
Abstract
Large-scale fabrication of precisely defined nanostructures with tunable functions is critical to the exploitation of nanoscience and nanotechnology for production of electronic devices, energy generators, biosensors, and bionanomedicines. Although self-assembly processes have been developed to exploit biological molecules for functional materials, the resulting nanostructures and functions are still very limited, and scalable synthesis is far from being realized. Recently, we have established a bacteriophage-based biomimetic process, called 'self-templating assembly'. We used bacteriophage as a nanofiber model system to exploit its liquid crystalline structure for the creation of diverse hierarchically organized structures. We have also demonstrated that genetic modification of functional peptides of bacteriophage results in structures that can be used as soft and hard tissue-regenerating materials, biosensors, and energy-generating materials. Here, we describe a comprehensive protocol to perform genetic engineering of phage, liter-scale amplification, purification, and self-templating assembly, and suggest approaches for characterizing hierarchical phage nanostructures using optical microscopy, atomic-force microscopy (AFM), and scanning electron microscopy (SEM). We also discuss sources of contamination, common mistakes during the fabrication process, and quality-control measures to ensure reproducible material production. The protocol takes ∼8–10 d to complete.
Similar content being viewed by others

Introduction
Designing new materials with well-defined structures and desirable functions is a challenge in materials science, particularly when these materials have nanometer-scale dimensions. Materials are often generated through rational, but laborious, design and performance characterization processes1. By contrast, nature creates various functional nanostructures through self-assembly of helical nanofibers such as collagens, chitins, and celluloses during the morphogenesis process. In this process, kinetic and environmental factors have a critical role in organizing helical nanofibers in a hierarchical structure. The timely secretion of the helical nanofibers and control of the molecular interactions within cellular microenvironments lead to self-templated assembly, in which the nanofibers self-organize into parallel, twisting, or rotating molecular orientations2,3. Furthermore, local environments, such as confined spaces, curvatures, and preexisiting structures, influence the orientation of incoming helical molecules4,5. These self-assembly processes occur in controlled environments that momentarily favor far from equilibrium states to form highly ordered materials. Secretion induces flow forces and makes the orientation of the fiber parallel to the flow direction. When secretion terminates, the helical building blocks assemble the most stable structures in the given conditions. The periodic on-and-off secretion of the helical building blocks spontaneously controls kinetic and thermodynamic factors. These states stabilize the nanometer- or micrometer-level self-assembled structures that combine to arrange macroscopic organized hierarchical structures6,7,8,9. Examples of such hierarchical structures include orthogonally arranged corneal tissues in the eyes, cholesterically rotating matrices on colored mammalian skins, and helicoidally arranged bone structures10,11,12. Because of the complexity of the basic biological building blocks and limited number of microscopic tools to investigate the in situ morphogenesis process, our understanding of the self-templating assembly processes of collagens, chitins, and celluloses on the molecular level is far from ideal. Recently, by mimicking self-templating assembly processes in nature, various functional nanomaterials have been developed using self-assembly of genetically engineered viral particles13,14,15.
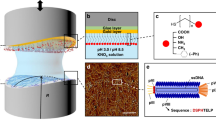
An ideal starting point for designing helical-nanofiber-based diverse structures is to exploit the helical nanofiber shape of M13 bacteriophage (phage) and its self-assembled liquid crystalline structures. M13 phage is a filamentous bacterial virus ∼880 nm in length and 6.6 nm in diameter (Fig. 1; refs. 16, 17). Similar to natural helical nanofibers (i.e., collagens, chitins, and celluloses), M13 phage possesses a fibrous shape with a helical surface, which is covered by 2,700 copies of the pVIII major coat proteins with a fivefold helical symmetry with an additional five copies of the minor coat proteins pIII and pIX located at either end (Fig. 1, left; ref. 18). Phage can be easily engineered with functional peptides through genetic modification of the coat proteins19. Using Escherichia coli (E. coli) bacterial host cells, chemically and physically identical monodisperse phages can be produced at large scales (Fig. 1, center). Because of its monodispersity, phage has been used as a model system to study liquid crystalline structures (i.e., nematic, cholesteric, and smectic phases) and their phase transitions15,20,21,22,23,24. Recently, we developed a facile meniscus-induced phage particle deposition process to mimic the self-templating process in nature (Fig. 1, right; ref. 13). Exploiting spontaneous evaporation of the phage solution at the meniscus, a phase transition of the phage can induce nematic, cholesteric, and smectic phases only near the meniscus where air, liquid, and solid meet. When substrates are pulled vertically from the meniscus during the evaporation process, the condensed phage solution adsorbs onto the substrate and crystallizes out of the meniscus. During this time, surface forces at the meniscus maintain phage in solution in a 'stick' position. Evaporative forces increase the dynamic surface energy at the solid–liquid–vapor interface until the energy surpasses a critical threshold, and the solution 'slips' so that the liquid front rapidly recedes. The oscillating stick-and-slip phenomenon mimics the periodic secretion and deposition process of natural helical fibers in cellular microenvironments. We can control the thermodynamic (phage solution concentration or ionic concentration of solution) and kinetic factors (pulling speed or surface chemistry) to modulate the interaction at the meniscus and the self-templating of hierarchical nanostructures at a large scale. For example, by controlling the phage concentration, we can control the number of particles engaged in the self-templating assembly. By controlling the ionic concentration, we can tune the electrostatic forces bringing phage particles together. Through control of the pulling speed, we can tune the force field that interacts with the substrate and particles adsorbed onto the surface. In addition, the wettability (i.e., hydrophilic or hydrophobic surface functionalization) of the solid substrate can be controlled to selectively deposit phage at precise locations on the substrate25,26. Using the self-templating film growth process, we can create diverse naturally existing structures (i.e., collagen- or chitin-like tissue structures) as well as structures that are never observed in the nature (i.e., nematic orthogonal twist, cholesteric helical ribbon, and smectic helicoidal nanofilament phases)13. After controlled modification of phage, the resulting self-templated phage material can be used for biosensors14,15,27, energy storage and generation28,29, tissue regeneration30,31, and inorganic–organic composite materials13,32.

(Left) Helical nanofiber-shaped phage structure. Phage is covered by ∼2,700 pVIII major capsid proteins with a fivefold helical symmetry and a twofold screw rotation axis. M13 phage can be modified near the N terminus of various capsid proteins such as pVIII (major, yellow), pIX (minor, green), and pIII (minor, orange) through genetic engineering. (Center) Large quantities of genetically engineered phage can be prepared through bacterial infection. The phage solution can be purified and concentrated through PEG precipitation. (Right) The resulting highly pure and concentrated phage suspension can be self-assembled into various nanostructured films. As the substrate is slowly pulled from phage suspensions, a liquid crystal phase transition of the phage solution is induced at the air/liquid/solid interface, which deposits the phage nanostructured film on the substrate in a self-templated manner. Left and right images adapted with permission from ref. 13, Nature Publishing Group.
Here, we describe a comprehensive protocol to generate engineered phages, for liter-scale amplification and purification, followed by self-assembly and characterization of hierarchical phage nanostructures.
Comparison with previously reported biomolecule self-assembly methods
Self-assembly of biomaterials into different geometries has been accomplished by several approaches, including ink-jet printing, laser writing, microcontact printing, and dip-pen nanolithography33,34,35,36,37,38. Whiteside and collaborators have developed a microcontact printing technology with an elastomeric stamp to fabricate various types of both microscale and nanoscale structures by transferring molecules on desired substrates, such as planar, curved, flexible, and soft substrates34. Microcontact printing provides not only flexibility in choice of materials but also compatibility in fabricating relatively large patterns. This method, however, requires relatively high-cost facilities, such as photolithography or electron beam lithography equipment, to fabricate master molds. Mirkin and colleagues35 invented dip-pen nanolithography based on the application of a scanning-probe cantilever tip as a precisely controlled molecular-patterning tool. Although this development has prompted new research into control of molecules and biological patterning, the need for expensive and delicate cantilevers and the technique's limited capability for large-scale patterning hinder widespread usage. Belcher and coworkers22 developed M13 phage self-assembly and various functional materials applications. Lee et al.22,39 showed that M13 phage could form self-standing liquid crystalline fibers and films. After genetic modification to enable interaction with various nanomaterials, the phage could organize organic and inorganic nanoparticles into phage-based nanomaterials22,40. Furthermore, through layer-by-layer deposition of M13 phage, assisted by cationic (linear polyethylenimine) and anionic (poly acrylic acid) polymer polyelectrolyte multilayers, large-scale self-supporting phage films could be fabricated41. The resulting films could be further functionalized through nucleation of target metallic and semiconductor nanocrystals useful for battery-electrode materials42. Although this method was used to successfully fabricate scalable functional 2D film with desired functions, the resulting structures were limited to directionally organized liquid crystalline 2D films with little hierarchical tunability.
Natural biomaterials are composed of basic building blocks such as amino acids, nucleotides, lipids, and sugars. These building blocks are used in bottom-up fabrication processes in a sophisticated and highly precise manner through self-organizing into refined structures ranging from the molecular to the whole-organism level. Recently, we have developed a novel self-assembly method, termed 'self-templating assembly', that mimics the natural helical nanofiber assembly process in the sense that phage particles are used as helical nanofiber building blocks to form liquid crystalline nanostructures that are transformed into hierarchically organized films at a large scale using a meniscus-induced self-assembly process13. After large-scale production of phage and purification, we prepared a phage solution and pulled a substrate vertically using a computer-controlled pulling machine. We exploited the meniscus forces to deposit the phage into hierarchically organized films at the air/solid/liquid meniscus, controlling the process by the kinetic (i.e., pulling speed and surface chemistry) and thermodynamic conditions (i.e., phage concentration and ionic concentration). This single-step process produces long-range, ordered structures with multiple levels of hierarchical organization and helical twists. The first critical factor during the self-templating process is the local transition of chiral liquid crystal phases at the meniscus. The second is the competition of the interfacial forces at the meniscus, which determines liquid crystal orientation, material flow, and higher-order twist. Through the interplay and control of these factors, we have created many novel, chiral liquid crystalline structures that are found in nature, as well as many new structures that are neither found in nature nor previously synthesized, such as nematic orthogonal twists, cholesteric helical ribbons, and smectic helicoidal nanofilaments13.
Application of self-assembled phage materials
Genetic control of exposed proteins on the phage and their resulting self-assembled structures generates nanomaterials with tunable properties, including optical, electrical, and biological properties, that can be used for biosensor, energy generator, or tissue-engineering materials13,14,26,30.
Biomimetic structural colored phage film and sensing applications The self-assembled phage films can be used as structural color matrices for sensing applications (Fig. 2a). In nature, there are many examples of structural biomaterials (i.e., collagens, chitins, and celluloses) exhibiting brilliant colors attributed to bundled nanostructures that coherently scatter light43,44,45,46. These animals can rapidly change their colors through structural modulation for communication, camouflage, and response to surroundings47,48. Genetically engineered M13 phages can self-assemble to create a biomimetic bundled nanostructure exhibiting brilliant structural color. We can further incorporate receptor-like molecular recognition elements to use them for color sensors14,15. Because phage provides a rapid mining tool to identify the specific binding peptide motifs of substrates for various targets through phage display, it allows for the recognition of desired target molecules at the molecular level within a relatively short amount of time49. Recently, we engineered the phage to bind desired chemicals through a phage-display process and self-assembled the resulting phage particles into bundled nanostructured film structures14. The resulting phage films exhibit a distinct color. Upon application of desired chemicals, the phage color matrices exhibited characteristic color changes14. For example, after we engineered phages to bind a trinitrotoluene (TNT) through phage display, the resulting TNT-binding phage matrices exhibited sensitive and selective color changes upon exposure to TNT. The resulting colorimetric phage film can be further engineered to respond against desired targets that can be applicable to various health and environmental sensor designs15.

(a) Phage-based colorimetric sensor matrices. Through genetic engineering, the phage protein structure can be engineered to display receptors for desired chemicals. The resulting engineered phage can self-assemble to form colorimetric thin films that can change colors upon binding of target molecules. Through color analysis and pattern recognition, we can detect various molecules sensitively and selectively. (b) Phage-based piezoelectric-energy-generating matrices. Because of its noncentrosymmetric coat protein arrangement that has no inversion centers, the M13 phage possesses intrinsic piezoelectric properties. Upon application of external forces, the phage and their self-assembled matrices can generate electric potentials. The photograph depicts a phage-based piezoelectric device turning on an LCD device in response to a finger tap. (c) Phage-based tissue-engineering matrices. The phage can be engineered to display high-density signaling peptides (1.5 × 1013 RGD peptides per cm2). The resulting phage can self-assemble to form tissue-engineering matrices to guide the cellular growth process of the desired cells. Composite fluorescent image (right) shows that the preosteoblast cells (MC-3T3 E1) grow to confluence (day 3) in the desired directions on the self-assembled RGD phage films. c adapted with permission from ref. 13, Nature Publishing Group.
M13-phage-based piezoelectricity generation Self-assembled phage films can be used as piezoelectric-energy-generating materials (Fig. 2b; ref. 26). Materials possessing a noncentrosymmetric structure without an inversion center can exhibit piezoelectric properties, meaning they convert mechanical energy into electrical energy. Many ordered biomaterials, such as DNA, amino acid and protein crystals, collagen, cellulose, and chitin exhibit piezoelectric properties50,51,52,53,54,55. The M13 phage is covered with 2,700 copies of the major capsid protein pVIII and has fivefold rotational and twofold helical screw symmetry with the 20° tilted angle to the phage axis18. The pVIII protein consists of 50 amino acids having an alpha-helical structure with a dipole moment directed from the N-terminus to the C-terminus direction. The M13 phage structure satisfies the piezoelectric material requirement26. Self-assembled phage monolayer films exhibit piezoelectricity upon mechanical stimulation because of the breaking of the piezoelectric symmetry26,56. To enhance piezoelectricity, we manipulated the morphology of self-assembled phage structures and investigated the structure–property relationships of these materials26. AFM characterization verified that the resulting phage films exhibited smectic structures (ordered in both orientation and position) with ∼1-μm layer spacing. Piezoelectric-coefficient mapping revealed that the effective piezoelectric coefficient, deff, of the ridge areas was higher than that of the groove areas. In addition, the observed piezoelectric response was dependent on the film thickness: the effective piezoelectric coefficient increased with increasing film thickness, reaching a saturated level of deff ≈ 3.9 pm/V for films thicker than ∼100 nm. Furthermore, the fabricated phage-based piezoelectric devices can produce currents of 6 nA and voltages of 400 mV, enough to turn on a microelectronic device (i.e., an LCD panel)26. The hierarchical self-assembly of phage-nanostructured film can further enhance the piezoelectric properties of phage in the future.
M13-phage-based tissue regeneration The genetically modified phage-based self-assembled matrices can offer a novel scaffold for tissue regeneration (Fig. 2c; refs. 13, 30, 57). A precise manipulation of structural arrangement and engineering of signaling motifs to detect and/or regulate cell behavior are the most vital issues in the design of tissue-regenerating materials. Hierarchical nanofibrous structures in nature synergistically support and guide cellular behavior through various chemical and physical stimuli with neighboring cells58,59,60,61. M13 phage is one of the most attractive nanofibrous basic building blocks for use in tissue-regenerating materials because phage engineering enables the construction of well-defined physical, chemical, and mechanical structures to direct the tissue-growth process62. Furthermore, phages can be replicated in large quantities through bacterial amplification. Bioinspired phage-based nanofibrous hierarchical structures act as a tissue regeneration scaffold to provide a cell-conducive environment30,62. These biomaterials provide large surface areas of nanometer-sized fibrous structures for cell engagement that leads to improvement of cell attachment, material integration, and enhancement of both cell proliferation and differentiation30,62. Furthermore, tuning the density of displayed signaling motifs at the nanometer-scaled phage surface allows quantitative examination of cell behaviors. Previously, we have demonstrated that the self-assembled matrices of genetically engineered M13 phage can be used as tissue-regenerating scaffolds30,63,64,65. Phages were genetically engineered to express cell-signaling motifs (cell-adhesive peptide RGD (arginine–glycine–aspartate) and the neural-cell-stimulating peptide IKVAV (isoleucine–lysine–valine–alanine–valine)) on the pVIII major capsid protein in a homogeneous and compact manner30. The resulting self-assembled phage matrices can serve as scaffolds for cell (neural progenitor cell and preosteoblast cell) proliferation and differentiation. We observed that the specificity of engineered phage matrices led to a greater migration and spread of cells than that observed in the case of wild-type phage. We also observed that the hierarchical phage structures and their alignment guided the cells to position and orient themselves in substrate-defined directions13,62. Self-assembled phages have also been used for hard tissue-engineering materials as well13. In nature, the glutamate-rich proteins have a critical role in biomineralizing hard tissue, such as in apatite and carbonate mineralization42,66. Four glutamate-engineered (4E-) phages can nucleate calcium phosphate and calcium carbonate biominerals based on the affinity between glutamates on the phage and positive ions13,32. The resulting phage with biomineralized inorganic materials can form mechanically strong and tough materials that mimic dental enamel tissues or abalone shell structures found in nature13,32. Therefore, the resulting self-assembled phage matrices can be used to design various soft and hard tissue-engineering materials.
Experimental design
Through the combination of the phage genetic engineering, liter-scale phage preparation, and phage self-assembly processes, a variety of nanostructured phage materials can be fabricated.
Phage genetic engineering. M13 phage can display desired peptides or proteins, such as the pVIII (major capsid) protein and pIX and pIII (minor capsid) proteins, on its major and/or minor protein coats (Fig. 1). Thus, there are many potential combinations of phage-coat protein modifications that can be implemented. Typically, researchers are interested in altering binding sites on the phage (i.e., at the pIII minor coat protein) to facilitate phage engineering with molecular recognition elements, biomolecules, or piezoresponsive materials to create application-specific hybrids22,49. In the following protocol, we describe the general case of an engineered M13 phage with quadruple glutamates on its pVIII proteins. It is important to note that the M13 genome is a single-stranded DNA polynucleotide. However, in this protocol, a double-stranded oligonucleotide from infected E. coli is used. To genetically engineer the surface proteins, the desired peptides are inserted between the first and sixth amino acids at the N terminus of the major coat protein, using an inverse PCR cloning method. The genome of the genetically engineered phages is confirmed using DNA sequencing. Depending on the desired phage application, we can expand protein-coat modification to pIII and/or pVIII. We have listed specific applications and examples in Table 1.
Liter-scale phage preparation. Large quantities of identical phages are prepared through infection of bacterial host cells (Fig. 3). A critical factor in the fabrication of highly functional phage films is the preparation of a highly pure solution of identical phages. In general, liter-scaled amplification of phages might include unwanted biomaterials (e.g., cell debris and PEG aggregates) during the amplification and purification steps. A pure M13 phage solution without preaggregation is also vital to the production of self-assembled phage nanostructures during the deposition process. In this protocol, we describe detailed procedures regarding the preparation of engineered phage stock solution to create self-assembled phage films.

(a) Phage amplification in 50 ml of LB medium and additional bacterial culture (Step 19). (b) Separation of E. coli from the phage suspension in LB medium through centrifugation (Step 21). (c) Amplified phage (supernatant of b) through bacterial infection in 1.6 liters of LB medium (Step 22). (d) Amplified phage in 800 ml of LB medium after centrifugation to precipitate the E. coli. (Steps 23 and 24). (e) Decanted phage supernatant (Step 24). (f) Phage pellet after centrifugation with PEG–NaCl precipitation and removal of supernatant. Inset: magnified image of precipitated phage pellet (Step 27). (g) Phage suspension in Tris-buffered saline (TBS) after centrifugation to remove the bacterial debris (Step 31). (h) Filtration of the phage suspension to remove aggregates after suspension in TBS (Step 32). (i) Highly pure and concentrated phage suspension in TBS after purification (Steps 36–42).
Phage self-assembly process. The fabrication design in this protocol is based on the self-assembly of genetically engineered phages into various nanostructures13. The self-assembling process organizes phage particles into supramolecular structures with multiple levels of hierarchical organization. Phages are assembled into large-area films by using a computer-controlled device, which vertically pulls solid substrates from phage suspensions at precisely controlled speeds. When the substrates are pulled from the phage suspensions, the evaporation rate is enhanced at the interface between air, liquid, and solid (meniscus), leading to the formation of phage films exhibiting liquid crystalline phases (nematic, cholesteric, and smectic phases) due to the local accumulation and deposition of phage particles onto the substrate by self-templating. By manipulating thermodynamic (concentration of the phage and ionic concentration of the solutions) and kinetic (pulling speed and surface chemistry of the substrates) factors, we have created tunable liquid crystalline nanostructures with various twisted orientations in a hierarchical manner similar to natural hierarchical structures (i.e., helicoidally twisted bone and teeth, collagen bundled structures), as well as hierarchical structures that have never been observed before (Table 2; ref. 13). We also provide guidelines (Box 1) for the characterization of micro- and nanostructures of the self-assembled phage matrices using reflective- or transmission-polarized optical microscopy, dark-field optical microsocpy, SEM, and AFM.

Materials
REAGENTS
-
M13KE phage (New England BioLabs, cat. no. N0316S)
-
Oligonucleotide primers (Integrated DNA Technologies, custom order)
-
QuikChange II site-directed mutagenesis kit (Agilent, cat. no. 200523)
-
Phusion high-fidelity DNA polymerase (New England BioLabs, cat. no. M0530S)
-
PstI (New England BioLabs, cat. no. R0140S)
-
T4 DNA ligase (New England BioLabs, cat. no. M0202S)
-
QIAquick gel extraction Kit (Qiagen, cat. no. 28704)
-
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
-
QIAprep Spin Miniprep Kit (Qiagen, cat. no. 27104)
-
E. coli (strain: XL-1 Blue; Stratagene, cat. no. 200236)
-
XL1-Blue electroporation-competent cells (Agilent, cat. no. 200228)
-
Miller LB broth (EMD Chemicals, cat. no. 1.10285.0500)
-
PEG 8000 (Fisher Scientific, cat. no. BP233-1)
-
Sodium chloride (Macron Chemical, cat. no. 7581-06)
-
Tris base (J.T. Baker, cat. no. 4109-02)
-
Ethanol (Koptec, cat. no. V1016)
-
Magnesium chloride (Fisher Scientific, cat. no. BP214-500)
-
DMF (N,N-dimethylformamide; Sigma-Aldrich, cat. no. D4551-500ML)
-
Tetracycline (Sigma-Aldrich, cat. no. 87128)
-
Gold-coated Si wafer (100-nm Au, 5-nm Ti on Si; Platypus Technologies, cat. no. AU.1000.SL2)
-
Agar (Merck, cat. no. 1.01614.1000)
-
X-gal (5-bromo-4-chloro-3-indoyl-b-D-galactoside; APEX,cat. no. 20-108)
-
IPTG (isopropyl-b-D-thiogalactoside; APEX, cat. no. 20-109)
-
Glycerol (Merck, cat. no. 8.18709.1000)
-
Deionized water
-
SOC medium (Invitrogen, cat. no. 1544034)
EQUIPMENT
-
Centrifuge (Beckman Coulter, model no. Avanti J-26 XP, cat. no. 393124)
-
Fixed-angle centrifuge rotor for 1 liter (Beckman Coulter, model no. JLA-8.1000, cat. no. 969328)
-
Fixed-angle centrifuge rotor for 50 ml (Beckman Coulter, model no. JA-20, cat. no. 334831)
-
Electron-beam evaporator (Kurt J. Lesker, model no. PVD 75)
-
Benchtop centrifuge (Eppendorf, model no. 5424R, cat. no. 22620401)
-
Incubator 37 °C (VWR, model no. 1575)
-
Temperature controlled shaker/incubator (New Brunswick Scientific, model no. I2500)
-
PCR machine (Bio-Rad, model no. PTC1148)
-
Electroporator (Eppendorf, model no. 2510)
-
Syringe pump (KD Scientific, model no. 780270)
-
RS232C cable (Parallax, cat. no. 2109245)
-
Modular adaptor (CableWholesale, cat. no. 31D1-1640BK)
-
Scanning electron microscope (FEI, model no. Quanta 3D FEG)
-
Atomic-force microscope (Asylum Research, model no. MFP-3D)
-
Optical microscope (Olympus, model no. IX 71)
-
UV-visible spectrophotometer (Beckman Coulter, model no. DU800)
-
Cylinder of compressed N2 with air blow gun
-
Diamond-tipped glass scribe
-
Wafer tweezers
-
Serological pipettes (VWR, cat no. 89130-900)
-
Micropipette
-
Syringe (BD, cat. no. 302830)
-
25-mm diameter 0.45-μm polyethersulfone (PES) filter (VWR, cat. no. 28415-505)
-
Centrifuge bottles (1 liter; Beckman Coulter, cat. no. A98813)
Critical
Sterilize the centrifuge bottles with UV irradiation for at least 2 h before use.
-
Centrifuge tubes (50 ml; Beckman Coulter, cat. no. 334831)
Critical
Autoclave the centrifuge tubes before use.
-
Petri dish (100 × 15 mm; Falcon, cat. no. 351029)
-
Round-bottom tubes (14 ml; Falcon, cat. no. 352059)
-
Conical tubes (50 ml; Falcon, cat. no. 352070)
-
Eppendorf tubes (1.5 ml; VWR, cat. no. 89000-028)
Critical
Autoclave the Eppendorf tubes before use.
-
Glass 4-liter flasks (Pyrex, cat. no. 4980-4L)
Critical
Autoclave the flasks before use.
-
Glass 250-ml Erlenmeyer flasks with baffles (Kimble-Chase, cat. no. 25630-250)
Critical
Autoclave the flasks before use.
-
Pipette tips with filter (20, 200, and 1,000 μl; Genesee Scientific, cat. nos. 24-404, 24-412, and 24-430)
Critical
Autoclave the pipette tips before use.
REAGENT SETUP
Tetracycline solution
-
To prepare 45 mM (1,000×) tetracycline solution (20 mg/ml), dissolve 100 mg of tetracycline in a mixture of 2.5 ml of ethanol and 2.5 ml of deionized water. This solution can be stored at −20 °C for up to 1 year.
Critical
Thoroughly vortex and homogenize the solution before use.
Tetracycline–LB agar plate
-
Dissolve 18.75 g of Miller LB broth and 11.25 g of agar in deionized water. Add deionized water up to 750 ml and autoclave. Cool the solution to 50 °C and add 750 μl of 1,000× tetracycline solution. Pour the medium into a Petri dish (diameter: 10 mm, height: 15 mm). The plates can be stored at 4 °C in the dark for up to 4 weeks.
Critical
Store the plates upside down to minimize the possible chance of contamination.
IPTG–X-gal solution
-
To prepare 210 mM IPTG–98 mM X-gal (1,000×) solution, dissolve 0.625 g of IPTG and 0.5 g of X-gal in 12.5 ml of DMF. Divide the solution into 0.75-ml portions. This solution can be stored at −20 °C for up to 1 year.
IPTG–X-gal LB agar plate
-
Dissolve 18.75 g of Miller LB broth and 11.25 g of agar in deionized water. Add deionized water up to 750 ml and autoclave. Cool the solution to 50 °C and add 750 μl of 1,000× IPTG–X-gal solution. Pour the medium into a Petri dish (diameter: 10 mm, height: 15 mm). The plates can be stored at 4 °C in the dark for up to 4 weeks.
Critical
Store the plates upside down to minimize the possible chance of contamination.
Overnight-cultured XL1-Blue bacteria
-
Overnight-cultured XL1-Blue bacteria should be prepared the day before Step 10 and Step 19. 1 d before, use a 50-ml culture tube to grow a single colony of uninfected XL1-Blue bacteria in 5 ml of LB medium with 5 μg/ml tetracycline solution at 37 °C for 12–16 h by shaking at 225–250 r.p.m.
Critical
For a 5-ml Overnight culture (OC) culture, a 15-ml culture tube is too narrow. This will result in insufficient aeration of the culture.
Critical
The OC will be used for subsequent infection by engineered phages; therefore, avoid any phage contamination while making the OC. It is recommended to make the OC in a clean bench.
Top agar
-
Dissolve 25 g of LB Miller broth, 7 g of agar, and 1 g of MgCl2·6H2O in deionized water. Add deionized water up to 1 liter and autoclave. Divide the solution into 25-ml aliquots. This solution can be stored at room temperature (15–28 °C) for up to 6 months. Melt the top agar completely using a microwave and cool to 45 °C before use.
PEG–NaCl solution
-
To prepare 20% (wt/vol) PEG–2.5 M NaCl solution, dissolve 262.98 g of NaCl and 360 g of PEG 8000 in deionized water. Add deionized water up to 1.8 liters and autoclave. Shake to mix the solution and homogenize the separated solution layers. This solution can be stored at room temperature for up to 2 months.
TBS buffer
-
To prepare Tris-buffered saline (TBS, 50 mM Tris-HCl, 150 mM NaCl) solution, dissolve 6.06 g of Tris-base and 8.77 g of NaCl in 800 ml of deionized water. Adjust the pH to 7.5 with 1 M HCl. Add deionized water up to 1 liter and autoclave. This solution can be stored at room temperature for up to 6 months.
LB medium
-
Dissolve 5 g of Miller LB broth in 0.2 liters of deionized water and autoclave. Divide into 5-ml aliquots. For liter-scale amplification, prepare the medium as needed; for example, 1.25 g of Miller LB broth in 50 ml (250-ml Erlenmeyer flask) and 40 g of Miller LB broth in 1.6 liters (4-liter flask).
Critical
Freshly prepare the medium before use.
Critical
Flowchart showing use of LB medium, TBS buffer and PEG–NaCl solution is shown in Figure 4.
Figure 4: The workflow for phage PEG precipitation steps in phage purification. 
For all PEG–NaCl precipitation steps, the target ratio of PEG–NaCl to supernatant is 1:6.

EQUIPMENT SETUP
Computer-controlled pulling device
-
The pulling device is a motorized syringe pump connected to a computer with an RS232C cable and a modular adaptor (Fig. 5). The tweezers are installed lengthwise along the vertical direction, with respect to the horizontal plane, on the moving part of the syringe pump. The syringe pump is controlled with a custom computer program (available from the authors upon request), which sets the direction (i.e., vertically up or down), speed (i.e., 0.1 μm/min–30 mm/min), and time (which is dependent on the pulling speed) of the pulling routine.
Figure 5: Photographs of computer-controlled pulling device. 
Phage is self-assembled onto a substrate by using a computer-controlled pulling device to vertically pull substrates from highly pure and concentrated phage suspensions at precisely controlled speeds. The device consists of a computer, syringe pump, and substrate holders. The enlarged photograph exhibits the process of pulling substrate from phage suspensions.

Procedure
Genetic engineering of phage
Timing 3 d
Critical Step
Sterilization consists of either autoclaving and storing in a dry place (preferably an oven at <70 °C), or wiping down with 70% (vol/vol) ethanol and at least 2 h of UV irradiation before use67.
-
1
Design mutagenic oligonucleotide primers to modify the PstI restriction site (CTGCAG) in LacZα (M13KE).
Critical Step
For effective mutagenesis reaction, mutagenic oligonucleotide primers should be between 25 and 45 bases in length, with a melting temperature (Tm) of ≥78 °C. The recommended sequences of primers are 5′-CAAGCTTGCATGCCAGCAGGTCCTCGAATTCACTG-3′ and 5′-CAGTGAATTCGAGGACCTGCTGGCATGCAAGCTTG-3′.
-
2
Change T6246 to A6246 in the M13KE vector using the QuikChange II site-directed mutagenesis kit by following the manufacturer's instructions.
-
3
Design forward and reverse synthetic oligonucleotide primers for the genetic modification of the pVIII protein. Add the PstI restriction site (CTGCAG, in bold) in the forward primer, followed by the desired insert sequence. Add the PstI restriction site in the reverse primer to make the M13 plasmid linear by PCR (reverse primer: 5′-CACCCTCTGCAGCGAAAGACAGC-3′).
Critical Step
In the case of quaternary glutamate (EEEE, 4E) insertion into the pVIII protein, the sequence of the forward primer is 5′-ATATATCTGCA G AAGAAGAGGAACCCGCAAAAGCGGCCTTTAACT CCC-3′ or 5′- ATATATCTGCA G ARGARGARGARCCCGCAAAAGCGG CCTTTAACTCCC-3′ (desired sequence is in italics, R = A/G).
-
4
Prepare the PCR mix as follows using the primers designed in Step 3:
Table 5 -
5
Use the following PCR program:
Table 6 Pause point
The PCR product can be stored at −20 °C until you are ready for Step 6.
-
6
Separate the PCR product using gel electrophoresis with a 0.6% (wt/vol) agarose gel. Extract and purify the PCR product (size ∼7.3 kb) using a QIAquick Gel Extraction Kit (according to the manufacturer's instructions).
-
7
Perform a restriction digestion of the PCR product by mixing the PstI restriction enzyme (40 U) with 100 μl of the purified product, and incubate the mixture for 4 h at 37 °C. Inactivate PstI activity by incubation at 80 °C for 20 min. Purify the PstI-digested M13KE vector using a QIAquick PCR Purification Kit (according to the manufacturer's instructions).
-
8
Ligate the PstI-digested M13KE vector plasmid for recircularization with T4 DNA ligase. Mix 10 μl of PstI-digested M13KE vector plasmid (all the purified vector from Step 7) and T4 DNA ligase (200 units) and incubate the mixture at 16 °C for 18 h.
-
9
For optimal efficiency of the electrotransformation, add 1 μl of the ligation product to 50 μl of XL1-Blue electroporation-competent cells and transform at a voltage setting of 1,700 V by the electroporation method (according to the manufacturer's instructions). Incubate the transformed XL1-Blue cells in 950 μl of SOC medium (prewarmed to 37 °C before use) at 37 °C for 1 h by shaking at 225–250 r.p.m.
Critical Step
To increase the transformation efficiency, transfer the competent cell–DNA mixture to a prechilled electroporation cuvette, and immediately add 950 μl of SOC medium (prewarmed to 37 °C) after electroporation to resuspend the cells. Expected efficiency depends on the inserted peptide sequence; efficiency is ≥1 × 108 plaque-forming units per μg for M13KE DNA.
-
10
Mix 1–20 μl of the transformed bacteria with 200 μl of overnight-cultured XL-1 Blue bacteria (Reagent Setup), and pour it with 3 ml of top agarose onto IPTG–X-gal LB agar plates and incubate them at 37 °C.
-
11
Pick engineered phage plaques from the plates and culture each plaque in 1 ml of LB medium containing 10 μl of OC XL-1 Blue bacteria at 37 °C for ∼4–6 h.
Critical Step
M13KE contains the LacZ gene, so the infected plaques appear blue on the IPTG–X-gal plate.
Critical Step
The blue plaque formation on IPTG–X-gal LB agar plates is the result of growing a genetically modified phage. Only well-separated blue plaques (i.e., each plaque contains a single type of phage) are picked for further culture and identification of phage.
-
12
Extract the engineered phage vector from E. coli using a QIAprep Spin Miniprep Kit. Confirm the inserted sequences using a −96 gIII sequencing primer (5′-CCCTCATAGTTAGCGTAACG-3′) by DNA sequencing.
Verification of a clonal population of mutant phage
Timing 2 d
-
13
Add 15 μl of phage stock from Step 11 (or a single, well-separated blue plaque from an IPTG–X-gal screening plate in Step 10) and 15 μl of OC XL-1 Blue bacteria to 1.5 ml of LB medium in a 14-ml round-bottom culture tube. Culture the tube overnight, or for 8–16 h at 37 °C, on an orbital shaker operating at 225 r.p.m.
Critical Step
A concentration of >0.01 mg/ml (3.2 × 1011 virus per ml) of phage stock is preferred.
-
14
Transfer the contents of the culture tube to a centrifuge tube (1.5 ml) and centrifuge the tube at 18.4 k g at 4 °C (Table 3) for 10 min to pellet the bacteria.
Table 3 Centrifugal speeds. -
15
Transfer the upper 90% of the phage-containing supernatant to a fresh centrifuge tube and discard the remaining 10%. The typical phage concentration of the supernatant is ∼0.01–0.2 mg/ml, as determined by titration (i.e., counting blue plaques on serial-diluted-phage-incubated IPTG–X-gal LB agar plates (Step 10)). Keep the E. coli pellet for DNA sequence analysis.
Pause point
The phage stock can be stored at 4 °C for several months. For longer storage, the same volume of sterile glycerol can be added and the phage stock can be stored at −20 °C for up to 5 or more years.
-
16
Extract the double-stranded DNA from the bacterial pellet using a QIAprep Spin Miniprep Kit.
-
17
Evaluate the phage genome sequences using −96 gIII sequencing primer (5′-CCCTCATAGTTAGCGTAACG-3′) by DNA sequencing. The remaining double-stranded phage DNA can be stored for genetic engineering for up to 1 year at −20 °C.
Critical Step
The −96 gIII primer covers DNA sequencing of the gIII and gVIII sites. It is important to sequence both the cloning sites (gIII and gVIII) to ensure that your phage particle contains the desired mutation and also to evaluate the contamination by checking the existence of minor DNA peaks.
Liter-scale amplification
Timing 2 d
Critical Step
Depending on the experimental setup, the scale of phage amplification can be varied. However, incremental increases of medium from small to large scale are recommended. Performance of phage genome identification to evaluate contamination is necessary after each transferring step of the medium. This protocol describes the use of three separate 4-liter flasks.
-
18
Prepare two 250-ml baffled flasks with 50 ml of LB medium each.
-
19
Transfer 500 μl of phage stock solution from Step 15 and 500 μl of OC XL-1 Blue to one of the 250-ml baffled flasks. In the second flask, grow 50 ml of uninfected bacterial culture by adding 500 μl of OC XL-1 Blue. Culture the flasks for 5–9 h at 37 °C by shaking at 225 r.p.m. (Fig. 3a).
-
20
Remove 1 ml of phage-infected culture for DNA sequencing (Steps 14–17).
-
21
Divide the remaining phage-infected culture into two 50-ml centrifuge tubes and centrifuge them for 20 min at 12.1 k g at 4 °C (Table 3; Fig. 3b). Decant the supernatant into new tubes without disturbing the bacterial pellet.
-
22
To three separate 4-liter flasks containing 1.6 liters of LB medium each, add 14 ml of amplified phage supernatant (from Step 21) and 14 ml of fresh uninfected OC XL1 Blue bacteria (from Step 19). Incubate the flasks by shaking them for ∼12–16 h at 37 °C and 225 r.p.m. (Fig. 3c).
Critical Step
When solutions are transferred, the serological pipette and solutions must not come into contact with the sides of the flasks. Liquid containing amplified phage and bacteria should be directly transferred to the LB medium. It is important to note that wild-type contamination, as well as mutations of the original sequence, is often observed during this step. Typically, phage titers reach up to 1013 viral particles per ml in 50-ml scale cultures16, whereas lower titers are observed in 1-liter or larger-scale cultures. The phage concentration in LB medium can be determined by titration (i.e., counting blue plaques on serial-diluted phage-incubated IPTG–X-gal LB agar plates (Step 10)).
Phage purification
Timing 2 d
-
23
Dispense the culture evenly into sterilized 1-liter centrifuge bottles, keeping 1 ml of cultured solution for DNA sequencing (Steps 14–17) (Fig. 3d).
Critical Step
It is important to sequence both cloning sites (gIII and gVIII) to ensure that your phage particle contains the desired mutation and also to evaluate wild-type or mutation contamination by checking for the existence of minor DNA peaks.
-
24
Centrifuge for 20 min at 15.9 k g at 4 °C (Table 3) to pellet the bacteria. Decant the supernatant into 1-liter centrifuge bottles. Each bottle should have a final volume of 800 ml of clear phage supernatant (Fig. 3d,e).
-
25
Add 133 ml of PEG–NaCl solution to each 1-liter centrifuge bottle to a final volume of 0.933 liters. Shake the bottles to mix and incubate them at 4 °C for 12–16 h.
Critical Step
For all PEG–NaCl precipitation steps, the target ratio of PEG–NaCl to supernatant is 1:6. The workflow for phage PEG precipitation steps is shown in Figure 4. There are a number of factors that contribute to the overall yield; we have found that centrifugation is often the critical step during PEG precipitation.
-
26
Centrifuge the bottles for 20 min at 15.9 k g at 4 °C (Table 3).
-
27
Remove all of the supernatant and centrifuge the bottles again for 15 min at 15.9 k g at 4 °C (Table 3) to concentrate the precipitated phage at the bottom of the bottles. Discard the remaining supernatant (Fig. 3f).
Critical Step
The pellet of the phage at this step appears opaque white.
-
28
Add 10 ml of TBS solution to each bottle. Close the bottle and agitate it for 10 min to break up and resuspend the phage pellet. Pipette the solution up and down to break up the remaining clumps. Agitate the bottle again for 10 min to ensure a homogeneous suspension of phage particles. Transfer the solution to new 50-ml centrifuge tubes and vortex.
Critical Step
TBS has been widely used as a buffer for M13 bacteriophage to obtain the best stability. Alternatively, PBS can also be used (e.g., for chemical modification).
-
29
Add an additional 15 ml of TBS to the phage pellet in the original 1-liter centrifuge bottle. Pipette the solution up and down to capture as many phage particles as possible. Transfer the solution to 50-ml centrifuge tubes with the previous 10 ml of phage solution in TBS and vortex to homogenize the solution.
Critical Step
To break up clumps, it is critical to vortex the tubes vigorously and pipette the solution up and down repeatedly to make a homogeneous phage suspension.
-
30
Incubate the phage solution in 50-ml centrifuge tubes at 4 °C for 5 h.
-
31
Centrifuge the tubes for 10 min at 12.1 k g at 4 °C (Table 3; Fig. 3g). The pellet now contains the remaining bacteria or PEG and phage aggregates. Discard the pellet.
-
32
Filter the supernatant from Step 31 using a 0.45-μm polyethersulfone (PES)-membrane syringe filter (25-mm diameter in our setting) (Fig. 3h).
-
33
Add 4.2 ml of PEG–NaCl to each 25 ml of phage-containing supernatant, and incubate the tubes at 4 °C for ∼12–16 h.
-
34
Centrifuge the tubes for 20 min at 12.1 k g at 4 °C (Table 3). Discard the supernatant. Centrifuge one more time to collect all phage at the bottom of the tubes. Discard the remaining supernatant.
Critical Step
The pellet of the phage in this step appears opaque white.
-
35
Resuspend the pellets from six 50-ml centrifuge tubes in 10 ml of TBS, pipette the suspension up and down, and vortex the tubes to homogenize. Divide the homogenized suspension into ten 1.5-ml centrifuge tubes.
Critical Step
To break up clumps, it is important to vortex the tubes and pipette the suspension up and down vigorously.
-
36
Centrifuge the tubes for 20 min at 18.4 k g at 4 °C (Table 3). Transfer the supernatant to ten clean 1.5-ml centrifuge tubes. The pellet now contains the remaining bacteria or PEG and phage aggregates. Discard the pellet.
-
37
Add 167 μl of PEG–NaCl to each tube and incubate the tubes at 4 °C for ∼1.5–2 h.
Critical Step
Incubate the tubes for <2 h. Longer incubation will affect the quality of the phage film.
-
38
Centrifuge the tubes for 10 min at 18.4 k g at 4 °C (Table 3) to pellet the phage and discard the supernatant.
-
39
Resuspend the combined pellets of 10 tubes in 4 ml of TBS, pipette the suspension up and down, and vortex the tube to homogenize. Divide the homogenized suspension into four 1.5-ml centrifuge tubes. The target concentration of the final phage solution will be 10 mg/ml in TBS.
Critical Step
The required resuspension volume can be determined by the pellet size. In addition, the final dispersion buffer can be varied according to the purpose of the subsequent experiments. It is imperative to sequence the phage to ensure that the genome is correct, and it does not contain any mutations.
-
40
Centrifuge the tube for 20 min at 18.4 k g at 4 °C (Table 3). Transfer the supernatant to a clean 1.5-ml centrifuge tube. The pellet now contains the remaining bacteria or PEG and phage aggregates. Discard the pellet.
-
41
Incubate the supernatant for 1 h at 4 °C, and centrifuge it for 20 min at 18.4 k g at 4 °C (Table 3). Transfer the supernatant to clean 1.5-ml centrifuge tubes.
-
42
Centrifuge the supernatant for 20 min at 18.4 k g at 4 °C (Table 3). Transfer the supernatant to clean 1.5-ml centrifuge tubes (Fig. 3i).
-
43
Dilute the phage-containing solution 100 times with TBS buffer. Measure the concentration of phage particles with UV-visible spectroscopy using equation (1).

100: dilution factor, A269: absorbance at 269 nm for M13 phage, A320: absorbance at 320 nm for background.
Critical Step
OD represents the optical densities at 269 and 320 nm, respectively. Usually, 4 ml of dispersion has a 6 –10 mg/ml phage concentration. To prevent analytical error, a plaque-forming assay should be used periodically to ensure accuracy.
-
44
Once you have a final concentration of phage in solution, verify the phage genome using DNA sequencing (Steps 14–17). Prepare the desired concentration of the phage suspension in a desired buffer system for self-assembly experiments (Table 2).
Pause point
Purified phages can be stored for 1 year at 4 °C. Before use, centrifuge the stock for 20 min at 18.4 k g at 4 °C (Table 3).

Self-assembly
Timing 1–24 h, depending on the pulling speed
-
45
Prepare the desired substrates. For example, cut a gold-coated Si wafer to the desired size (typically 2 × 0.5 cm) using a diamond-tipped glass scribe.
-
46
Clean the wafer by blowing it with nitrogen gas.
-
47
To accomplish this, we use a preprogrammed syringe pump (KD Scientific) controlled through an RS232C cable (Equipment Setup; Fig. 5). Set the program according to the vendor's instructions.
-
48
Mount the gold-coated Si wafer on the setup.
Critical Step
Undesired movement of the substrate will produce substantial defects during the self-assembly process.
-
49
Prepare highly pure phage suspensions at the desired concentrations (Table 2) in 1.5-ml centrifuge tubes. Typically, we prepare ∼1 ml of the phage suspension (with TBS solution for color-phage sensor).
-
50
Before the self-assembly process, centrifuge the phage suspensions for 10 min to remove any remaining bacteria, aggregated PEG, and phage.
-
51
Lower and dip the wafer into the phage solution.
-
52
Run the designed pulling program. The film should be pulled vertically at a speed between ∼10 and 300 μm/min. Depending on the parameters, the resulting phage structures should be different (Table 2).
Critical Step
By tuning parameters such as phage concentration, pulling speed, ionic concentration, phage surface chemistry, and substrate surface, a variety of self-assembled structures, such as nematic stripe, nematic orthogonal twist, cholesteric helical ribbon, and smectic helicoidal nanofilament structures, can be fabricated. The self-assembly conditions for representative phage nanostructures are described in Table 2.
-
53
(Optional) Fabricate bundled phage nanofilaments that exhibit different colors by using a solution of 4–6 mg of phage/ml and by setting the pulling speed from 10 to 120 μm/min with 10-μm/min increments. With the incremental increase of pulling speed, the colors on each layer are shifted toward the blue visible spectrum (Fig. 2a).
Troubleshooting
Troubleshooting advice can be found in Table 4.
Timing
Steps 1–12, genetic engineering of phage: 3 d
Steps 13–17, verification of a clonal population of mutant phage: 2 d
Steps 18–22, liter-scale amplification: 2 d
Steps 23–44, phage purification: 2 d
Steps 45–53, self-assembly: 1–24 h, depending on the pulling speeds
Box 1, film characterization: 2–5 h
Anticipated results
Upon completion of the experiment described in this protocol, one should successfully obtain a highly pure genetically engineered phage stock solution. Using a sequence-verified clone, liter-scale amplification of phage is possible. The M13 phage replicates and produces large numbers of the desired phage; final yields between 10 and 120 mg of phage are typically obtained from 4.8-liter scaled amplifications. A highly purified phage solution is key to reproducible preparation of high-quality phage films. During the self-assembly process, the substrate is slowly pulled from the phage suspensions, leading to a self-templating assembly process occurring at the meniscus (Fig. 5). By tuning various parameters, such as phage concentration, pulling speed, ionic concentration, phage surface chemistry, and substrate surface, a variety of self-assembled structures such as nematic stripe, cholesteric helical ribbon, nematic orthogonal twist, and smectic helicoidal nanofilament structures can be fabricated, as shown in Figure 6. The detailed fabrication conditions (concentration of phage, pulling speed, and ionic concentration) for representative phage hierarchical structures are described in Table 2 (ref. 13). These nanostructured phage films can be used for piezoelectric energy harvesting, colorimetric sensors, and tissue regeneration13,14,26,30. Furthermore, through the investigation of the governing thermodynamic and kinetic parameters of the phage self-assembly process, we can decipher the natural self-assembly processes to build far-from-equilibrium structures. We can also design next-generation functional materials with biomimetic self-templating assembly approaches.

(a) Photograph of nematic striped pattern (scale bar, 100 μm). Inset shows an AFM image of the phage-fiber bundled structures on a ridge (scale bar, 5 μm). Phage fibers on a ridge were aligned to the pulling direction. (b) Photograph of a nematic orthogonal-twist structure (scale bar, 100 μm). Inset shows an AFM image of the phage-fiber bundled structures on a ridge (scale bar, 5 μm). Phage fibers on a ridge were perpendicularly aligned to the pulling direction. Left-handed rotation of phage-fiber bundles occurs when progressing from a groove to ridge area. (c, bottom) Photograph of cholesteric helical ribbon (CHR) structure (scale bar, 300 μm). Curved meniscus induces the rolling of phage supramolecular structures. (Top) SEM images of the CHR structure on the left and right sides, which show right- and left-handedness, respectively (scale bars, 100 μm). (d) Polarized optical microscopy image (cross-polar) of smectic helicoidal nanofilament (SHN) structure (scale bar, 50 μm). Inset: AFM image exhibited SHN structure composed of left-handed and right-handed helicoidal nanofilaments (scale bar, 5 μm). Image adapted with permission from ref. 13, Nature Publishing Group.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Crookes-Goodson, W.J., Slocik, J.M. & Naik, R.R. Bio-directed synthesis and assembly of nanomaterials. Chem. Soc. Rev. 37, 2403–2412 (2008).
Stewart, G.T. Liquid crystals in biology - i. Historical, biological and medical aspects. Liq. Cryst. 30, 751 (2003).
Neville, A.C. Biology of Fibrous Composites: Development Beyond the Cell Membrane (Cambridge University Press, 1993).
Nelson, C.M., VanDuijn, M.M., Inman, J.L., Fletcher, D.A. & Bissell, M.J. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science 314, 298–300 (2006).
Place, E.S., Evans, N.D. & Stevens, M.M. Complexity in biomaterials for tissue engineering. Nat. Mater. 8, 457–470 (2009).
Giraudguille, M.M. Twisted plywood architecture of collagen fibrils in human compact-bone osteons. Calcif. Tissue Int. 42, 167–180 (1988).
Giraud-Guille, M.M., Mosser, G. & Belamie, E. Liquid crystallinity in collagen systems in vitro and in vivo. Curr. Opin. Coll. Int. Sci. 13, 303–313 (2008).
Sharma, V., Crne, M., Park, J.O. & Srinivasarao, M. Structural origin of circularly polarized iridescence in jeweled beetles. Science 325, 449–451 (2009).
Revol, J.F., Bradford, H., Giasson, J., Marchessault, R.H. & Gray, D.G. Helicoidal self-ordering of cellulose microfibrils in aqueous suspension. Int. J. Biol. Macromol. 14, 170–172 (1992).
Daxer, A. & Fratzl, P. Collagen fibril orientation in the human corneal stroma and its implication in keratoconus. Invest. Ophthalmol. Vis. Sci. 38, A4–A4 (1997).
Prum, R.O. & Torres, R.H. Structural colouration of mammalian skin: convergent evolution of coherently scattering dermal collagen arrays. J. Exp. Biol. 207, 2157–2172 (2004).
Wegst, U.G.K., Bai, H., Saiz, E., Tomsia, A.P. & Ritchie, R.O. Bioinspired structural materials. Nat. Mater. 14, 23–36 (2015).
Chung, W.J. et al. Biomimetic self-templating supramolecular structures. Nature 478, 364–368 (2011).
Oh, J.W. et al. Biomimetic virus-based colourimetric sensors. Nat. Commun. 5, 3043 (2014).
Lee, J.H. et al. Phage-based structural color sensors and their pattern recognition sensing system. ACS Nano 11, 3632–3641 (2017).
Rakonjac, J., Bennett, N.J., Spagnuolo, J., Gagic, D. & Russel, M. Filamentous bacteriophage: biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 13, 51–75 (2011).
Wang, Y.A. et al. The structure of a filamentous bacteriophage. J. Mol. Biol. 361, 209–215 (2006).
Bhattacharjee, S., Glucksman, M.J. & Makowski, L. Structural polymorphism correlated to surface-charge in filamentous bacteriophages. Biophys. J. 61, 725–735 (1992).
Sambrook, J., Fritsch, E.F. & Maniatis, T. Molecular Cloning: A Laboratory Manual 2nd edn, (Cold Spring Harbor Laboratory Press, 1989).
Onsager, L. The effects of shape on the interaction of colloidal particles. Ann. N.Y. Acad. Sci. 51, 627–659 (1949).
Adams, M.E., Dogic, Z., Keller, S.L. & Fraden, S. Entropically driven microphase transitions in mixtures of colloidal rods & spheres. Abstr. Pap. Am. Chem. S. 216, U643 (1998).
Lee, S.W., Mao, C.B., Flynn, C.E. & Belcher, A.M. Ordering of quantum dots using genetically engineered viruses. Science 296, 892–895 (2002).
Dogic, Z. & Fraden, S. Cholesteric phase in virus suspensions. Langmuir 16, 7820–7824 (2000).
Dogic, Z. & Fraden, S. Smectic phase in a colloidal suspension of semiflexible virus particles. Phys. Rev. Lett. 78, 2417–2420 (1997).
Yoo, S.Y., Chung, W.J., Kim, T.H., Le, M. & Lee, S.W. Facile patterning of genetically engineered m13 bacteriophage for directional growth of human fibroblast cells. Soft Matter 7, 363–368 (2011).
Lee, B.Y. et al. Virus-based piezoelectric energy generation. Nat. Nanotechnol. 7, 351–356 (2012).
Lee, J.H. et al. Biomimetic sensor design. Nanoscale 7, 18379–18391 (2015).
Lee, Y.J. et al. Fabricating genetically engineered high-power lithium-ion batteries using multiple virus genes. Science 324, 1051–1055 (2009).
Nam, Y.S. et al. Biologically templated photocatalytic nanostructures for sustained light-driven water oxidation. Nat. Nanotechnol. 5, 340–344 (2010).
Merzlyak, A., Indrakanti, S. & Lee, S.W. Genetically engineered nanofiber-like viruses for tissue regenerating materials. Nano Lett. 9, 846–852 (2009).
Rong, J.H. et al. Oriented cell growth on self-assembled bacteriophage m13 thin films. Chem. Commun. 41, 5185–5187 (2008).
Tom, S., Jin, H.-E., Heo, K. & Lee, S.-W. Engineered phage films as scaffolds for caco3 biomineralization. Nanoscale 8, 15696–15701 (2016).
Serra, P. et al. Laser direct writing of biomolecule microarrays. Appl. Phys. A 79, 949–952 (2004).
Wilbur, J.L., Kumar, A., Kim, E. & Whitesides, G.M. Microfabrication by microcontact printing of self-assembled monolayers. Adv. Mater. 6, 600–604 (1994).
Piner, R.D., Zhu, J., Xu, F., Hong, S. & Mirkin, C.A. 'Dip-pen' nanolithography. Science 283, 661 (1999).
Roth, E.A. et al. Inkjet printing for high-throughput cell patterning. Biomaterials 25, 3707–3715 (2004).
Lee, H., Rho, J. & Messersmith, P.B. Facile conjugation of biomolecules onto surfaces via mussel adhesive protein inspired coatings. Adv. Mater. 21, 431–434 (2009).
Holliday, B.J. & Mirkin, C.A. Strategies for the construction of supramolecular compounds through coordination chemistry. Angew. Chem. Int. Ed. Engl. 40, 2022–2043 (2001).
Lee, S.W. & Belcher, A.M. Virus-based fabrication of micro- and nanofibers using electrospinning. Nano Lett. 4, 387–390 (2004).
Lee, S.W., Lee, S.K. & Belcher, A.M. Virus-based alignment of inorganic, organic, and biological nanosized materials. Adv. Mater. 15, 689–692 (2003).
Yoo, P.J. et al. Spontaneous assembly of viruses on multilayered polymer surfaces. Nat. Mater. 5, 234–240 (2006).
Nam, K.T. et al. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science 312, 885–888 (2006).
Kolle, M. et al. Mimicking the colourful wing scale structure of the Papilio blumei butterfly. Nat. Nanotechnol. 5, 511–515 (2010).
Vukusic, P. & Sambles, J.R. Photonic structures in biology. Nature 424, 852–855 (2003).
Prum, R.O. & Torres, R. Structural colouration of avian skin: convergent evolution of coherently scattering dermal collagen arrays. J. Exp. Biol. 206, 2409–2429 (2003).
Kinoshita, S. & Yoshioka, S. Structural colors in nature: the role of regularity and irregularity in the structure. Chemphyschem 6, 1442–1459 (2005).
Vigneron, J.P. et al. Switchable reflector in the Panamanian tortoise beetle Charidotella egregia (Chrysomelidae: Cassidinae). Phys. Rev. E 76, 031907 (2007).
Young, R.E. & Mencher, F.M. Bioluminescence in mesopelagic squid: diel color change during counterillumination. Science 208, 1286–1288 (1980).
Smith, G.P. & Petrenko, V.A. Phage display. Chem. Rev. 97, 391–410 (1997).
Hagan, M.F., Majumdar, A. & Chakraborty, A.K. Nanomechanical forces generated by surface grafted DNA. J. Phys. Chem. B 106, 10163–10173 (2002).
Farrar, D. et al. Permanent polarity and piezoelectricity of electrospun α-helical poly(α-amino acid) fibers. Adv. Mater. 23, 3954–3958 (2011).
Dong, X.X., Ospeck, M. & Iwasa, K.H. Piezoelectric reciprocal relationship of the membrane motor in the cochlear outer hair cell. Biophys. J. 82, 1254–1259 (2002).
Minary-Jolandan, M. & Yu, M.F. Nanomechanical heterogeneity in the gap and overlap regions of type I collagen fibrils with implications for bone heterogeneity. Biomacromolecules 10, 2565–2570 (2009).
Csoka, L. et al. Piezoelectric effect of cellulose nanocrystals thin films. ACS Macro Lett. 1, 867–870 (2012).
Fukada, E. & Sasaki, S. Piezoelectricity of alpha-chitin. J. Polym. Sci. Pol. Phys. 13, 1845–1847 (1975).
Shin, D.M. et al. Bioinspired piezoelectric nanogenerators based on vertically aligned phage nanopillars. Energy Environ. Sci. 8, 3198–3203 (2015).
Wang, J.L. et al. Phage nanofibers induce vascularized osteogenesis in 3D printed bone scaffolds. Adv. Mater. 26, 4961–4966 (2014).
Huber, A.B., Kolodkin, A.L., Ginty, D.D. & Cloutier, J.F. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu. Rev. Neurosci. 26, 509–563 (2003).
Milner, R. & Campbell, I.L. The integrin family of cell adhesion molecules has multiple functions within the CNS. J. Neurosci. Res. 69, 286–291 (2002).
Rutka, J.T., Apodaca, G., Stern, R. & Rosenblum, M. The extracellular-matrix of the central and peripheral nervous systems - structure and function. J. Neurosurg. 69, 155–170 (1988).
Oster, S.F., Deiner, A., Birgbauer, E. & Sretavan, D.W. Ganglion cell axon pathfinding in the retina and optic nerve. Semin. Cell. Dev. Biol. 15, 125–136 (2004).
Merzlyak, A. & Lee, S.W. Engineering phage materials with desired peptide display: rational design sustained through natural selection. Bioconjug. Chem. 20, 2300–2310 (2009).
Yoo, S.Y., Merzlyak, A. & Lee, S.W. Facile growth factor immobilization platform based on engineered phage matrices. Soft Matter 7, 1660–1666 (2011).
Yoo, S.Y., Kobayashi, M., Lee, P.P. & Lee, S.-W. Early osteogenic differentiation of mouse preosteoblasts induced by collagen-derived dgea-peptide on nanofibrous phage tissue matrices. Biomacromolecules 12, 987–996 (2011).
Chung, W.J., Merzlyak, A. & Lee, S.W. Fabrication of engineered m13 bacteriophages into liquid crystalline films and fibers for directional growth and encapsulation of fibroblasts. Soft Matter 6, 4454–4459 (2010).
Nam, K.T., Lee, Y.J., Krauland, E.M., Kottmann, S.T. & Belcher, A.M. Peptide-mediated reduction of silver ions on engineered biological scaffolds. ACS Nano 2, 1480–1486 (2008).
Sanders, E.R. Aseptic laboratory techniques: plating methods. J. Vis. Exp. 63, e3064 (2012).
Chung, W.J., Merzlyak, A., Yoo, S.Y. & Lee, S.W. Genetically engineered liquid-crystalline viral films for directing neural cell growth. Langmuir 26, 9885–9890 (2010).
Yoo, S.Y. et al. M13 bacteriophage and adeno-associated virus hybrid for novel tissue engineering material with gene delivery functions. Adv. Healthc. Mater. 5, 88–93 (2016).
Zhu, H.B. et al. Controlled growth and differentiation of mscs on grooved films assembled from monodisperse biological nanofibers with genetically tunable surface chemistries. Biomaterials 32, 4744–4752 (2011).
Jin, H.E., Farr, R. & Lee, S.W. Collagen mimetic peptide engineered m13 bacteriophage for collagen targeting and imaging in cancer. Biomaterials 35, 9236–9245 (2014).
Moon, J.S. et al. M-13 bacteriophage based structural color sensor for detecting antibiotics. Sensor Actuat. B-Chem. 240, 757–762 (2017).
Moon, J.S. et al. Bioinspired m-13 bacteriophage-based photonic nose for differential cell recognition. Chem. Sci. 8, 921–927 (2017).
Moon, J.S. et al. Identification of endocrine disrupting chemicals using a virus-based colorimetric sensor. Chem. Asian J. 11, 3097–3101 (2016).
Kim, W.G. et al. Biomimetic self-templating optical structures fabricated by genetically engineered m13 bacteriophage. Biosens. Bioelectron. 85, 853–859 (2016).
Yoo, S.Y., Oh, J.W. & Lee, S.W. Phage-chips for novel optically readable tissue engineering assays. Langmuir 28, 2166–2172 (2012).
Acknowledgements
This work was funded by the US Army Environmental Quality Research Program (611102T2500). Permission was granted by the Chief of Engineers to publish this information. We acknowledge funding support from the Tsinghua-Berkeley Shenzhen Institute. This work was also supported in part by the Samsung Electronics Global Research Outreach Program. This work was supported under the framework of international cooperation program managed by the National Research Foundation of Korea (NRF; 2016K2A9A1A01951919 and NRF-2014S1A2A2027641). H.-E.J. is supported by a NRF grant funded by the Korea government (MSIP) (2016R1C1B1008824).
Author information
Authors and Affiliations
Contributions
J.H.L., C.M.W., H.-E.J., and S.-W.L. wrote the paper, and all other authors provided substantial editorial revisions. Representative experiments on genetic engineering of the phage and verification of a clonal population of mutant phage were designed or performed by J.H.L., H.-E.J., and S.-W.L. Similar contributions by J.H.L., C.M.W., H.-E.J., E.B., A.R.P., E.J.P., and S.-W.L. were made with regard to liter-scale amplification and phage purification. The experiments on phage self-assembly involved J.H.L., C.M.W., and S.-W.L. The procedure for the film-characteristics experiment involved J.H.L. and S.-W.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Lee, J., Warner, C., Jin, HE. et al. Production of tunable nanomaterials using hierarchically assembled bacteriophages. Nat Protoc 12, 1999–2013 (2017). https://doi.org/10.1038/nprot.2017.085
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2017.085
This article is cited by
-
High-throughput fabrication of antimicrobial phage microgels and example applications in food decontamination
Nature Protocols (2024)
-
Monomeric streptavidin phage display allows efficient immobilization of bacteriophages on magnetic particles for the capture, separation, and detection of bacteria
Scientific Reports (2023)
-
M13 phage: a versatile building block for a highly specific analysis platform
Analytical and Bioanalytical Chemistry (2023)
-
Genome-wide investigation of maize RAD51 binding affinity through phage display
BMC Genomics (2022)
-
M13 Bacteriophage-Based Bio-nano Systems for Bioapplication
BioChip Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
