNP detection in the tumor necrosis factor (TNF) promoter region. For a schematic of the technique described below, see Figure 11.
Materials
Reagents
-
Chrysene (Aldrich Zone Purified, cat. no. C8008)
-
Glacial acetic acid
-
Sodium dichromate (Aldrich, cat. no. 483060)
-
2,3-Dichloro-1,4-naphthoquinone (Aldrich, cat. no. D67200)
-
1,2-Phenylenediamine (Aldrich, cat. no. P23938)
-
Pyridine
-
Concentrated nitric acid (not fuming yellow or red)
-
Water (deionized and filtered, i.e., Millipore purified to greater than 18 Ω resistivity)
-
Ethanol
-
Diethyl ether
-
Rhodium chloride hydrate (RhCl3, Pressure Chemicals, cat. no. 3750)
-
Hydrazine monohydrochloride (Aldrich, cat. no. 207942)
-
2,2′-Bipyridyl (Aldrich, cat. no. D216305)
-
Triflic acid (5 g ampule, Fluka, cat. no. 91736)
-
Concentrated ammonium hydroxide (NH4OH)
-
Sodium hydroxide (NaOH)
-
Acetonitrile (MeCN)
-
Methanol (MeOH)
-
Trifluoroacetic acid (TFA; Aldrich, cat. no. T6508)
-
Potassium nitrate (KNO3 ; Aldrich, cat. no. P6803)
-
Sephadex SP-C25 ion exchange resin (Aldrich, cat. no. C25120)
-
Magnesium chloride (MgCl2)
-
(+)-KSb-tartrate (Aldrich, cat. no. 60063)
-
Sodium phosphate (NaH2PO4, referred to as NaPi)
-
Polynucleotide kinase (PNK; Roche, 10 activity U μl−1, cat. no. 10174645001)
-
PNK buffer (Roche, supplied with PNK)
-
[32P]ATP (end-labeling grade, MP Biomedicals)
-
Ultrapure SequaGel sequencing system diluent (National Diagnostics, cat. no. EC-833)
-
Ultrapure SequaGel sequencing system concentrate (National Diagnostics, cat. no. EC-833)
-
Ultrapure SequaGel sequencing system buffer (National Diagnostics, cat. no. EC-833)
-
Ultrapure TEMED (National Diagnostics, cat. no. EC-503)
-
Ammonium persulfate (MP Biomedicals, cat. no. 190556)
-
Denaturing formamide loading dye (see REAGENT SETUP)
-
Saran Wrap (or equivalent product)
-
ExoI (New England Biolabs, cat. no. M0293S)
-
Calf intestinal alkaline phosphatase (Roche, cat. no. 10108138001)
-
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
-
Tris–HCl
-
Dithioerythritol
-
XhoI (Roche, cat. no. 10703770001)
-
ClaI (Roche, cat. no. 10404217001)
-
SNaPshot fluorescent labeling kit (Applied Biosystems, cat. no. 4323159)
-
Chloroform
-
Ethyl acetate
-
Ammonium hexafluorophosphate (NH4PF6,Aldrich, cat. no. 09820)
-
Formamide (Aldrich, cat. no. F9037)
-
Xylene cyanol (Aldrich, cat. no. X4126)
-
Bromophenol blue (Aldrich, cat. no. B0126)
-
Triethylammonium acetate (Aldrich, cat. no. 90358)
-
Deionized formamide (Applied Biosystems, cat. no. 4311320)
Equipment
-
Assortment of round-bottom flasks (50, 100, 250 and 500 ml)
-
Reflux condensers (14/20 and 24/40 joints)
-
Heating mantle
-
60-ml medium glass frit
-
500-ml vacuum flask
-
50-ml Schlenk flask
-
Silica TLC plates (Baker)
-
Flash column (approximately 20 cm × 1.5 cm)
-
Very long glass column (approximately 1.7 m × 1.5 cm)
-
Two medium-sized glass columns (approximately 0.5 m × 1.5 cm)
-
Tubing (Tygon)
-
Tweezers
-
X-ray developer (Konica Minolta, cat. no. SRX-101A)
-
1.7-ml centrifuge tubes
-
Lucite sample boxes for 1.7-ml centrifuge tubes
-
Reverse-phase C18 cartridges (e.g., Waters Sep-Pak)
-
Micro Bio-Spin 6 chromatography column (Bio-rad)
-
Evacuated centrifuge (SpeedVac)
-
Glass gel plates, notched and unnotched (35 cm × 45 cm, Fisher Biotech)
-
Gel running apparatus (23 cm × 40 cm × 50 cm, Fisher Biotech)
-
Power supply for gel running apparatus (Fisher Biotech, cat. no. FB-EC-4000P)
-
Assortment of micropipettes (10, 20, 200 and 1,000 μl)
-
Light source (see EQUIPMENT SETUP)
-
Phosphorimager screen (35 cm × 43 cm, Molecular Dynamics)
-
Phosphorimager (Storm 820, GE Healthcare)
-
Photoshop software (Adobe)
-
ImageQuant software (Molecular Dynamics)
-
Thermocycler
-
Peristaltic pump
-
ABI Prism 310 instrument (Applied Biosystems) for capillary electrophoresis (see EQUIPMENT SETUP)
-
Microcentrifuge (e.g., Eppendorf 5415D)
Reagent setup
-
Denaturing formamide loading dye is made from 80% formamide, 10 mM sodium hydroxide, 0.025% xylene cyanol and 0.025% bromophenol blue, in 1× TBE buffer.
Equipment setup
-
Light source Both Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+ cleave DNA at a variety of wavelengths; although this allows for a number of light source options, the cleavage efficiency is highly wavelength dependent. For the most part, irradiations are performed using either a solar simulator (Oriel Instruments, wavelength output 320–450 nm) equipped with a UV filter or at 340 or 442 nm on a 1,000 W Hg/Xe arc lamp equipped with a monochromator, a 295-nm UV-cutoff filter and an IR filter (Oriel Instruments). Typical irradiation times are 15 min on the solar simulator and 30–60 min on the lamp. Other options exist, however. Cleavage can also be induced using a 302-nm transilluminator or a 365-nm 'black-light' situated 3–4 cm above an open sample tube. However, in these two cases, approximately 10 times more irradiation time (when compared with the more powerful lamp and solar simulator) is required to prompt substantial photocleavage. Neither white fluorescent light nor incandescent light provides sufficient UV power for strand cleavage with Rh(bpy)2(chrysi)3+ or Rh(bpy)2(phzi)3+. Regardless of the source, all irradiations are performed in 1.7-ml centrifuge tubes. In a case where the light shines vertically from above, transparent Lucite samples boxes are simple and effective sample holders. In a case where the light shines horizontally, sample holders more typical of laser setups can be employed.
-
ABI Prism 310 instrument Although other options exist for capillary electrophoresis, this procedure has been optimized for use with an ABI Prism 310 instrument. A sample instrument configuration is capillary, 47 cm × 50 μm; SNaPshot dye, Hex; polymer, POP-4; dye set, E5; molecular weight marker, Genescan 500 TAMRA; module, GS STR POP4 (1 ml); injection time, 5 s; injection voltage, 15.0 V; run voltage, 15.0 V; run temperature, 60 °C; run time, 35 min; analysis matrix, matrix E5; analysis size standard, gs-500 liz; analysis parameters, 0–500 bp.
Procedure
Synthesis of 5,6-chrysene quinone
Timing 1 d
-
1
Combine 10.0 g (44 mmol) chrysene and 110 ml glacial acetic acid in a 250-ml round-bottom flask and stir vigorously.
Caution
Chrysene is a known carcinogen.
-
2
Add 46 g sodium dichromate slowly to the stirring slurry.
-
3
Affix a reflux condenser and heat the slurry to reflux.
Critical Step
The reaction can be monitored by color change. The reaction is complete when no more white solid can be seen in the refluxing mixture.
Pause point
Continue refluxing for 24 h. Over the course of the heating, the color of the solution will slowly change from bright orange with white solid to dark green/brown with orange precipitate.
-
4
After 24 h, stop heating. Remove the reflux condenser once boiling has stopped, and before the mixture reaches room temperature (22 °C) pour it into 100 ml rapidly boiling water in a beaker.
-
5
While it is still hot, filter the solution through a medium glass frit.
Caution
Hot-filtering, although necessary in this case, is always dangerous. Be careful.
-
6
Wash the collected red solid three times with 100 ml boiling water.
Critical Step
The filtration must be performed when the solution is boiling or nearly boiling. Any cooling will result in the precipitation of insoluble chromium byproducts that are very difficult to remove.
-
7
No purification is required. Very small amounts of chrysene may be observed via 1H NMR, although these will not react in subsequent steps and thus will be eliminated.
Pause point
The product can be stored as a solid at ambient temperature indefinitely.
Synthesis of 3,4-benzo[a]phenazine quinone
Timing 3.5 h
-
8
Dissolve 4.5 g (20 mmol) 2,3-dichloro-1,4-napthoquinone and 2.0 g (20 mmol) o-phenylene diamine in 150 ml pyridine. This must be done by placing both materials into the flask and then by adding pyridine.
-
9
Attach a reflux condenser and bring the solution to reflux.
-
10
After 1 h, allow the solution to cool to room temperature.
-
11
Filter the cooled solution. This should yield a dark brown-red solid. The solid can be re-crystallized from hot pyridine or used as it is.
Pause point
This intermediate can be stored for up to a week at ambient temperature before further use. Longer periods of storage may be possible.
-
12
After weighing the brown-red solid, place it in a second round-bottom flask and add to it 10 ml glacial acetic acid and 1 ml concentrated nitric acid. Add 0.66 ml water per 1 g solid subsequently.
Caution
Concentrated nitric acid is caustic and an oxidizer. Handle with care.
-
13
Heat the resultant solution in a boiling water bath for 1 h.
Critical Step
The reaction is complete when only yellow-orange precipitate remains.
-
14
Filter this solution. The solid should be yellow.
-
15
Rinse the resulting solid with 15 ml ethanol and 15 ml diethyl ether.
Caution
Do not let the washes contact the nitric acid solution. Use a new flask.
-
16
The final product can be purified by re-crystallization from 7:3 chloroform/ethyl acetate (vol/vol), but further purification is usually not necessary.
Pause point
The product can be stored as a solid at ambient temperature indefinitely.
Synthesis of [Rh(bpy)2Cl2]Cl
Timing 15 h
-
17
Dissolve 0.64 g RhCl3 (2.8 mmol) and 50 mg hydrazine monohydrochloride in 12.5 ml deionized water in a 50-ml round-bottom flask.
-
18
Add a solution of 0.85 g (5.6 mmol) 2,2′-bipyridyl in 20 ml ethanol and deoxygenate the resultant solution by the repeated application of vacuum followed by back-filling with Ar gas.
-
19
Bring the reaction mixture to reflux and heat until all materials have dissolved (approximately 20 min) and formed a yellow-orange solution.
-
20
While it is still hot, filter the reaction mixture through a medium glass frit.
Caution
Hot-filtering, although necessary in this case, is always dangerous. Be careful.
-
21
Chill the filtrate overnight at 4 °C to promote crystallization.
-
22
Isolate the yellow product crystals via filtration with a medium glass frit.
Pause point
The product can be stored as a solid at ambient temperature indefinitely.
Synthesis of [Rh(bpy)2(OTf)2]OTf
Timing 17 h
-
23
Add 500 mg Rh(bpy)2(Cl)2+ (1.0 mmol) to a 50-ml Schlenk flask with a 14/20 joint on top and a sidearm, and deoxygenate the flask by evacuating it and refilling with Ar(g) three times.
-
24
Crack open the ampule of triflic acid carefully and using a glass Pasteur pipette add 5 g (excess) triflic acid to the reaction vessel under positive argon pressure.
Caution
Triflic acid is very reactive and pyrophoric (self-ignites spontaneously at room temperature). Handle quickly and with care.
-
25
After adding the triflic acid, close the flask with a rubber septum, pierce the septum with a needle and purge the flask with argon for 30–60 s.
-
26
Allow the reaction mixture to stir for 16 h. Purge occasionally to remove HCl generated by the reaction.
-
27
After 16 h, cool 300 ml diethyl ether to −78 °C in a dry-ice/acetone bath. Place the bath on a stir plate and add a stir bar to the flask.
-
28
Using a Pasteur pipette, add the reaction mixture drop by drop to the rapidly stirring, cold diethyl ether. A yellowish-white powder will precipitate. No purification is necessary.
Critical Step
It is imperative that the diethyl ether solution is stirring, so that each new drop of reaction mixture falls into cold diethyl ether and thus prompts product precipitation.
Pause point
Although it is best to proceed directly to the next step, this product can be stored for a few days in a dessicator.
Synthesis of [Rh(bpy)2(NH3)2](X)3 (where X = PF6− or OTf−)
Timing 1 h to 1 d depending on the choice of counter-ion
-
29
Combine 500 mg Rh(bpy)2(OTf)2 + (0.6 mmol) and 20–50 ml concentrated NH4OH in a 250-ml round-bottom flask fitted with a reflux condenser. The starting material should be relatively insoluble in NH4OH.
-
30
Bring the mixture to reflux and boil it until all the material has gone into solution (5–10 min).
-
31
Depending on the desired counter-ion, the product can be isolated by following either option (A) or option (B).
-
A
Precipitate from NH 4 OH solution with NH 4 PF 6
-
i
Add excess NH4PF6 to the ammonia solution.
Pause point
Chill the mixture overnight at 4 °C to facilitate precipitation.
-
ii
Filter through a medium frit to isolate the product.
-
i
-
B
Remove solvent under vacuum
-
i
Remove all NH4OH by rotary evaporation at room temperature. This will yield the triflate salt of the product.
Pause point
The product can be stored as a solid at ambient temperature indefinitely.
-
i
-
A
Synthesis of [Rh(bpy)2(chrysi)](Cl)3
Timing 20 h
-
32
In a 100-ml round-bottom flask, dissolve 195 mg [Rh(bpy)2(NH3)2](X)3 (approximately 0.2 mmol) and 57 mg 5,6-chrysene quinone (0.22 mmol) in 50 ml MeCN with rapid stirring under ambient conditions.
-
33
Add 2 ml aqueous sodium hydroxide (0.4 M) and close the vessel to prevent evaporation.
-
34
After 3 h, halt the reaction by bringing the pH of the solution to 7 by adding a stoichiometric amount of HCl. By this point, the reaction should have changed color dramatically from orange/yellow to dark red. Alternatively, one can monitor the reaction by TLC using silica F plates in a solvent system of 3:1:1 MeCN/H2O/MeOH (vol/vol/vol) with 0.1 M KNO3.
-
35
Remove MeCN in vacuo by rotary evaporation at ambient temperature.
-
36
Re-dissolve the reaction mixture in a minimum volume of water.
Pause point
The reaction mixture can stand at room temperature while the ion exchange column is being prepared.
-
37
Equilibrate 20 g Sephadex SP-C25 ion exchange resin with MgCl2 by making a slurry of the resin in approximately 500 ml 0.05 M MgCl2.
-
38
With this equilibrated mixture, fill a column of 1–1.5-inch diameter with approximately 5 inches of resin.
-
39
Flush the excess MgCl2 solution out of the column with 250 ml H2O.
-
40
Flush the column with 500 ml deionized water.
-
41
Load the Rh(bpy)2(chrysi)3+ by pushing the diluted reaction mixture through the column. The rhodium complex should stick to the uppermost layer of resin, creating a red band at the top of the column.
-
42
Once all the reaction mixture has been loaded onto the column, wash the column with 250 ml deionized water.
-
43
Elute the metal complex by slowly increasing the MgCl2 concentration of the eluant in 500-ml batches. Start with 0.01 M and increase by 0.01 M increments until 0.10 M MgCl2; at this point, switch to 0.05 M increases until the metal complex (red band) has been eluted (most likely approximately 0.3 M MgCl2).
Critical Step
It is very important that the MgCl2 concentration gradient is shallow. Too steep a gradient will cause the ion exchange column to 'crack', leading to band smearing and poor separation.
-
44
Concentrate the product-containing fractions on a reverse-phase cartridge (e.g., Waters, 2 g C18 Sep-Pak Cartridge) primed with MeOH.
-
45
Wash the cartridge thoroughly with water.
-
46
Elute the product with 1:1:0.001 H2O/MeCN/TFA (vol/vol/vol).
-
47
Remove the solvent by rotary evaporation or lyophilization. Note that a representative UV-visible spectrum of the product is shown in Figure 7.
Pause point
The finished product can be stored as a solid indefinitely.
Synthesis of [Rh(bpy)2(phzi)](Cl)3
Timing 20 h
-
48
In a 100-ml round-bottom flask, dissolve 100 mg [Rh(bpy)2(NH3)2](X)3 (approximately 0.1 mmol) and 35 mg 3,2-benzo[a]phenazine quinone (0.125 mmol) in 50 ml MeCN with rapid stirring under ambient conditions.
-
49
Add 2 ml aqueous sodium hydroxide (0.4 M) and close the vessel to prevent evaporation.
-
50
After 3 h, halt the reaction by bringing the pH of the solution to 7 by adding water and a stoichiometric amount of HCl.
-
51
Remove the MeCN in vacuo by rotary evaporation at ambient temperature.
-
52
Re-dissolve the product in a minimum volume of water.
Pause point
The reaction mixture can stand at room temperature while the ion exchange column is being prepared.
-
53
Equilibrate 20 g Sephadex SP-C25 ion exchange resin with MgCl2 by making a slurry of the resin in approximately 500 ml 0.05 M MgCl2.
-
54
With this equilibrated mixture, fill a column of 1–1.5-inch diameter with approximately 5 inches of resin.
-
55
Flush the excess MgCl2 solution out of the column with 250 ml water.
-
56
Flush the column with 500 ml deionized water.
-
57
Load the Rh(bpy)2(phzi)3+ by pushing the diluted reaction mixture through the column. The rhodium complex should stick, yielding a dark yellow/brown band at the top of the ion exchange column.
-
58
Once all the reaction mixture has been loaded onto the column, wash the column with 250 ml deionized water.
-
59
Elute the metal complex by slowly increasing the MgCl2 concentration of the eluant in 500-ml batches. Start with 0.01 M and increase by 0.01 M increments until 0.10 M MgCl2; at this point, switch to 0.05 M increases until the metal complex (dark band) has been eluted (most likely approximately 0.3 M MgCl2).
Critical Step
It is very important that the MgCl2 concentration gradient is shallow. Too steep a gradient will cause the ion exchange column to 'crack', leading to band smearing and poor separation.
-
60
Concentrate the product-containing fractions on a reverse phase cartridge (e.g., Waters, 2 g C18 Sep-Pak Cartridge) primed with MeOH.
-
61
Wash the cartridge thoroughly with water.
-
62
Elute the product with 1:1:0.001 water/MeCN/TFA (vol/vol/vol).
-
63
Remove the solvent by rotary evaporation or lyophilization. Note that a representative UV-visible spectrum of the product is shown in Figure 7.
Pause point
The finished product can be stored as a solid indefinitely.
Enantiomeric separation of Rh(bpy)2(chrysi)+
Timing 3.5 d
-
64
Fill one very long column (1.70 m × 1.5 cm) and two smaller columns (0.5 m × 1.5 cm, hereafter referred to as guard columns) with Sephadex SP-C25 ion exchange resin equilibrated with water.
-
65
Prepare 4 l solution of 0.15 M (+)-KSb-tartrate in water. Filter this solution.
Caution
(+)-KSb-tartrate is toxic. Handle with care and dispose of properly.
-
66
Elute all three columns thoroughly with the 0.15 M (+)-KSb-tartrate solution. Do not recycle this eluant.
Critical Step
Upon elution of the (+)-KSb-tartrate solution, the resin will contract a little (20%); this may necessitate the addition of more resin to fill the columns all the way.
-
67
Set aside one of the guard columns and set up the other two columns and eluant-recycling pump as shown in Figure 8. Make sure all three columns are full of both the chiral eluant and ion exchange resin.
-
68
Dissolve 0.4 g rac-[Rh(bpy)2(chrysi)]Cl3 in 5 ml water.
-
69
Load the rhodium solution onto the top of the large column carefully.
Critical Step
To obtain a good enantiomeric separation, it is very important that the initial rhodium band on the column is very small. To ensure this, use minimum water when first dissolving the rhodium complex and be careful when first loading the rhodium solution onto the top of the large column.
-
70
Turn on the pump and allow the eluant to cycle.
Pause point
The separation will take approximately 2–3 d. During cycling, monitor the column regularly (every 4 h during the day).
Critical Step
Make sure all connections are secured with clamps or wire to prevent leaks.
-
71
After approximately 1 d, separation should begin to become apparent. The first (lower, faster) band is Λ-Rh(bpy)2(chrysi)3+; the second (upper, slower) band is Δ-Rh(bpy)2(chrysi)3+. Detach the first guard column after it 'catches' the lower band and replace it with the second guard column. Cap the first guard column to prevent it from drying.
Critical Step
Do not let the guard columns or the main column dry out during the separation. This will cause the compound to stick permanently to the resin.
-
72
Continue eluting the column (using the pump) until the second guard column fully catches the second band.
-
73
After the second guard column has fully caught the second band, detach and cap it to prevent any evaporation.
Caution
All the eluant and resin used will contain metallic antimony (Sb), which is toxic. Elute the large column with water and dispose of the Sb-containing eluant appropriately. Similarly, the resin will be contaminated with Sb and should be disposed of properly.
-
74
Wash the two guard columns with 0.05 M MgCl2 solution to remove the remaining (+)-KSb-tartrate.
Caution
Be sure to dispose of the Sb-containing eluant properly.
Critical Step
Be careful to keep all columns and product-containing eluants well labeled and separate during product isolation to prevent inadvertent re-racemization.
-
75
Remove the compound from the guard columns by washing the columns with 0.5 M MgCl2 until all compounds have been eluated.
-
76
Concentrate the two product fractions on 5 g Waters Sep-Pak C18 cartridges previously primed with 2 × 10 ml MeOH and 1 × 10 ml water.
-
77
Wash the cartridges with 200 ml water.
-
78
Elute the products with a mixture of 1:1:0.001 MeCN/H2O/TFA (vol/vol/vol).
-
79
Freeze the eluants with liquid N2 and lyophilize to dryness. Owing to some permanent product adhesion to the ion exchange resin, only approximately 80% mass yield can be expected from the separation. The two enantiomers should be obtained in approximately 1:1 ratio. Representative circular dichroism spectra of both the Λ- and Δ-enantiomers are shown in Figure 9.
Mismatch detection via photocleavage with 5-32P-labeled DNA and denaturing PAGE
Timing 3 d
-
80
Make a 100 μM stock solution of the single-stranded DNA sequences (forward and complement) to be investigated.
-
81
Label the 5′ end of each of the strands with [32P]ATP: combine 15 μl water, 2 μl PNK buffer, 1 μl ssDNA stock solution, 1 μl PNK solution and 1 μl [32P]ATP solution in a 1.7-ml microcentrifuge tube. Vortex and spin the reaction mixture in a microcentrifuge.
Caution
Be sure to follow all standard radioactivity safety procedures to limit your exposure.
Critical Step
The mismatch-specific metallointercalators—Rh(bpy)2(chrysi)3+, in this case—will cleave only one strand of the mismatched DNA. Therefore, cleavage will be evident only using one of the two labeled strands. Although the location of cleavage has been determined for most sequence contexts5, it is best to label both and determine cleavage on both strands.
-
82
Incubate the labeling reaction for 2 h at 37 °C.
-
83
After 2 h, add 80 μl water, vortex and purify using a Micro Bio-Spin 6 chromatography column.
Caution
Do not centrifuge the spin columns at rates higher than 3,000 r.p.m.: they will break.
-
84
Dry the samples on a lyophilizer or vacuum centrifuge.
-
85
Purify the 5′-labeled oligonucleotides via 20% PAGE: Pour a 20% polyacrylamide gel. Take up the dried oligonucleotide samples in 10 μl denaturing formamide loading dye. Load the sample onto the gel, and run the gel for 60–90 min at 90 W using 1× TBE as the running buffer. Remove one gel plate, leaving the gel affixed to the other gel plate. Visualize the gel via X-ray. Cut out the parts of the gel that correspond to full-length, labeled DNA with a clean razor blade. Using tweezers, place these parts into a clean centrifuge tube. Add 1 ml 100 mM triethylammonium acetate (pH 7.0) to each centrifuge tube containing gel.
Pause point
Incubate at 37 °C overnight.
-
86
Remove triethylammonium solution and place in a clean 1.7-ml centrifuge tube. SpeedVac to remove solvent. Take up dried sample in 100 μl water and purify using a Micro Bio-Spin 6 chromatography column. Dry the samples on a vacuum centrifuge. Take up labeled oligonucleotide in 50 μl 10 mM NaPi buffer, pH 7.1.
Pause point
Labeled DNA can be stored for weeks at 4 °C; however, it is best to perform the experiments before the label decays too much.
-
87
Prepare an aqueous buffer solution of 100 mM NaCl and 20 mM NaPi, pH 7.1.
-
88
Make a 2 μM duplex stock solution for the irradiation experiments: combine 2 μl of each unlabeled DNA single strand (forward and complement), 4 μl of one of the two labeled oligonucleotide solutions and 92 μl 100 mM NaCl per 20 mM NaPi buffer.
-
89
Anneal the 2 μM duplex stock solution by heating to 90 °C followed by gradual cooling.
Critical Step
It is important that the solution cool to room temperature slowly to ensure that proper hybridization has taken place. Our suggested method is to place the centrifuge tube containing the solution on a 90 °C heat block for 5 min, turning off the heat block and allowing the block to cool to ambient temperature with the sample tube still in it over the course of approximately 90 min.
-
90
Prepare a stock of 2 μM Rh(bpy)2(chrysi)3+ in water.
-
91
Prepare irradiation samples: in 1.7-ml centrifuge tubes, combine 10 μl DNA stock solution (with either forward or reverse strand 5-labeled) and 10 μl rhodium stock solution to create a final 20 μl sample containing 1 μM DNA (concentration in strands, with radiolabel) and 1 μM Rh(bpy)2(chrysi)3+ in a buffer of 50 mM NaCl and 10 mM NaPi, pH 7.1. Note that in addition to samples containing both DNA and Rh(bpy)2(chrysi)3+, it is also important to make control samples under various conditions (e.g., DNA and intercalator in the absence of irradiation, DNA alone in the presence of light and DNA alone in the absence of light).
-
92
Irradiate the samples (for more information, see EQUIPMENT SETUP).
-
93
Dry all samples under vacuum.
-
94
Determine the c.p.m. for each sample using a scintillation counter.
-
95
Dissolve each sample in enough denaturing formamide dye in such a way that there are 10,000 c.p.m. per μl (e.g., dissolve a sample containing 100,000 counts in 10 μl loading dye).
Pause point
Samples dissolved in denaturing formamide dye can be stored at room temperature for days.
-
96
Load 5 μl of each sample onto a 20% denaturing polyacrylamide gel.
-
97
Run the gel at 90 W for approximately 1 h (or the amount of time it takes the parent band, estimated using the running dyes, to travel approximately two-fifths of the way down the plate).
-
98
Separate the gel plates, being careful that the gel stays on just one of the plates.
-
99
Remove the gel from the plate using a large piece of film.
-
100
Cover the gel with Saran Wrap.
-
101
Place the gel in a blanked phosphorimager screen and expose it for 4 h.
-
102
After 4 h, remove the gel from the phosphorimager screen and develop it on a phosphorimager.
-
103
Analyze the gel as a *.gel file in the ImageQuant program or as a *.tiff file in Photoshop (Adobe). See Figure 10 for the result of a typical PAGE experiment.
SNP detection with Rh(bpy)2(phzi)3+
Timing 3 d
-
104
Amplify both genes in question, one known to have the correct sequence and the other suspected to contain an SNP, by PCR using primers containing restriction enzyme sites (in this case, sites for ClaI and XhoI; see ref. 15 for detailed instructions for amplication and restriction).
-
105
Add 6 U ExoI and 20 U calf alkaline phosphatase to each PCR and incubate for 1 h at 37 °C to degrade excess primers and dNTPs.
-
106
To determine the success and purity of the PCR, run out a small amount of both PCR products on a 2% agarose gel pre-stained with ethidium bromide. Only a single band should be present.
Critical Step
The purity of this initial PCR is very important to the success of the overall assay, so optimization may be required if the reaction appears messy or frequent stops are visible.
-
107
Purify both PCR products using a QIAquick PCR purification column, eluting with 10 mM Tris–HCl (pH 8.5).
Pause point
At this point, the PCR products can be stored for up to a month at 4 °C.
-
108
Denature the DNA thermally under these low-ionic-strength conditions by heating to 99 °C for 30 min followed by immediate and rapid cooling to 4 °C.
-
109
Combine equimolar concentrations of each PCR product in a buffer of 60 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl and 1 mM dithioerythritol.
-
110
Re-anneal the pooled sample by heating to 95 °C for 10 min and linearly decreasing the temperature to 4 °C over the course of 150 min. This procedure generates the heterozygous duplexes that contain the photocleavable mismatch.
-
111
Add 5 U ClaI and 5 U XhoI to the pooled sample and incubate the duplex with the restriction enzymes for 1 h at 37 °C, followed by denaturing the enzymes at 80 °C for 20 min.
-
112
Prepare a sample of 400 nM Rh(bpy)2(phzi)3+ in water.
-
113
In a 1.7-ml centrifuge tube, combine 10 μl pooled DNA stock and 10 μl Rh(bpy)2(phzi)3+ stock to yield a final reaction mixture containing 200 nM Rh(bpy)2(phzi)3+, 30 mM Tris–HCl (pH 7.5), 5 mM MgCl2, 50 mM NaCl and 0.5 mM dithioerythritol.
-
114
Irradiate the samples (for more information on irradiation, see EQUIPMENT SETUP).
-
115
Dry the samples in vacuo.
Pause point
Dried samples can be left overnight at room temperature or for a few days at 4 °C.
-
116
Add fluorescent tags to the strands by single base extension using the Applied Biosystems SNaPshot kit. The two strands, forward and reverse, will receive different fluorescent tags, resulting in two different 'color' signals on the capillary electrophoresis trace, one 'color' for the forward strand and a different 'color' for the reverse strand.
-
117
To degrade excess nucleotides from the SNaPshot kit, add 2 U calf intestinal alkaline phosphatase to each sample and incubate 1 h at 37 °C, followed by denaturing the enzyme at 80 °C for 20 min.
-
118
Precipitate each sample with ethanol and remove leftover solvent in vacuo. Add 9 volumes 100% ethanol to 1 volume liquid, cool in dry ice for 15 min and spin down cold in a microcentrifuge to precipitate.
-
119
Re-dissolve each sample in 1 μl water and 1 μl molecular weight standard (see EQUIPMENT SETUP for more information on the molecular weight standard).
-
120
Add 24 μl deionized formamide to each sample and heat to 95 °C for 5 min.
-
121
Cool the samples to 4 °C for 5 min.
-
122
Load samples on an ABI Prism 310 capillary electrophoresis instrument and analyze results. For more information on the configuration of the ABI instrument, see EQUIPMENT SETUP. Note that sample capillary electrophoresis traces for assayed wild-type and SNP-containing genes are shown in Figure 12.
Figure 12: Sample SNP detection gel electrophoresis traces. 
Panel (a) illustrates the result obtained using two homozygous plasmids. Without an SNP, no mismatch is formed upon denaturing and re-annealing the two plasmids. Therefore, only full-length products (in this case, 436 bases), the parent band, can be observed. In contrast, panel (b) shows the result when two different templates, one containing an SNP, are mixed to create a mismatched site. In this case, the denaturing and re-annealing process creates a photocleavable CC mismatch. Therefore, after irradiation at 442 nm for 30 min with 200 nM Rh(bpy)2(phzi)3+, a fragment corresponding to the photocleaved strand, 170 bases in length, marking the SNP site, can be seen in addition to the full-length parent band. Adapted with permission from ref. 15.

Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Synthesis of 5,6-chrysene quinone: Steps 1–3, 24 h; Steps 4–7, 45 min
Synthesis of 3,4-benzo[a]phenazine quinone: Steps 8–10, 90 min; Steps 11–16, 2 h
Synthesis of [Rh(bpy)2Cl2]Cl: Steps 17–18, 30 min; Steps 19–20, 45 min.; Steps 21–22, 14 h
Synthesis of [Rh(bpy)2(OTf)2]OTf: Step 23, 10 min; Steps 24–26, 16 h; Steps 27–28, 1 h
Synthesis of [Rh(bpy)2(NH3)2](X)3: Steps 29–30, 15 min; Step 31A(i)–(iii), 16 h; Step 31B, 45 min
Synthesis of [Rh(bpy)2(chrysi)](Cl)3: Steps 32–35, 3 h; Steps 36–43, 5 h; Steps 44–46, 10 min; Step 47, 12 h
Synthesis of [Rh(bpy)2(phzi)](Cl)3: Steps 48–51, 3 h; Steps 52–59, 5 h; Steps 60–62, 10 min; Step 63, 12 h
Enantiomeric Separation of Rh(bpy)2(chrysi)3+: Steps 64–68, 3 h; Steps 69–71, 3 d; Steps 73–79, 3 h
Mismatch detection with Rh(bpy)2(chrysi)3+ analyzed by PAGE: Step 80, 10 min; Steps 81–84, 2.5 h; Steps 85–86, 1 d; Steps 87–88, 30 min; Step 89, 1.5 h; Steps 90–92, 1.5 h; Step 93, 1 h; Steps 94–95, 30 min; Steps 96–97, 1.5 h; Steps 98–103, 5 h
SNP detection with Rh(bpy)2(phzi)3+ analyzed by capillary electrophoresis: Step 104, 4 h; Step 105, 1 h; Step 106, 3 h; Step 107, 30 min; Step 108, 30 min; Steps 109–110, 3 h; Step 111, 1 h; Steps 112–115, 2 h; Step 116, 30 min; Step 117, 1.5 h; Steps 118–121, 30 min; Step 122, 2 h
Anticipated results
Analytical data
5,6-Chrysene quinone
Yield, 85%; orange powder. 1H NMR (CD2Cl2) δ 9.4 (d, 1H), 8.85 (d, 1H), 8.8 (d, 1H), 8.5-7.5 (m, 7H); MS (m/z) [M+H+] calcd 258.1, found 259.
3,4-Benzo[a]phenazine quinone
Yield, 60%; yellow solid (intermediate red powder). 1H NMR (CD2Cl2) δ 8.8 (d, 1H), 8.3-8.1 (m, 3H), 8.1-7.8 (m, 3H), 7.7 (t, 1H); MS (m/z) [M+H+] calcd 261, found 261.
[Rh(bpy) 2 (Cl) 2 ]Cl
Yield, 60%; yellow crystalline solid. 1H NMR (d6 -DMSO): δ 9.71 (d, 2H), 9.01 (d, 2H), 8.90 (d, 2H), 8.63 (t, 2H), 8.33 (t, 2H), 8.17 (t, 2H), 7.82 (d. 2H), 7.59 (t, 2H); FAB-MS (m/z) [M+H+] calcd 484.98, found 485.96.
[Rh(bpy) 2 (OTf) 2 ]OTF
Yield, 30–40%, yellow-white powder. 1H NMR (d6 -DMSO): δ 9.17 (d, 2H), 9.08 (d, 2H), 8.90 (d, 2H), 8.80 (t, 2H), 8.4 (m, 4H), 7.8 (d, 2H), 7.7 (t. 2H); FAB-MS (m/z) [M+H+] calcd 713.1, found 712.9.
[Rh(bpy)2(NH3)2](X)3
Yield, 30% via precipitation, 95% by rotary evaporation, White powder. 1H NMR (d6 -acetone): δ 9.44 (d, 2H), 9.05 (d, 2H), 8.89 (d, 2H), 8.75 (t, 2H), 8.45 (t, 2H), 8.30 (t, 2H), 8.03 (d. 2H), 7.75 (t, 2H), 5.06 (broad singlet, 6H); FAB-MS (m/z) [M+H+] calcd 449.3, found 449.
[Rh(bpy) 2 (chrysi)](Cl) 3
Yield, 80%, brownish-red powder. UV/VIS (Fig. 7) λmax 303 nm (ε=57,000 M−1), 315 nm (ε=52,200 M−1), 391 nm (ε=10,600 M−1). 1H NMR (d4 -MeOH): δ 8.94 (t, 2H), 8.86 (t, 2H), 8.80 (d, 1H), 8.77 (d, 1H), 8.56 (split t, 2H), 8.44 (m, 5H), 8.4 (d, 1H), 8.15 (m, 1H), 8.03 (m, 1H), 7.95 (m, 3H), 7.86 (d, 1H), 7.81 (d, 1H), 7.64 (m, 5H); FAB-MS (m/z) [M+H+] calcd 671.1, found 671.
Δ-[Rh(bpy) 2 (chrysi)](Cl) 3
Circular dichroism (H2O, see Fig. 9): 233 (34), 264 (26), 286 (−12), 308 (−42), 318 (−100), 341 (6).
Λ-[Rh(bpy) 2 (chrysi)](Cl) 3
Circular dichroism (H2O, see Fig. 9): 233 (−34), 264 (−26), 286 (12), 308 (42), 318 (100), 341 (−6).
[Rh(bpy) 2 (phzi)](Cl) 3
Yield 50%, brownish-yellow powder. UV-visible (Fig. 7) λmax 304 nm (ε=65,800 M−1), 314 nm (ε=67,300 M−1), 343 nm (ε=39,300 M−1). 1H NMR (d6 -DMSO): δ 14.88 (s, 1H), 14.70 (s, 1H), 9.03 (m, 4H), 8.90 (d, 2H), 8.72 (d, 1H), 8.6 (t, 2H), 8.54 (d, 1H), 8.47 (t, 2H), 8.32 (d, 1H), 8.2 (d, 1H), 8.11 (t, 1H), 8.03 (m, 3H), 7.94 (t, 1H), 7.84 (t, 1H), 7.75 (t, 3H), 7.69 (d, 1H); ESI-MS (m/z): [M+2H+] calcd 671, found 671.
References
Erkkila, K.E., Odom, D.T. & Barton, J.K. Recognition and reaction of metallointercalators with DNA. Chem. Rev. 99, 2777–2796 (1999).
Kielkopf, C.L., Erkkila, K.E., Hudson, B.P., Barton, J.K. & Rees, D.C. Structure of a photoactive rhodium complex intercalated into DNA. Nat. Struct. Biol. 7, 117–121 (2000).
Jackson, B.A. & Barton, J.K. Recognition of DNA base mismatches by a rhodium intercalator. J. Am. Chem. Soc. 119, 12986–12987 (1997).
Jackson, B.A., Alekseyev, V.Y. & Barton, J.K. A versatile mismatch recognition agent: specific cleavage of a plasmid DNA at a single base mispair. Biochemistry 38, 4655–4662 (1999).
Jackson, B.A. & Barton, J.K. Recognition of base mismatches in DNA by 5,6-chrysenequinone diimine complexes of rhodium(III): a proposed mechanism for preferential binding in destabilized regions of the double helix. Biochemistry 39, 6176–6182 (2000).
Modrich, P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 25, 229–253 (1991).
Kolodner, R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 10, 1433–1442 (1996).
Harfe, B.D. & Jinks-Robertson, S. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 34, 359–399 (2000).
Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309 (2006).
Kolodner, R.D. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem. Sci. 20, 397–401 (1995).
Arzimanoglou, I.I., Gilbert, F. & Barber, H.R. Microsatellite instability in human solid tumors. Cancer 82, 1808–1820 (1998).
Loeb, L.A., Loeb, K.R. & Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 100, 776–781 (2003).
Junicke, H. et al. A rhodium(III) complex for high-affinity DNA base-pair mismatch recognition. Proc. Natl. Acad. Sci. USA 100, 3737–3742 (2003).
Syvänen, A. Acessing genetic variation: genotyping single nucleotide polymorphisms. Nat. Rev. Genet. 2, 930–942 (2001).
Hart, J.R., Johnson, M.D. & Barton, J.K. Single-nucleotide polymorphism discovery by targeted DNA photocleavage. Proc. Natl. Acad. Sci. USA 101, 14040–14044 (2004).
Pierre, V.C., Kaiser, J.T. & Barton, J.K. Insights into finding a mismatch through the structure of a mispaired DNA bound by a rhodium intercalator. Proc. Natl. Acad. Sci. USA 104, 429–434 (2007).
Mürner, H., Jackson, B.A. & Barton, J.K. A versatile synthetic approach to rhodium(III) diimine metallointercalators: condensation of o-quinones with coordinated cis-ammines. Inorg. Chem. 37, 3007–3012 (1998).
Krotz, A.H., Kuo, L.Y. & Barton, J.K. Metallointercalators: syntheses, structures, and photochemical characterizations of phenanthrenequinone diimine complexes of rhodium(III). Inorg. Chem. 32, 5963–5974 (1993).
Ferrari, M., Carrera, P. & Cremonesi, L. Different approaches of molecular scanning of point mutations in genetic diseases. Pure Appl. Chem. 68, 1913–1918 (1996).
Kwok, P.Y., Deng, Q., Zakeri, H., Taylor, S.L. & Nickerson, D.A. Increasing the information content of STS-based genome maps: identifying polymorphisms in mapped STSs. Genomics 31, 123–126 (1996).
Taillon-Miller, P., Piernot, E.E. & Kwok, P.Y. Efficient approach to unique single-nucleotide polymorphism discovery. Genome Res. 9, 499–505 (1999).
Zhou, W. Mapping genetic alterations in tumors with single nucleotide polymorphisms. Curr. Opin. Oncol. 15, 50–54 (2003).
Schork, N.J., Fallin, D. & Lanchbury, J.S. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin. Genet. 58, 250–264 (2000).
Rieder, M.J., Taylor, S.L., Tobe, V.O. & Nickerson, D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 26, 967–973 (1998).
Petitjean, A. & Barton, J.K. Tuning the DNA reactivity of cis-platinum: conjugation to a mismatch-specific metallointercalator. J. Am. Chem. Soc. 126, 14728–14729 (2004).
Schatzschneider, U. & Barton, J.K. Bifunctional rhodium intercalator conjugates as mismatch-directing DNA alkylating agents. J. Am. Chem. Soc. 126, 8630–8631 (2004).
Zeglis, B.M. & Barton, J.K. A mismatch-selective bifunctional rhodium-oregon green conjugate: a fluorescent probe for mismatched DNA. J. Am. Chem. Soc. 128, 5654–5655 (2006).
Brunner, J. & Barton, J.K. Targeting DNA mismatches with rhodium intercalators functionalized with a cell-penetrating peptide. Biochemistry 45, 12295–12302 (2006).
Hart, J.R., Glebov, O., Ernst, R.J., Kirsch, I.R. & Barton, J.K. DNA mismatch-specific targeting and hypersensitivity of mismatch-repair-deficient cells to bulky rhodium(III) intercalators. Proc. Natl. Acad. Sci. USA 103, 15359–15363 (2006).
Acknowledgements
We are grateful to the National Institutes of Health (GM33309) for their financial support. We also thank ABI for their support. In addition, we are grateful to our co-workers, J. Hart, I. Lau, V. Pierre and R. Ernst, for their help in working out experimental details.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Zeglis, B., Barton, J. DNA base mismatch detection with bulky rhodium intercalators: synthesis and applications. Nat Protoc 2, 357–371 (2007). https://doi.org/10.1038/nprot.2007.22
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.22
This article is cited by
-
Tuning electron delocalization of hydrogen-bonded organic framework cathode for high-performance zinc-organic batteries
Nature Communications (2023)
-
Inorganic Nanoparticles in Cancer Therapy
Pharmaceutical Research (2011)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
