
Abstract
Hi-C is a powerful method that provides pairwise information on genomic regions in spatial proximity in the nucleus. Hi-C requires millions of cells as input and, as genome organization varies from cell to cell, a limitation of Hi-C is that it only provides a population average of genome conformations. We developed single-cell Hi-C to create snapshots of thousands of chromatin interactions that occur simultaneously in a single cell. To adapt Hi-C to single-cell analysis, we modified the protocol to include in-nucleus ligation. This enables the isolation of single nuclei carrying Hi-C–ligated DNA into separate tubes, followed by reversal of cross-links, capture of biotinylated ligation junctions on streptavidin-coated magnetic beads and PCR amplification of single-cell Hi-C libraries. The entire laboratory protocol can be carried out in 1 week, and although we have demonstrated its use in mouse T helper (TH1) cells, it should be applicable to any cell type or species for which standard Hi-C has been successful. We also developed an analysis pipeline to filter noise and assess the quality of data sets in a few hours. Although the interactome maps produced by single-cell Hi-C are sparse, the data provide useful information to understand cellular variability in nuclear genome organization and chromosome structure. Standard wet and dry laboratory skills in molecular biology and computational analysis are required.
Similar content being viewed by others
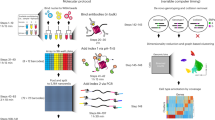


Introduction
In the interphase nucleus of higher eukaryotes, the genome is nonrandomly organized within the 3D space to form long-range and trans-chromosomal interactions. This organization is an emerging area of importance, as the function and activity of loci are strongly correlated with their 3D conformations1,2. To explore such chromosome interactions at a genome-wide level, chromosome conformation capture (3C)-based high-throughput methods, in particular Hi-C, have been extensively used3,4,5. Hi-C measures pairwise contact frequencies between all loci in the genome using populations of millions of cells. These data have led to advances in the understanding of higher-order organization of the genome, such as chromatin loop formation and the discovery of topologically associating domains6,7,8, and they have been used to build 3D models of chromatin3,9,10. However, it is important to note that Hi-C data represent an ensemble average of millions of cells, and decades of microscopy studies have shown that nuclear organization is not uniform, even among cells of a homogeneous population11. Thus, the actual genome organization of an individual cell cannot be determined from ensemble Hi-C.
Development of the protocol
In conventional Hi-C, cross-linked chromatin in millions of cells is digested with a restriction enzyme, followed by biotin fill-in labeling of the digested ends. After nuclear lysis and dilution of chromatin complexes, DNA fragments in the same cross-linked complex are ligated and sheared, and ligation junctions are recovered on streptavidin-coated beads before paired-end sequencing12. To develop single-cell Hi-C, we modified the original Hi-C protocol12 to perform the ligation in the nucleus13, allowing us to isolate single cells in which the Hi-C procedure was nearly complete. Cells were then transferred to individual tubes for cross-link reversal, DNA fragmentation, capture of ligation junctions and Hi-C library construction (Fig. 1). Despite these technical differences, we confirmed that the single-cell Hi-C protocol produces data similar to conventional ensemble Hi-C by comparing pooled single-cell Hi-C data from multiple cells13. Our more recent analyses, and those of others, suggest that in-nucleus ligation actually improves Hi-C results14,15.

Adapted with permission from ref. 13, Nature Publishing Group.
We also developed computer scripts (scell_hicpipe) to filter spurious read-pairs and assess the quality of single-cell Hi-C data sets; the most recent versions of these scripts and a test data set are available at https://bitbucket.org/tanaylab/schic_pipeline.git. The rationale behind these scripts is described in detail in the supplementary information of Nagano et al.13, which can be found here: http://www.nature.com/nature/journal/v502/n7469/extref/nature12593-s1.pdf.
The scell_hicpipe takes mapped paired-end reads and filters out the read-pairs that are likely to be noise (e.g., read-pairs appearing only once in the sequence output), and it statistically assesses the probability that the data set derives from a single cell. In our recent study13, we chose the data sets that satisfy the following three conditions for further in-depth analyses: at least 10,000 unique contacts; the percentage of read-pairs that were sequenced only once (and therefore filtered) was <30%; and the data set was as expected from a single cell—i.e., the number of autosomal restriction fragment ends (fends) interacting with more than two other distinct fends should be less than the expected number according to a binomial model. Of 74 data sets created in the study, ten satisfied these criteria.
Here we provide a detailed protocol for single-cell Hi-C based on our previously published methods13. The protocol also describes basic data analysis using the scell_hicpipe scripts via the command line on a Linux operating system.
Applications, advantages and limitations of single-cell Hi-C
The interactome obtained with this protocol is not an average among multiple cells but a snapshot from a single cell at the time of cell fixation. Therefore, the data provide the opportunity to analyze snapshots of chromosome conformations from individual cells capturing cellular heterogeneity, which may reflect their dynamic behavior. For example, we showed in our single-cell Hi-C study on mouse TH1 cells that individual chromosomes maintain topological domain organization at the megabase scale, but that chromosome structures vary from cell to cell at larger scales13. An important point to note is that single-cell Hi-C data are sparse, and a potential concern is that the variability in interactome maps derived from individual cells could be because of nonuniform sampling of the interactome from each cell. However, proper statistical analyses can rule out such explanations for the observations.
A distinct advantage of single-cell or single-molecule Hi-C data (as in the case of the X chromosome in male cells) is the ability to apply 3D modeling approaches. For example, the interaction data from the single-copy male X chromosome was used to derive distance restraints to calculate 3D models of chromosomes, upon which genomic and epigenomic information can be projected for the study of spatial patterns of such features13.
Currently, a major limiting factor for the single-cell Hi-C technique is the sparsity of data or genome coverage. We detected up to ∼30,000 unique interaction pairs per single cell. The theoretical number of distinct mappable interaction pairs in a mouse diploid cell is ∼1.2 million, indicating that the genome coverage from the richest data sets was ∼2.5%. Although coverage was low, it was uniform, suggesting that it was unlikely to be due to biased retrieval from the genome. Chromosome structure modeling confirmed this, as combining individual cell data sets led to compact ball-shaped structures with increased violations, rather than a refinement of the models from the initial data sets13.
The sparsity of genome coverage leads to concerns over the success rate of the experimental protocol. We showed that the richest data sets—i.e., those with the highest number of restraints (unique read pairs)—generated the most accurate and precise 3D models. In this regard, we chose ten single-cell data sets out of the 74 single cells processed for in-depth statistical analyses, and six of these were used for 3D modeling analyses in our previous study13. The low rate of useful libraries is a technical limitation of this protocol. A laboratory automation system to process larger numbers of single cells would obviously increase the number of successful data sets with this protocol. Future work aimed at streamlining or improving the efficiency of individual steps in the single-cell Hi-C protocol may also help increase genomic coverage per cell and, in so doing, improve success rates.
However, it is worth noting that despite the technical limitations, there are currently no published alternatives to this single-cell Hi-C protocol.
Experimental design
Cell type. In general, single-cell Hi-C should be possible in any cell preparation in which 3C or Hi-C has been successful. For cells in which 3C or conventional Hi-C information is not available, it is possible to check the experimental conditions by testing the efficiencies of restriction digestion16, biotin labeling and ligation (Box 1) using several million cells.
Cell fixation and permeabilization. In this protocol, cells are fixed in 2% formaldehyde for 5 min at room temperature (20–25 °C). However, different conditions (e.g., 1–2% formaldehyde for 10 min at room temperature) can also be applied. Generally, stronger fixation conditions (i.e., higher formaldehyde concentration or longer fixation time) may increase the risk of higher background noise in single-cell Hi-C data. For dissociation to single cells and cell permeabilization, additional steps such as trypsin treatment or Dounce homogenizing may be necessary for some cell types17.
First restriction enzyme (RE1). The resolution of Hi-C depends on the frequency of restriction enzyme digestion sites in the genome. Therefore, four-cutter enzymes, which have significantly more cut sites in the genome than six-cutter enzymes, have the potential to create higher-resolution interactome maps. The RE1 digestion of the Hi-C protocol is carried out in the presence of detergents, and it is important to note that not every enzyme is suitably active under these conditions. We have used either BglII or DpnII for single-cell Hi-C (BglII in the protocol below), but other 3C and Hi-C methods have used EcoRI (ref. 18), HindIII (ref. 19), NcoI (ref. 12) and NlaIII (ref. 20). If an enzyme has been used successfully in standard 3C or Hi-C protocols, it is reasonable to assume that the enzyme will work for single-cell Hi-C. Nevertheless, the efficiency of RE1 digestion should be checked when initiating Hi-C studies and in particular when using a restriction enzyme that has not been used for 3C or Hi-C previously. The procedure to check the digestion efficiency has been previously described16.
Library barcoding and sequencing. Each single-cell library is barcoded or indexed so that multiple libraries can be mixed in the same sequencing lane and demultiplexed once the sequence data are collected. Because of the greatly reduced complexity of single-cell Hi-C libraries compared with ensemble Hi-C libraries, several single-cell libraries can be multiplexed and still sequenced to reasonable depth. In our recent study13 in which RE1 was BglII (approximately one million digestion sites in mouse haploid genome), we loaded up to 12 single-cell Hi-C libraries per lane of an Illumina Genome Analyzer IIx that is capable of ∼30 million paired-end reads. We found this to be sufficient to determine the quality of single-cell Hi-C libraries, and to achieve near saturation of depth for most libraries13. Single-cell libraries of particularly high quality (i.e., sufficient number of fend-pairs, high statistical probability for single-cell sourced data and high percentage of valid read-pair; see ANTICIPATED RESULTS section below) can be resequenced to obtain additional read-pairs, which results in more informative data sets.

Materials
REAGENTS
-
Fresh single-cell suspension (>107 cells are preferable to start with)
Caution
If the cells are directly from animals, approved governmental and institutional regulations for animal work must be adhered to, and experiments should be reported according to the ARRIVE (Animal Research: Reporting in vivo Experiments) guidelines21.
Caution
If you are using cell lines in your research, they should be regularly checked to ensure that they are authentic and are not infected with mycoplasma.
-
Full medium appropriate for the cell of choice (we used RPMI 1640 with 5% (vol/vol) FBS for mouse ex vivo TH1 cells for our previous study13)
-
Formaldehyde (16% solution; Agar Scientific, cat. no. R1026)
Caution
Follow the appropriate health and safety regulations to handle formaldehyde solution, as it is toxic.
-
PBS, pH 7.4 (10×; Life Technologies, cat. no. 70011-036)
-
Water (molecular biology grade; Sigma-Aldrich, cat. no. W4502)
-
Glycine (freshly prepared 2 M solution)
-
Tris-HCl, pH 8.0 (1 M; Life Technologies, cat. no. 15568-025)
-
NaCl (5 M; Life Technologies, cat. no. 24740-011)
-
NP-40 (also known as IGEPAL CA-630; Sigma-Aldrich, cat. no. I8896)
-
cOmplete, EDTA-free (Roche Diagnostics, cat. no. 11873580001)
-
SDS, 20% (wt/vol) solution (Bio-Rad Laboratories, cat. no. 161-0418)
-
Triton X-100 (20% (vol/vol); Sigma-Aldrich, cat. no. T8787)
-
BglII (50,000 U/ml; New England Biolabs, cat. no. R0144M); NEBuffer 3 (10×) supplied with the enzyme. Alternative enzymes can be used instead, as discussed in Experimental design
-
dCTP (10 mM; Life Technologies, cat. no. 18253-013)
-
dGTP (10 mM; Life Technologies, cat. no. 18254-011)
-
dTTP (10 mM; Life Technologies, cat. no. 18255-018)
-
dATP (10 mM; Life Technologies, cat. no. 18252-015)
-
Biotin-14-dATP (0.4 mM; Life Technologies, cat. no. 19524-016)
-
DNA polymerase I, large (Klenow) fragment (5,000 U/ml; New England Biolabs, cat. no. M0210S)
-
NEBuffer 2 (10×; New England Biolabs, cat. no. B7002S)
-
T4 DNA ligase reaction buffer (10×; New England Biolabs, cat. no. B0202S)
-
BSA (100× (10 mg/ml); New England Biolabs, cat. no. B9001)
-
T4 DNA ligase (1 U/μl; Life Technologies, cat. no. 15224-025)
-
Agarose, low gelling temperature (Sigma-Aldrich, cat. no. A9414)
-
DNeasy blood and tissue kit (Qiagen, cat. no. 69504)
-
HotStarTaq DNA polymerase (Qiagen, cat. no. 203203)
-
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
-
DNA LoBind tube (1.5 ml; Eppendorf, cat. no. 0030 108.051)
-
Tris-HCl, pH 7.5 (1 M; Life Technologies, cat. no. 15567-027)
-
EDTA, 0.5 M, pH 8.0 (Life Technologies, cat. no. 15575-020)
-
Dynabeads M-280 streptavidin (Life Technologies, cat. no. 11205D)
-
AluI (10,000 U/ml; New England Biolabs, cat. no. R0137S (comes with NEBuffer 4 (10×)))
-
Klenow fragment (3′–5′ exo–; 5,000 U/ml; New England Biolabs, cat. no. M0212S)
-
T4 DNA ligase (2,000,000 U/ml; New England Biolabs, cat. no. M0202M)
-
Library amplification primers (for Illumina platform): (F) 5′-AATGATAC GGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T-3′; (R) 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCA TTCCTGCTGAACCGCTCTTCCGATC*T-3′; note that the asterisk (*) indicates 5′–3′ phosphorothioate linkage)
Critical
These oligonucleotides should be 'HPLC-purification grade'.
-
Platinum Pfx DNA polymerase (2.5 U/μl; Life Technologies, cat. no. 11708-013 (comes with Pfx amplification buffer (10×) and 50 mM MgSO4))
-
Agencourt AMPure XP (Beckman Coulter, cat. no. A63881)
Critical
AMPure XP is stored at 4 °C, but make sure that AMPure XP added to samples is at room temperature. For convenience, make an aliquot to equilibrate it to room temperature in advance.
-
Agarose electrophoresis gel in TBE buffer (2% (wt/vol))
-
Glycerol, for molecular biology (Sigma-Aldrich, cat. no. G5516)
-
Orange G (Sigma-Aldrich, cat. no. O3756)
-
MinElute gel extraction kit (Qiagen, cat. no. 28604)
-
Illumina library quantification kit (KAPA Biosystems, cat. no. KK4824)
EQUIPMENT
-
Centrifuge (Heraeus, Megafuge 3.0R)
-
Cell strainer (40 μl; Corning, cat. no. 352340)
-
Hemocytometer (Weber Scientific)
-
Slide glass
-
Petri dish
-
Pasteur pipettes (Fig. 2a; see Equipment Setup)
Figure 2: A drawn Pasteur pipette and aspirator tube assembly. 
Cells are collected by slight suction and capillary action using a mouth-controlled assembly modified to hold a drawn Pasteur pipette. (a) The Pasteur pipette is drawn to a final diameter of ∼80 μm. The final diameter is not crucial, but it greatly aids in the fine control of the cell during collection. The numbers on the ruler shown are in cm. (b) A length of the rubber tubing connecting a red mouth piece and an adapter onto which the Pasteur pipette is fixed through the silicone tubing (see Equipment Setup).
-
Aspirator tube assemblies for calibrated microcapillary pipettes (Sigma-Aldrich, cat. no. A5177; Fig. 2b; see Equipment Setup)
-
Silicone tubing (6.0 mm bore, 2.0-mm-thick wall)
-
Phase contrast microscope (Nikon TMS)
-
Rotating wheel (Stuart SB3)
-
Liquid nitrogen in a Dewar flask (Dilvac)
-
Benchtop centrifuge (Eppendorf 5417R)
-
Thermomixer (Eppendorf, cat. no. EF4283)
-
Stereoscopic microscope with more than 60× magnification (Nikon SMZ800)
-
Hybridization oven (Techne HB-1D)
-
Low-retention filter tips (Starlab, cat. nos. S1180-3810, S1180-1810, S1180-8810 and S1182-1830)
-
DynaMag-2 magnet (Life Technologies, cat. no. 12321D)
-
Thermal cycler (MJ Research PTC-200)
-
DynaMag-96 side magnet (Life Technologies, cat. no. 12331D)
-
Gel tank (Flowgen Mini Rapide)
-
2100 Bioanalyzer system (Agilent Technologies)
-
Real-time PCR system (Bio-Rad CFX96)
-
Illumina Genome Analyzer IIx (or HiSeq system; Illumina)
-
Perl software package (https://www.perl.org): the scripts used in this protocol have been tested with v5.10.1
-
R software package (http://www.r-project.org) with gplots installed: the scripts used in this protocol have been tested with R v3.1.2
-
Single-cell Hi-C pipeline (scell_hicpipe; version 1.2); download from https://bitbucket.org/tanaylab/schic_pipeline.git

REAGENT SETUP
Critical
The volumes given below are to process up to ten million cells and to prepare 12 single-cell Hi-C libraries. These volumes can be scaled up if the number of processed cells is increased, or they can be scaled up or down depending on the number of single-cell samples. However, always prepare extra (∼10%) to ensure that all samples receive the exact amount indicated in the PROCEDURE.
NP-40, 20% (vol/vol)
-
To prepare 1 ml of 20% (vol/vol) NP-40, add 800 μl of water to 200 μl of NP-40, and mix it well.
Critical
Freshly prepare the solution on the day of the cell permeabilization.
Permeabilization buffer
-
Permeabilization buffer is 10 mM Tris-HCl, 10 mM NaCl and 0.2% (vol/vol) NP-40, cOmplete EDTA-free; to prepare a volume of 50 ml, add 500 μl of 1 M Tris-HCl (pH 8.0), 100 μl of 5 M NaCl and 500 μl of 20% (vol/vol) NP-40 to 48.9 ml of water. Then, add one tablet of cOmplete EDTA-free, mix it well to dissolve the tablet and chill the buffer on ice.
Critical
Freshly prepare the buffer on the day of the cell permeabilization.
Triton X-100, 20% (vol/vol)
-
To prepare 1 ml of 20% (vol/vol) Triton X-100, add 800 μl of water to 200 μl of Triton X-100 and mix it well.
Critical
Freshly prepare the solution for each restriction digestion.
NEBuffer 3, 1.2×
-
To prepare 1.3 ml of 1.2× NEBuffer 3, add 156 μl of NEBuffer 3 (10×) to 1.144 ml of water and mix it well.
Critical
Freshly prepare the solution for each restriction digestion.
Binding and washing (BW) buffer, 1×
-
BW buffer, 1× is 5 mM Tris-HCl, 0.5 mM EDTA and 1 M NaCl; to prepare a volume of 50 ml, add 250 μl of 1 M Tris-HCl (pH 7.5), 50 μl of 0.5 M EDTA and 10 ml of 5 M NaCl to 39.7 ml of water and mix the contents well. We usually freshly prepare the buffer.
BW buffer, 2×
-
BW buffer, 2× is 10 mM Tris-HCl, 1 mM EDTA and 2 M NaCl; to prepare a volume of 1 ml, add 10 μl of 1 M Tris-HCl (pH 7.5), 2 μl of 0.5 M EDTA and 400 μl of 5 M NaCl to 588 μl of water and mix it well. We usually freshly prepare the buffer.
EB buffer
-
EB buffer is 10 mM Tris-HCl; to prepare a volume of 15 ml, add 150 μl of 1 M Tris-HCl (pH 7.5) to 14.85 ml of water. We usually freshly prepare the buffer.
Oligonucleotides for sequencing adapters
-
(For Illumina sequencing platform) Pairs of two oligonucleotides (forward, F) and (reverse, R) shown in Table 1 should be annealed to prepare the sequencing adapter without the identification tag, or with each corresponding identification tag. p indicates 5′-phosphate modification, and the asterisk (*) indicates 5′–3′ phosphorothioate linkage. Dissolve each adapter to 100 μM in water, and make aliquots of 15 μl. These aliquots can be stored at −20 °C for up to 2–3 years.
Table 1 Oligonucleotides for sequencing adapters. Critical
These oligonucleotides should be 'HPLC-purification grade'.
Annealing of sequencing adapters (15 μM)
-
Mix 15 μl of 100 μM (F) strand with 15 μl of 100 μM (R) strand of the same tag and add 70 μl of water. Transfer the mixture to a 0.2-ml PCR tube and run the following program in a thermal cycler.
Table 4 Critical
Keep it on ice until it is used; however, if it is not used on the same day, make small aliquots (3–10 μl) and store them at −20 °C for up to 6 months.
Orange G loading dye
-
(1 ml) Mix 270 μl of water, 10 μl of 1 M Tris-HCl (pH 7.5), 120 μl of 0.5 M EDTA and 600 μl of glycerol; add 1.5 mg of orange G and mix it well. Store the dye at −20 °C for up to 6 months.
EQUIPMENT SETUP
Pasteur pipette with the aspirator tube assembly
-
Draw the Pasteur pipettes by hand in a flame to a diameter of ∼80 μm, and break each one with a diamond pen to create an open end at a right angle to the pipette bore. The tip size and the appearance can be checked under a stereo microscope. Remove the silicone capillary holder (at the left end in Fig. 2b) from the original aspirator tube assembly and, to the adapter, add a 50-mm piece of silicone tubing (6.0 mm bore, 2.0-mm-thick wall). Insert the Pasteur pipette into the tubing to complete the assembly.
Procedure
Cell fixation
Timing 30 min to 1 h
-
1
Prepare a fresh single-cell suspension (preferably containing 107 cells or more) in 21.875 ml of full medium at room temperature in a 50-ml centrifuge tube.
-
2
Add 3.125 ml of 16% formaldehyde, to obtain a final formaldehyde concentration of 2% in 25 ml.
-
3
Fix for exactly 5 min at room temperature with gentle mixing on a rotating wheel.
Caution
As formaldehyde is toxic, it should be handled in a fume hood and discarded according to relevant institutional and local regulations.
-
4
Quench fixation by adding 1.7 ml of 2 M glycine (final glycine concentration will be 0.127 M) and mix gently by inverting the tube.
-
5
Centrifuge the tube at ∼300g for 8 min at 4 °C.
-
6
Remove the supernatant and resuspend the pellet in 50 ml of cold PBS (1×).
Critical Step
To completely remove the supernatant without disturbing the pellet, do not try to remove all of the supernatant at once. After removing most of the supernatant by pipetting or decanting, with 2–3 ml left, briefly recentrifuge the tube to collect all of the remaining supernatant droplets at the bottom of the tube and carefully remove the remaining supernatant with a 200- or 1,000-μl pipette. This also applies to Step 8 below.
-
7
Centrifuge the tube at ∼300g for 8 min at 4 °C.
-
8
Remove the supernatant as described in Step 6 and proceed to cell permeabilization (Step 9).
Pause point
The cell pellet can be frozen in liquid nitrogen and stored at −80 °C for several months.
Cell permeabilization and the first restriction digestion
Timing 3 h + incubation for 12–16 h
-
9
Resuspend the fixed cell pellet from Step 8 in 50 ml of ice-cold permeabilization buffer.
Critical Step
If it is frozen, the cell pellet should be left on ice until it thaws. Resuspension is most effective by first adding a small volume (2–3 ml) of permeabilization buffer and vigorously tapping the tube, and then adding the remaining buffer.
-
10
Incubate the tube for 30 min on ice, and mix it by inversion every 5–10 min.
Critical Step
Mixing is important to ensure high-quality nuclei prep.
-
11
Centrifuge the tube at ∼600g for 6 min at 4 °C.
-
12
Remove most of the supernatant, leaving ∼1 ml, resuspend the cells with the remaining supernatant, and transfer it to a 1.5-ml tube.
-
13
Centrifuge the tube at 600g for 6 min at 4 °C in a benchtop centrifuge.
-
14
Remove the supernatant and wash the pellet twice (once in 800 μl and once in 400 μl of 1.2× NEBuffer 3).
Critical Step
Substitute 1.2× NEBuffer 3 buffer with appropriate buffer when you are using a restriction enzyme other than BglII as RE1.
Critical Step
Resuspending the cell pellet is not necessary at this step. It may even cause cell loss, especially when you are working with a low number of cells.
Critical Step
Buffer exchange is most efficient by first removing ∼90% of the supernatant; briefly recentrifuge the tube (keeping the same tube orientation so as not to disturb the existing cell pellet) and carefully remove the remaining supernatant with a 20-μl pipette. This applies to all subsequent steps involving supernatant removal from 1.5-ml tubes after centrifugation.
-
15
Add 6 μl of 20% SDS (final concentration 0.3%, wt/vol), resuspend the cell pellet and shake at 950 r.p.m. for 1 h at 37 °C on a thermomixer.
-
16
Add 40 μl of 20% Triton X-100 (final concentration 1.8%, vol/vol) and shake at 950 r.p.m. for 1 h at 37 °C on a thermomixer.
-
17
Add 1,500 U of BglII (30 μl of 50 U/μl solution) and shake at 950 r.p.m. at 37 °C on a thermomixer for 12–16 h.
Critical Step
Replace BglII with the appropriate enzyme when using a restriction enzyme other than BglII as RE1, and adapt the reaction temperature accordingly.
Biotin labeling and Hi-C ligation
Timing ∼90 min + incubation for 4 h to overnight
-
18
(Optional) If the reaction buffer used for the RE1 digestion is not compatible with Klenow enzyme, a buffer change to NEBuffer 2 will be required. Centrifuge the tube from Step 17 at 600g for 6 min at 4 °C, remove the supernatant as described in Step 14 and resuspend the cell pellet in 476 μl of NEBuffer 2 (1×). NEBuffers 1–4 are compatible with Klenow enzyme, but some special buffers such as DpnII buffer (New England Biolabs) are not.
-
19
Add the following components to the sample and mix them by gentle pipetting:
Table 5 -
20
Incubate the sample at 37 °C on a thermomixer for 45 min with intermittent gentle shaking (700 r.p.m. for 10 s every 30 s).
-
21
Centrifuge the tube at 600g for 6 min at 4 °C.
-
22
Remove the supernatant, leaving ∼50 μl including the pellet.
-
23
Add the following components to the sample and mix them by gentle pipetting:
Table 6 -
24
Incubate the sample at 16 °C for 4 h or more (overnight is fine). Note that successfully labeled DNA fragment ends (at Step 20) do not reconstitute cleavable BglII restriction sites when ligated during this step.
Single-cell isolation and cross-link reversal
Timing ∼1 h + incubation for 12–16 h
-
25
Centrifuge the tube at 600g for 6 min at 4 °C.
-
26
Remove the supernatant as described in Step 14 and resuspend the pellet with 1 ml of PBS (1×). Figure 3 shows a typical cell suspension at this stage.
Figure 3: Example image of the cell suspension after Hi-C ligation. 
Microscopic inspection enables confirmation that cells are singletons and have normal morphology. Arrowheads, single cell; double arrowhead, doublet; arrow, debris.
Pause point
The cell suspension can be stored at 4 °C for several days.
-
27
Pass the cell suspension through a 40-μm cell strainer and determine the cell density using a hemocytometer. Use an aliquot of 2–5 × 106 cells to check the efficiency of biotin incorporation and ligation, as described in Box 1 and Figure 4.
Figure 4: Analysis of Hi-C ligation junctions. 
(a) Position of PCR primers (arrows) on genomic DNA BglII fragments. (b) One possible orientation of orange and green fragments after Hi-C ligation, in which the two primers designed yield a PCR product. The ligation junction at the middle of the amplicon can be digested with ClaI if biotin labeling is successful. If biotin labeling is unsuccessful, the junction is resistant to ClaI digestion, and it can be digested with BglII. (c) Two examples of gel results showing restriction digestion of Hi-C amplicons after high-efficiency biotin labeling (published previously13). The comparison with ligase minus control (−) confirms that the PCR product is Hi-C ligation-dependent, and near complete digestion with ClaI confirms high efficiency of biotin fill-in. NotI digestion serves as a non-digestion control. Primer sequences for amplicon 1: 5′-CCCTTGTCTTTCCTATGTCTCACCTG-3′;5′-GATGAGGGCTGAAGGAGAATTAAAG-3′. Primer sequences for amplicon 2: 5′-TGTTGTTCTGTTTCCTCGAAAGAC-3′; 5′-GTGATTTTTACGCCTGGAACGTA-3′. Figure adapted with permission from ref. 13, Nature Publishing Group.
Critical Step
The cell suspension can be stored at 4 °C for several days. The cell density should be checked before every use. If cell clumps are evident, the suspension should be passed through the cell strainer again and the cell density should be re-assessed.
-
28
Dilute an aliquot of cells from Step 27 in PBS (1×) to give a suspension of 150 cells/μl in ∼100 μl.
-
29
To a new 1.5-ml tube, add 1 ml of PBS (1×) and 8 mg of low-melting agarose, heat it at 70 °C in a water bath until the agarose dissolves and then keep the mixture molten at 37 °C.
-
30
To a new 1.5-ml tube, add 5 μl of cell suspension from Step 28, 20 μl of PBS (1×) and 25 μl of agarose solution from Step 29, and mix well.
-
31
Using a glass Pasteur pipette (preheated to 37 °C), take up the cell suspension in agarose and make microdrops (diameter <0.5 mm; volume <1 μl) on a new ice-cold glass slide and leave the droplets to solidify.
-
32
Place the slide under PBS (1×) in a Petri dish and, under a stereoscopic microscope, identify droplets that contain only single cells.
-
33
Use a fresh Pasteur pipette connected to a mouthpiece to transfer an isolated single-cell droplet to a new 1.5-ml DNA LoBind tube containing 25 μl of PBS (1×).
Caution
Mouth pipetting is actively discouraged in many laboratories. A potential alternative is to use a micromanipulator, although this can be cumbersome and time-consuming.
Critical Step
It is fine to transfer pieces of agarose with a cell. The Pasteur pipette should be changed after handling each cell to avoid contamination.
-
34
Briefly spin the tube containing the isolated single cell and incubate it at 65 °C for 12–16 h to reverse cross-links. In our hands, omission of proteinase K at this step did not affect results.
Critical Step
Make sure that the tube lid is tightly closed.


Purification of Hi-C DNA with magnetic beads
Timing ∼2 h
Critical Step
It is recommended to use low-retention pipette tips for handling beads and single-cell samples after this point.
-
35
Once cross-link reversal is complete, place the tubes from Step 34 on ice.
-
36
Mix Dynabeads M-280–streptavidin well to obtain a homogeneous suspension.
-
37
Transfer 25 μl of beads suspension (per single-cell sample) to a fresh 1.5-ml tube.
Critical Step
For example, take 300 μl of bead suspension if you are handling 12 single-cell samples simultaneously.
-
38
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant. Remove the tube from the magnetic stand, add 1× BW buffer and gently tap the tube to resuspend the beads.
Critical Step
The volume of 1× BW buffer per wash should be equal to or more than the original bead suspension.
-
39
Repeat Step 38 twice more.
-
40
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant. Resuspend the beads in 27 μl (per single-cell sample) of 2× BW.
Critical Step
For example, resuspend the beads with 324 μl of 2× BW buffer if you are handling 12 single-cell samples simultaneously.
-
41
Transfer 25 μl of the bead suspension to each tube containing 25 μl of single-cell sample (from Step 35), and gently tap the tube to mix.
Critical Step
Do not pipette the mixture up and down at this point because DNA is very dilute, and it is prone to loss because of sticking to the pipette tip.
-
42
Incubate the tube at room temperature for 1 h on a rotating wheel at 2–5 r.p.m.
-
43
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
44
Remove the tube from the magnetic stand, add 200 μl of 1× BW buffer to each tube and gently tap the tube to resuspend the beads. Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
45
Repeat Step 44 twice more.
-
46
Remove the tube from the magnetic stand, add 200 μl of EB buffer to each tube and gently tap the tube to resuspend the beads.
Second restriction enzyme (AluI) digestion
Timing ∼1.5 h
-
47
In a new tube, set up AluI reaction mixture as tabulated below:
Table 7 Critical Step
For multiple samples, always prepare an excess (∼10%) to ensure that all samples have the exact amount of mixture indicated in the protocol.
-
48
Collect the suspension from Step 46 at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
49
Remove the tube from the magnet, resuspend the beads with 50 μl of AluI reaction mixture (from Step 47) by pipetting gently and incubate the mixture at 37 °C for 1 h on a rotating wheel at 2–5 r.p.m.
Critical Step
The reaction can take place on a small rotating wheel in a hybridization oven.
-
50
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
51
Remove the tube from magnetic stand, add 200 μl of 1× BW buffer to each tube and gently tap the tube to resuspend the beads. Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
52
Repeat Step 51 twice more.
-
53
Remove the tube from the magnetic stand, add 200 μl of EB buffer to each tube and gently tap the tube to resuspend the beads.
A-tailing
Timing ∼1 h
-
54
In a new tube, set up the A-tailing reaction mixture as tabulated below:
Table 8 Critical Step
For multiple samples, always prepare an excess (∼10%) to ensure that all samples receive the exact amount of mixture indicated in the protocol.
-
55
Collect the suspension from Step 53 at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
56
Remove the tube from the magnet, resuspend the beads with 50 μl of A-tailing reaction mixture (from Step 54) by pipetting gently, and incubate the tube at 37 °C for 30 min on a rotating wheel at 2–5 r.p.m.
Critical Step
The reaction can take place on a small rotating wheel in a hybridization oven.
-
57
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
58
Remove the tube from the magnetic stand, add 200 μl of 1× BW buffer to each tube and gently tap the tube to resuspend the beads. Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
59
Repeat Step 58 twice more.
-
60
Remove the tube from the magnetic stand, add 200 μl of EB buffer to the tube, and gently tap the tube to resuspend the beads.
Adapter ligation
Timing ∼1 h
-
61
In a new tube, set up the adapter ligation mixture as tabulated below and prepare sequencing adapters on ice:
Table 9 Critical Step
For multiple samples, always prepare an excess (∼10%) to ensure that all samples receive the exact amount of mixture, as indicated in the protocol.
Critical Step
When the adapter without the identification tag is used, all reads theoretically start with TCT, because both ends of the library inserts are created by AluI digestion followed by A-tailing. The identification tags shown in the MATERIALS section appear as the first three bases of the read, followed by TCT, in the sequencing results. This means that the sequencing signals may be less variable when sequencing the first 3–6 bases, depending on the choice of identification tags, which affects cluster recognition by HiSeq sequencer (when using a Genome Analyzer IIx, one can avoid this problem by a specific technique22). When using HiSeq, make sure that each digit of the tag has substantial variation among the libraries to be loaded in the same lane. The adapters with identification tags should only be used when multiple libraries are to be mixed and sequenced in the same lane.
-
62
Collect the suspension from Step 60 at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
63
Remove the tube from the magnet, add 48 μl of ligation mixture and 2 μl of sequencing adapter (with an identification tag of choice), resuspend the beads by pipetting gently and incubate them at room temperature for 30 min on a rotating wheel at 2–5 r.p.m.
Pause point
The reaction can be extended overnight if necessary.
-
64
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
65
Remove the tube from the magnetic stand, add 200 μl of 1× BW buffer to each tube and gently tap the tube to resuspend the beads. Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
66
Repeat Step 65 twice more.
-
67
Remove the tube from the magnetic stand, add 200 μl of EB buffer to each tube and gently tap the tube to resuspend the beads.
Library amplification
Timing ∼1.5 h
-
68
In a new tube, set up the amplification mixture as tabulated below:
Table 10 Critical Step
For multiple samples, always prepare the mixture in excess (∼10%) to ensure that all samples receive the exact amount of mixture, as indicated in the protocol.
-
69
Collect the suspension from Step 67 at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 1 min, and then carefully remove the supernatant.
-
70
Remove the tube from the magnet, add 50 μl of amplification mixture (from Step 68), resuspend the beads by pipetting gently and transfer them to a new 0.2-ml PCR tube.
-
71
Run the amplification reaction in a thermal cycler, using the program below:
Table 11 Pause point
The samples can be kept at −20 °C for several days.
Library purification
Timing ∼1 h
-
72
Briefly centrifuge the amplified library (from Step 71) in a 0.2-ml PCR tube, put on the magnet for 96-well plates for 1–2 min and then carefully transfer 45 μl of the supernatant to a new 1.5-ml tube.
Critical Step
To avoid contamination of the supernatant with beads, take care not to disturb or touch the bead pellet with the pipette tip.
-
73
Add 81 μl of AMPure XP to 45 μl of library, mix it well and leave the tube for 5 min at room temperature.
Critical Step
AMPure XP is stored at 4 °C, but make sure that the AMPure XP added is at room temperature. For convenience, make an aliquot (81 μl per sample) to equilibrate to room temperature in advance.
-
74
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 2–3 min, and then carefully remove the supernatant.
-
75
While keeping the tube on the magnet, add 200 μl of 70% (vol/vol) ethanol. Leave it for 30 s and then carefully remove the supernatant.
Critical Step
The 70% (vol/vol) ethanol should be prepared fresh, and it should be used at room temperature.
-
76
Repeat Step 75 twice more.
-
77
Remove the tube from the magnetic stand and collect the remaining liquid at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand, and remove the remaining liquid at the bottom, taking care not to disturb or touch the bead pellet.
-
78
Open the lid of the tube on the magnet and leave it for 5–15 min until the bead pellet dries.
Critical Step
The tube can be covered with a clean paper towel. A sign of dry pellets is a loss of glossy surface and visible cracks.
-
79
Once the pellet is dry, immediately add 17 μl of 10 mM Tris-HCl (pH 8.5) to the pellet, remove the tube from the magnet, resuspend the pellet well by gentle pipetting and leave it for 5 min.
-
80
Collect the suspension at the bottom of the tube by gentle and brief centrifugation. Put the tube on a magnetic separation stand for 2–3 min, and then carefully transfer 15 μl of the supernatant to a new 1.5-ml tube.
Critical Step
To avoid contamination of the supernatant with beads, take care not to disturb or touch the bead pellet with the pipette tip.
Size selection of the library
Timing ∼5 h
-
81
Mix the sample from Step 80 with 3 μl of orange G loading dye and run it on a 2% (wt/vol) agarose gel until the orange G dye has migrated ∼3 cm.
Caution
As ethidium bromide is toxic, it should be handled with care and discarded according to relevant institutional and local regulations.
Critical Step
Include ethidium bromide in the gel and running buffer at a concentration of 0.5 μg/ml.
-
82
Cut out and collect the gel slice corresponding to 300–700 bp.
Critical Step
The typical result of the run is a fairly faint smear between 300 and 2,000 bp and a dense band near 130 bp, but the intensity of the smear varies. An intense smear is not necessarily indicative of a good library. Care should be taken not to collect the band near 130 bp, because it corresponds to the primer dimer that outcompetes sequencing of informative molecules in the library. Figure 5 shows an example gel image with smears of various intensities.
Figure 5: Example gel image of single-cell Hi-C library smears. 
Arrows on the left indicate the positions for 2,027, 1,353, 603 and 271 bp from the top, respectively. In our experience, the faint smears like those in the third and eighth lanes from the left are likely to give better results. The bands at the ends of smears are primer dimers, which should be removed as thoroughly as possible at Steps 82 and 83.
-
83
Retrieve DNA from the gel fragment using the MinElute gel extraction kit according to the manufacturer's recommendations with the following modifications:
Table 12
Manufacturer's original instructions
Modification
Reason
Solubilize a gel slice in buffer QG at 50 °C for 10 min with vortexing every 2–3 min
Solubilize a gel slice in buffer QG at room temperature for 30 min on a rotating wheel
To minimize DNA damage
To bind DNA to MinElute column, apply the sample and centrifuge only once
To bind DNA to MinElute column, apply the sample flow-thorough from the first spin once more
To maximize DNA recovery
Wash MinElute column with buffer PE once
Wash MinElute column with buffer PE three times
To make sample DNA purer
To elute, wait for 1 min after adding buffer EB to MinElute column
To elute, wait for 10 min after adding buffer EB to MinElute column
To maximize DNA recovery
Elute DNA from MinElute column once
Elute DNA from MinElute column twice (10 μl each; ∼19 μl in total)
To maximize DNA recovery

Bioanalyzer analysis and quantitative PCR
Timing ∼3 h
-
84
Assess the quality and size distribution of the library with an Agilent 2100 Bioanalyzer system. As the library was selected for 300–700 bp at Step 82, the size distribution should be close to this size range (Fig. 6).
Figure 6: Example electropherograms of single-cell Hi-C libraries generated by the Agilent 2100 Bioanalyzer system. 
(a) A typical example with obvious primer-dimer contamination at ∼130 bp. (b) The electropherogram of the library shown in a after re-purification, which is now suitable for sequencing. Please note that peaks at 35 and 10,380 bp are size markers and are not included in the sequencing sample.
Critical Step
If there is another peak ∼130 bp (Fig. 6a), it means that the library is contaminated with the primer dimer. Even if the primer dimers are minor in the library (e.g., 10%), they are overrepresented in the sequence results because they are quite efficient in cluster formation. This reduces the sequencing depth of the real library of interest. If the library includes a significant amount of large DNA (e.g., >1.5 kb), they may form large clusters overlapping with neighboring ones in the flowcell. This may make the sequence signals from these clusters of low quality or unreliable. To remove these out-of-range molecules from the library, repeat the size selection (Steps 81–83) once more (Fig. 6b).
-
85
If the size distribution of the library is acceptable for sequencing, quantify the library using KAPA Illumina library quantification kit (which comes with all necessary reagents) and real-time PCR system.

Cluster formation and sequencing
Timing up to 1 week
-
86
Sequence the library, 40- to 50-bp paired-end reads, according to the manufacturer's recommendations. This will generate two FASTQ files: one file contains the read 1 sequences and the second file contains the read 2 sequences.
Critical Step
When sequencing with the Genome Analyzer IIx, reasonable sequencing depth is achieved by loading ∼12 single-cell libraries with distinct indexes onto the same lane.
Bioinformatics processing: customizing the single-cell Hi-C pipeline
Timing 2 d
-
87
Independently map the two FASTQ files to an appropriate reference genome. In our previous study13, we used the Maq aligner program with standard parameters, retaining pairs in which both reads mapped uniquely and with high mapping quality (MapQ ≥ 30).
-
88
Download, set up, test and run scell_hicpipe via the command line on a Linux operating system, as described in Box 2. The description in Box 2 explains how to process a premade test data set to verify that scell_hicpipe runs correctly on your system. However, it is more common that the user will want to process other data sets, which should be done as described in Steps 89–93.
-
89
Prepare input data file type 1, an ASCII tab-delimited file of mapped single-cell Hi-C paired-end reads, with columns ordered as follows: Read1 chromosome name; Read1 co-ordinate; Read1 strand; Read2 chromosome name; Read2 co-ordinate; and Read2 strand.
Copy this file into the 'input' folder.
-
90
Prepare input file type 2, an ASCII tab-delimited file listing chromosome names and lengths, with columns ordered as follows: Chromosome name; and Chromosome length (in bp).
Copy this file into the 'input' folder.
-
91
Prepare an ASCII tab-delimited file of all the fragment ends. Each file should have columns ordered as follows: Index number (integer value starting and 1 and incrementing by 1); Restriction fragment number; Strand;
Chromosome name; Co-ordinate; Fragment length; and Fend length.
In addition, create a file listing only the predetermined valid fragment ends, which should be in the same format as above. Finally, create a file listing the nonvalid fends, which should simply be a one-column list of the index numbers of those fragments ends not considered valid.
Copy these three files into a folder named 'fends', and copy this folder into the 'input' folder.
-
92
Create a configuration file for each data set, as described in Box 3.
-
93
To process the reads and to perform the quality control, type the following on the command line:make CFG=input/[name of configuration file]
The resulting files will be written to a folder named 'output'. To produce the contact maps, enter the following on the command line:plot_cmap CFG=input/[name of configuration file]
The figures will be written to the folder: output/figures/cmaps/.


Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
Steps 1–8, cell fixation: 30 min to 1 h
Steps 9–17, cell permeabilization and the first restriction digestion: 3 h + incubation for 12–16 h
Steps 18–24, biotin labeling and Hi-C ligation: 1 h to 90 min + incubation for 4 h to overnight
Steps 25–34, single-cell isolation and cross-link reversal: ∼1 h, depending on the speed of single-cell isolation and the number of single cells to pick up + incubation for 12–16 h
Steps 35–46, sample binding to the magnetic beads: ∼2 h, depending on the number of single cells to work on simultaneously
Steps 47–53, second restriction enzyme digestion: ∼1.5 h, depending on the number of single cells to work on simultaneously
Steps 54–60, A-tailing: ∼1 h, depending on the number of single cells to work on simultaneously
Steps 61–67, adapter ligation: ∼1 h, depending on the number of single cells to work on simultaneously
Steps 68–71, library amplification: ∼1.5 h, depending on the number of single cells to work on simultaneously
Steps 72–80, library purification: ∼1 h, depending on the number of single cells to work on simultaneously
Steps 81–83, size selection of the library: ∼5 h, depending on the number of single cells to work on simultaneously
Step 84, Agilent 2100 Bioanalyzer analysis: ∼1 h, depending on the number of single cells to work on simultaneously
Step 85, quantitative PCR of the library: ∼2 h, depending on the number of single cells to work on simultaneously
Step 86, cluster formation and sequencing: several days up to 1 week
Step 87–93, bioinformatics analysis—pre-processing data and using the single-cell Hi-C pipeline: 2 d
Box 1, testing biotin incorporation and Hi-C ligation: 2–3 d
Box 2, download, set up, test and run the single-cell Hi-C pipeline: 1 d
Box 3, creating a configuration file for each data set: 1 h
Anticipated results
Sequenced single-cell Hi-C libraries will need to be demultiplexed. In this protocol, the indices constitute the first four base pairs of the forward read in combination with the first four base pairs of the reverse read. This information is obtained by processing the FASTQ files generated after paired-end sequencing. The barcode sequences should be removed before mapping, as these will most likely not align to the reference genome and their presence would substantially reduce mapping efficiency. The reads are then mapped to the genome to determine the corresponding BglII sites. This enables removal of unmappable reads, noninformative read-pairs (e.g., read-pairs derived from self-ligation of a single BglII fragment and so on) and abnormal read-pairs (e.g., where each read is not at the first AluI site from the corresponding BglII site), to end up with normal ligation read-pairs (see Nagano et al.13 for details). The actual read-pair counts and classification for several successful single-cell Hi-C libraries are shown in Figure 7.

Cells 1, 3, 5, 8 and 9 represent combined data sets formed by adding the original and re-sequencing data of the same libraries. Data sets including re-sequencing data have more read-pair numbers, but they do not necessarily have more unique fend-pairs in Table 3, showing the saturation of sequencing depth. See the original publication13 for the definitions of read-pair classifications. Adapted with permission from ref. 13, Nature Publishing Group.
Because the single-cell library is amplified through PCR before sequencing, a single ligation event between two different BglII sites in the single cell from which the library originates is usually supported by multiple, identical, read-pairs. The processing of read-pairs in a Hi-C data set thus comes down to the pairing of two BglII fragment ends (fends), which is denoted as a 'fend-pair'. In theory, a particular BglII fend on an autosome should not be found in more than two different fend-pairs in a single-cell data set from a diploid cell in the G1 phase of the cell cycle. In male cells, fends from the X chromosome should not be present in more than one fend-pair. The absence of overrepresented fends does not guarantee a single-cell source of the library, and the presence of a small number of over-represented fends does not necessarily mean that the source cell number is greater than one. A data set with a minimal number of over-represented fends probably derives from a single cell. We found that fend-pairs with only one read are probably noise rather than the results of authentic chromatin proximity in the source cell, and we recommend their removal from the data set. We found that data sets with few single fend-pairs appeared to be of better quality compared with data sets that had many singly occurring fend-pairs. Taking these observations into account, we set three minimum threshold criteria to measure the quality of the single-cell Hi-C data set: (A) More than 10,000 fend-pairs; (B) less than 30% of total read-pairs appear only once in the data set; and (C) the number of autosomal fends that are found in more than two fend-pairs is less than the expected number according to the binomial model.
The scell_hicpipe pipeline gives these data in a summary file found in the 'qc' folder, which in turn is located in the 'output' folder. In the summary file, the column 'Unique amplified fends pairs' lists the number of unique contacts (condition A above). The column 'Perc. of single read fend pairs' reports the percentage of read-pairs that were sequenced only once (condition B above). The results from the binomial test (condition C above) are written to columns labeled as 'Num 2 chrs fends with 3 obs hits', 'Predicted (binomial) num 2 chrs fends with 3 hits' and 'Pval (binomial) of num 2 chrs fends with 3 hits'.
The actual fend-pair counts and quality control values for several single-cell Hi-C libraries published previously13 are shown in Table 3.
Another way to gauge the quality of a single-cell Hi-C data set is to create a genome-wide heatmap. A heatmap from a successful data set shows (i) an intense accumulation of cis-chromosomal interactions along the diagonal in the map, which decays with distance; and (ii) clear clusters of trans-chromosomal interactions, which indicate contacts between one chromosome and a limited number of other chromosomes. Typical examples of successful and unsuccessful single-cell Hi-C heatmaps are shown in Figure 8.

Each colored pixel indicates the number of filtered interactions per 10 × 10 Mb bin, in which yellow means low, orange means low medium, red means high medium and blue means highest density. (a) An example of a high-quality single-cell Hi-C data set13, in which intense cis-chromosomal interactions and sparse but focal clustering of trans-chromosomal interactions are observed. Reproduced with permission from ref. 13, Nature Publishing Group. (b) Heatmap with suspected multiple cell source, with ubiquitous trans-chromosomal interactions (as if each chromosome interacts with all other chromosomes) in addition to focal clustering. (c) Example of a poor quality data set, in which enriched cis-chromosomal interactions and focal clustering of trans-chromosomal interactions are not observed.
References
Fraser, P. & Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 447, 413–417 (2007).
Gibcus, J.H. & Dekker, J. The hierarchy of the 3D genome. Mol. Cell 49, 773–782 (2013).
De Wit, E. & de Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 26, 11–24 (2012).
Ethier, S.D., Miura, H. & Dostie, J. Discovering genome regulation with 3C and 3C-related technologies. Biochim. Biophys. Acta 1819, 401–410 (2012).
Tanay, A. & Cavalli, G. Chromosomal domains: epigenetic contexts and functional implications of genomic compartmentalization. Curr. Opin. Genet. Dev. 23, 197–203 (2013).
Sexton, T. et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012).
Dixon, J.R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Nora, E.P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012).
Dekker, J., Marti-Renom, M.A. & Mirny, L.A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403 (2013).
Furlan-Magaril, M., Várnai, C., Nagano, T. & Fraser, P. 3D genome architecture from populations to single cells. Curr. Opin. Genet. Dev. 31, 36–41 (2015).
Rapkin, L.M., Anchel, D.R.P., Li, R. & Bazett-Jones, D.P. A view of the chromatin landscape. Micron 43, 150–158 (2012).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013).
Nagano, T. et al. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 16, 175 (2015).
Rao, S.S.P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Sexton, T. et al. Sensitive detection of chromatin coassociations using enhanced chromosome conformation capture on chip. Nat. Protoc. 7, 1335–1350 (2012).
Dryden, N.H. et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by capture Hi-C. Genome Res. 24, 1854–1868 (2014).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295, 1306–1311 (2002).
Tolhuis, B., Palstra, R.J., Splinter, E., Grosveld, F. & de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell 10, 1453–1465 (2002).
Simonis, M., Kooren, J. & de Laat, W. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 4, 895–901 (2007).
Kilkenny, C., Browne, W.J., Cuthill, I.C., Emerson, M. & Altman, D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412 (2010).
Krueger, F., Andrews, S.R. & Osborne, C.S. Large scale loss of data in low-diversity illumina sequencing libraries can be recovered by deferred cluster calling. PLoS ONE 6, e16607 (2011).
Acknowledgements
The authors thank I. Clay, N. Cope and K. Tabbada for help and support. This work was supported by the Biotechnology and Biological Sciences Research Council, UK.
Author information
Authors and Affiliations
Contributions
T.N. and P.F. developed and optimized the protocol. Y.L., E.Y. and A.T. contributed to protocol optimization by developing the processing pipeline for single-cell Hi-C and analyzing the sequence data. S.W.W. contributed to optimize the pipeline. W.D. contributed the single-cell isolation procedure. T.N., S.W.W. and P.F. wrote the manuscript, with inputs from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Nagano, T., Lubling, Y., Yaffe, E. et al. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat Protoc 10, 1986–2003 (2015). https://doi.org/10.1038/nprot.2015.127
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2015.127
This article is cited by
-
3D genomics and its applications in precision medicine
Cellular & Molecular Biology Letters (2023)
-
Whole-genome doubling drives oncogenic loss of chromatin segregation
Nature (2023)
-
3D genome organization in the epithelial-mesenchymal transition spectrum
Genome Biology (2022)
-
Lymphocyte activation gene-3-associated protein networks are associated with HDL-cholesterol and mortality in the Trans-omics for Precision Medicine program
Communications Biology (2022)
-
Seeing the forest through the trees: prioritising potentially functional interactions from Hi-C
Epigenetics & Chromatin (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
