
Abstract
Established memories undergo a period of vulnerability following retrieval, a process termed ‘reconsolidation.’ Recent work has shown that the hypothetical process of reconsolidation is only triggered when new information is presented during retrieval, suggesting that this process may allow existing memories to be modified. Reconsolidation has received increasing attention as a possible therapeutic target for treating disorders that stem from traumatic memories, yet little is known about how this process changes the original memory. In particular, it is unknown whether reconsolidation can reorganize the neural circuit supporting an existing memory after that memory is modified with new information. Here, we show that trace fear memory undergoes a protein synthesis-dependent reconsolidation process following exposure to a single updating trial of delay conditioning. Further, this reconsolidation-dependent updating process appears to reorganize the neural circuit supporting the trace-trained memory, so that it better reflects the circuit supporting delay fear. Specifically, after a trace-to-delay update session, the amygdala is now required for extinction of the updated memory but the retrosplenial cortex is no longer required for retrieval. These results suggest that updating procedures could be used to force a complex, poorly defined memory circuit to rely on a better-defined neural circuit that may be more amenable to behavioral or pharmacological manipulation. This is the first evidence that exposure to new information can fundamentally reorganize the neural circuit supporting an existing memory.
Similar content being viewed by others


INTRODUCTION
Memory is not permanently stored in a fixed, unalterable state, but instead can be updated when new information is learned. Stable memory can be rendered labile with a retrieval trial (Jarome et al, 2011; Misanin et al, 1968; Nader et al, 2000; Parsons et al, 2006a; Sara, 2000), a process termed ‘reconsolidation.’ During the reconsolidation process, memories are believed to undergo a transient period of vulnerability, marked by an initial period of destabilization and protein degradation (Jarome et al, 2011; Lee, 2008; Lee et al, 2008; Nader and Hardt, 2009) followed by a period of restabilization and protein synthesis (Jarome et al, 2012; Nader et al, 2000; Parsons et al, 2006a). Although the purpose of this hypothetical reconsolidation process is unclear, recent work has suggested that reconsolidation may allow existing memory to update when new, relevant information is presented. Indeed, reconsolidation is only triggered when new information is presented at retrieval; when a retrieval trial is identical to what was used in training, the memory trace is not rendered labile (Diaz-Mataix et al, 2013; Jarome et al, 2015; Morris et al, 2006). Presumably, new information triggers destabilization of the memory trace to allow for modification (Goltseker et al, 2016; Haubrich et al, 2015; Jarome et al, 2015; Lee, 2008; Lee et al, 2008; Sevenster et al, 2012); when retrieval includes only familiar information, the memory remains stable and resistant to amnesic agents (Diaz-Mataix et al, 2013; Jarome et al, 2015; Rossato et al, 2007; Sevenster et al, 2012; Winters et al, 2011).
Understanding how memory is altered by the presentation of new information is key to potentially using reconsolidation-based strategies to treat and reduce traumatic fear-based associations. The process of memory reconsolidation has received increasing attention as a possible therapeutic target for treating anxiety disorders that stem from traumatic memories (Alberini and Ledoux, 2013) as it may result in more persistent changes to the original memory. Recent work has shown that combining retrieval and extinction procedures can result in robust and persistent attenuation of fear memory (Clem and Huganir, 2010; Graff et al, 2014; Monfils et al, 2009; Rao-Ruiz et al, 2011; Schiller et al, 2010). Further, manipulating the affective component of a fear memory by presenting an appetitive outcome during retrieval can also produce lasting decreases in fear responding to a conditioned stimulus (Haubrich et al, 2015). Thus, reconsolidation-dependent updating holds significant promise as an effective treatment for reducing traumatic memory and associated anxiety disorders.
Although accumulating evidence suggests that reconsolidation can modify memory content, it is unclear how this process changes the original memory at the circuit level. In particular, it is not yet known whether reconsolidation-dependent updating can change the neural circuit supporting an existing memory. Here, we tested whether we could force the reorganization of a memory circuit by updating complex trace fear memory with one trial of delay fear training. Although both trace and delay memories require the association of an initially neutral conditional stimulus (CS) with a naturally aversive unconditional stimulus (UCS), trace conditioning is more complex, with an empty period of time interposed between the two cues. We have recently shown that trace and delay associations rely on distinct neural circuits despite sharing similar memory content (Gilmartin and Helmstetter, 2010; Gilmartin et al, 2013; Kwapis et al, 2015a; Kwapis et al, 2014a; Kwapis et al, 2015b). For example, extinction of trace fear requires the participation of the retrosplenial and prelimbic cortices instead of the amygdala, which is required for extinction of delay fear (Kwapis et al, 2015a; Kwapis et al, 2014a). Further, the retrosplenial cortex (RSC) plays a key role in retrieving trace memory but is not required to retrieve memory for delay fear (Kwapis et al, 2014a; Kwapis et al, 2015b). These distinctions can be leveraged to test whether it is possible to simplify the neural circuit supporting trace fear by exposing trace-trained animals to a single trial of delay fear. If updating can reorganize the trace memory so that it relies on the delay circuit, after the memory restabilizes, the amygdala should be required for extinction (Kwapis et al, 2014a) and the RSC should no longer be involved in memory retrieval (Kwapis et al, 2014a; Kwapis et al, 2015b). Thus, we can test whether reconsolidation-dependent updating reorganizes the neural circuit supporting a memory in addition to revising its content.
MATERIALS AND METHODS
Subjects
Male Long-Evans rats were obtained from Harlan (Madison, WI) and individually housed in shoebox cages with free access to water and rat chow. The colony room was maintained on a 14 : 10-h light/dark cycle and all behavioral tests were conducted during the light portion of this cycle. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Milwaukee and were in accordance with the National Institutes of Health Guidelines.
Surgery
Animals were adapted to handling for 3 days before surgery. Cannulae were chronically implanted into the amygdala or RSC as previously described (Kwapis et al, 2014a; Kwapis et al, 2015b; Kwapis et al, 2009). Rats were anesthetized with isoflurane (induction, 4%; and maintenance, 2%) and placed in a stereotaxic frame. Bilateral stainless steel 26-gauge cannulae aimed at the basolateral amygdala (AP −2.9 mm, ML ±5.0 mm, and DV −7.2 mm) or RSC (AP −3.5 mm; ML ±0.5 mm; and DV −1.8 mm, using dual cannulae with a 1.0 mm C–C distance). Rats were allowed to recover for at least 7 days before behavioral testing began.
Infusion Procedures and Drugs
All rats received bilateral infusions of 0.5 μl/side into the amygdala or RSC over a 60 s period. After each infusion was complete, the injectors (33-gauge, extending 0.8 mm (RSC), or 0.5 mm (amygdala) beyond the guide) were kept in place for an additional 90 s to ensure proper diffusion. The protein synthesis inhibitor anisomycin (ANI; Tocris) was fully dissolved in HCl and diluted to a final concentration of 125 μg/μl with ACSF. The NMDA receptor antagonist D-APV (Tocris) was diluted with ACSF to a final concentration of 10 μg/μl.
Behavioral Procedures
Fear conditioning
Before training, rats were exposed to 3 days of the restraint procedure. During this time, rats were transported to the laboratory, wrapped in a towel and gently restrained for several minutes while the infusion pump was activated and the dummy cannulae were loosened.
Fear conditioning was conducted in Context A, a set of 4 identical chambers housed within sound attenuating boxes in which the floor was composed of stainless steel rods through which footshocks could be delivered. Context A was cleaned with a solution of 5% ammonium hydroxide between animals to provide these chambers with a distinct scent. On day 1, animals were trained with either delay or trace fear conditioning. For delay conditioning, a 6-minute baseline period was followed by 4 pairings of the white noise CS (10 s, 72 dB) and footshock UCS (1 s, 1 mA), with the UCS presentation occurring at the moment of CS offset. Delay trials were separated by a variable ITI of 110±20 s and animals were removed 4 min after the final UCS presentation. For trace conditioning, after the 6-min baseline period, the CS and UCS were paired 6 times with a variable ITI of 240 ±20 s between trials. Importantly, the CS and UCS were separated by an empty 20 s trace interval period for all trace conditioning trials. As with delay, trace fear conditioning ended with a stimulus-free 4-min post-shock period.
CS tests were conducted in Context B, a novel set of chambers located in a different room with a number of distinct features that allowed us to test CS fear independently of fear to the training context. The distinct features of Context B included a solid and opaque textured floor panel, infrared lighting, and a novel scent, 5% acetic acid. During the CS test, animals were given a 1-min baseline period followed by 3 discrete CS presentations (30 s; 72 dB) with a 60 s ITI.
Pre-exposure
In some experiments, animals were given 2 consecutive days of pre-exposure to the training context to attenuate the context-shock association. During both pre-exposure sessions, animals were exposed to the training context for 5-min. No stimuli were presented during pre-exposure.
Retrieval and updating
On day 2, animals were given either a CS-only retrieval session or an update session, consisting of a single trial of delay or trace conditioning. CS-only retrieval occurred in Context B. Following a 1-min baseline period, the CS was presented for 30 s. After an additional 1-min post-CS period, animals were removed from the chambers and infused as described above.
Updating sessions occurred in the training context (Context A) and consisted of a single trial of delay or trace conditioning. For updating sessions, it was imperative that the context and stimulus timing were identical to that of training, as our hypothesis was that only new information would trigger updating of the memory. Following a 6-min baseline period, animals were given one presentation of the CS followed by the UCS so that the stimuli co-terminated (delay) or so that a 20 s trace interval period was presented between the two stimuli (trace). Animals were removed from the chambers 60 s after the UCS presentation and were either infused immediately this session or returned to their home cages.
Extinction
Some animals were extinguished 24 h after the updating session. For these animals, either ACSF or APV was infused into the amygdala 5-min before the extinction session. For extinction, animals were placed into Context B and, after a 1-min baseline, were presented with 40 unreinforced CS presentations separated by 60 s. The entire extinction session was 60-min long and has previously been demonstrated to produce extinction in delay- or trace-trained animals (Kwapis et al, 2015a; Kwapis et al, 2014a).
Histology
After behavioral testing was complete, animals were killed with an overdose of isoflurane and transcardially perfused as previously described (Kwapis et al, 2014a). After cryoprotecting, freezing, and slicing the tissue into 40 μm slices, we mounted the tissue onto slides and stained with cresyl violet. Only rats with acceptable cannula placement in the amygdala or RSC were included in the analyses (Supplementary Figure S1).
Analyses
Sample sizes were estimated based on past experience in our lab and based on similar studies from other labs. Typically, 6–10 animals per group is sufficient to produce statistically reliable results. In all experiments, rats were randomly assigned to groups and all behavior, drug injections, and scoring was done in a blinded fashion.
Freezing behavior was used as the measure of conditional fear during all sessions. The average percent time freezing was calculated in real-time with the FreezeScan 1.0 software (Clever Sys, Inc., Reston, VA). The computer scoring parameters were chosen to closely match handscoring methods used previously in our lab to measure freezing behavior. For update and retrieval sessions, we calculated the average percent time spent freezing during each period (baseline, CS, trace interval, or post-stimulus) of the session. For the CS-only retrieval experiment, a few of the animals (n=5) showed 0% freezing to the 30 s CS presentation, even before the drug was administered. To ensure that this did not bias our data, we removed all animals that failed to freeze during the CS presentation and confirmed that our drug groups showed equivalent average freezing to the retrieval CS before drugs were infused.
For CS test sessions, we calculated the percent time spent freezing during an average of all 8 CS presentations (Experiment 4) or during the first 3 CS presentations (Experiments 1–3). We chose to average the first 3 CS presentations in experiments where extinction began to occur after the first 3 CS presentations, as we have previously done (Kwapis et al, 2011). For the RSC experiment (Experiment 4), we averaged all 8 CS presentations, as freezing did not begin to decrease until the very end of the test session. For the extinction session, the data were handscored by a trained observer to correct for resting behavior. It was common for rats to lie down during the long extinction training session, which would automatically be scored by the computer as freezing behavior. An observer blind to the experimental conditions scored the extinction session by hand to correct for this resting behavior as previously described (Kwapis et al, 2014a). The mean percent time spent freezing during the first 8 CS presentations of the extinction session was analyzed as an index of memory retrieval in the presence of APV.
All statistical comparisons were performed with two-sided tests and normality was tested with the Kolmogorov–Smirnov test. Rats were removed from analyses if their behavior deviated more than 2 s.d.’s from the group mean or if their cannulae placements failed to hit the target region. To identify drug effects within each type of training, we ran mixed-model ANOVAs with a repeated factor of Period (eg, BL vs CS) and a between-subjects factor of drug (ACSF vs ANI) followed by Sidak’s post hoc tests to compare ACSF and ANI within each period. In all analyses an α value of 0.05 was required for significance. All data are expressed as mean±SEM.
RESULTS
Experiment 1: Exposure to New Information Triggers Reconsolidation in the Amygdala
We first ensured that trace fear reconsolidates in the amygdala following standard, CS-only retrieval. The amygdala is involved in the formation of memory for trace fear conditioning (Gilmartin et al, 2012; Guimarais et al, 2011; Kochli et al, 2015; Kwapis et al, 2011), but is not required for trace fear extinction (Kwapis et al, 2014a). It is currently unknown whether the amygdala is involved in the reconsolidation of trace fear conditioning. To test this, we first trained rats with delay or trace fear conditioning (Supplementary Figure S2A) followed by a typical retrieval protocol in which the auditory CS was presented a single time in a novel context the following day (Figure 1a and b). Immediately after CS retrieval, we infused the protein synthesis inhibitor ANI into the amygdala of delay- or trace-trained rats (Figure 1b) to block reconsolidation (delay: ACSF, n=16; ANI, n=10; trace ACSF, n=13; ANI, n=11). Testing the following day revealed that ANI disrupted reconsolidation for both delay and trace memory (Figure 1c). Mixed-model ANOVAs on each type of conditioning revealed main effects for both drug (delay: F(1,24)=6.96, p<0.05; trace: F(1,22)=10.23, p<0.005) and period (delay: F(1,24)=97.11, p<0.0001; trace: F(1,22)=71.63, p<0.0001), with no significant interaction for either type of training (delay: F(1,24)=2.40, p>0.05; trace: F(1,22)=3.01, p>0.05). Follow-up post hoc tests determined that ANI-treated rats showed significantly less freezing than vehicle rats during the CS period for both delay (p<0.01) and trace (p<0.01) conditioning. Thus, trace fear, like delay, undergoes protein synthesis-dependent reconsolidation in the amygdala following retrieval. The amygdala is therefore involved in the consolidation (Kwapis et al, 2011) and reconsolidation (Figure 1) of trace fear, but is not required for the successful extinction of trace fear (Kwapis et al, 2014a).

Both trace and delay fear conditioning undergo reconsolidation in the amygdala following a standard, conditional stimulus (CS)-only retrieval trial in a novel context. (a) Experimental timeline. (b) Mean time spent freezing during each period (Baseline, BL; CS presentation, CS; post-CS, post) of the retrieval session. (c) Freezing during the CS test. Intra-amygdala ANI disrupted reconsolidation of both delay and trace fear conditioning. Data are presented as mean±SEM **p<0.01; n=10–16/group. ACSF, artificial cerebrospinal fluid (vehicle); AMY, amygdala; ANI, aniosmycin (protein synthesis inhibitor); BL, baseline; CS, conditional stimulus.
Experiment 2: The Reconsolidation Process is Only Initiated by New Information
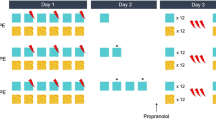
We next tested whether a single trial of delay fear would be sufficient to trigger protein synthesis-dependent reconsolidation in animals initially trained with trace fear conditioning (Figure 2a). As reconsolidation should only be initiated when new information is presented during the retrieval/update session (Diaz-Mataix et al, 2013; Jarome et al, 2015; Sevenster et al, 2012), we hypothesized that presenting a trace-trained animal with another presentation of trace conditioning during retrieval should not trigger reconsolidation. In contrast, presenting a trace-trained rat with a single trial of delay conditioning during retrieval should trigger protein synthesis-dependent reconsolidation, as new information about the CS–UCS relationship is available to incorporate into the memory.

Reconsolidation is only triggered when animals are exposed to new information during retrieval. (a) The experimental timeline. (b) Freezing during each period (Baseline, BL; CS presentation, CS; trace interval, TI, post-CS, post) of the update session. No differences in freezing were observed between drug conditions. (c) Freezing during the CS test. ANI only disrupted reconsolidation for animals given the shifted delay update. Data are presented as mean±SEM ***p<0.001; n=7–8/group. ACSF, artificial cerebrospinal fluid (vehicle); AMY, amygdala; ANI, aniosmycin (protein synthesis inhibitor); BL, baseline; CS, conditional stimulus.
To test this, following trace training (Supplementary Figure S2B), animals were presented with a single trial of delay conditioning, in which the CS and UCS co-terminated (shift) or were given another trial of trace conditioning identical to training (no shift; Figure 2a and b). ANI infused into the amygdala after updating selectively disrupted memory for the animals given the shifted delay update; when no new information was presented (trace–trace group) the memory was not affected by protein synthesis blockade (Figure 2c; Delay ACSF, n=7; ANI n=7; trace ACSF n=8; ANI n=7). For the trace-to-delay update group, a mixed-model ANOVA revealed a significant effect for drug (F(1,12)=15.92, p<0.01), period (F(1,12)=98.48, p<0.0001), and a significant drug x period interaction (F(1,12)=16.88, p<0.01). For the no shift, trace–trace update group, we observed a significant effect of period (F(1,13)=41.29, p<0.0001) but no significant main effect of drug (F(1,13)=0.29, p>0.05) or drug x period interaction (F(1,13)=0.50, p>0.05). Follow-up post hoc tests revealed that ANI reduced freezing during the CS period only in animals with the shifted delay update (p<0.0001) and did not affect CS freezing for animals in the no shift (trace–trace) condition (p>0.05). Consistent with previous reports (Diaz-Mataix et al, 2013; Jarome et al, 2015; Sevenster et al, 2012), this suggests that new information triggers reconsolidation-dependent updating of existing memory. When no new information is presented, the memory is not susceptible to protein synthesis inhibition. Importantly, this confirms that exposure to one trial of delay conditioning is sufficient to trigger memory updating in animals trained with trace fear conditioning.
Experiment 3: Updating Procedures Can Make Trace Fear Memory Rely on Neural Structures Unique to Delay Fear Memory
Our initial experiments demonstrate that fear memory undergoes protein synthesis-dependent reconsolidation in the amygdala following a change in CS–UCS timing from trace to delay. Next, we asked whether updating fear memory in this manner could also change the neural circuit supporting that memory. Previously, we have identified numerous key differences between the trace and delay fear circuits (Figure 3), including a requirement for the amygdala in delay, but not trace extinction and involvement of the RSC in trace, but not delay memory retrieval (Kwapis et al, 2015a; Kwapis et al, 2014a; Kwapis et al, 2015b). If the trace memory circuit is reorganized as a result of the delay update, it should resemble the delay fear circuit, so that the amygdala is now required for extinction and the RSC is no longer necessary for memory retrieval.

Key differences between the delay and trace fear circuits. (a) The AMY but not RSC is required for delay extinction. (b) The RSC but not AMY is required for the extinction and retrieval of trace fear memory. AMY, amygdala; RSC, retrosplenial cortex.
We first tested whether the amygdala is required to extinguish the trace-to-delay updated memory. As before, animals were trained with trace fear conditioning (Supplementary Figure S2C) and updated with one trial of delay (shift) or trace (no shift) the following day (Figure 4a and b). The memory was allowed 24 h to restabilize before we further interrogated the circuit. To test whether the amygdala is required for extinction of the updated memory, the following day, we blocked NMDA receptors in the amygdala with APV during extinction (Figure 4c; Delay ACSF, n=8; APV, n=7; trace ACSF, n=8; APV, n=6). Despite relatively weak within-session extinction (Supplementary Figure S3A), all groups did show reduced freezing at the end of extinction compared to the beginning of extinction (Supplementary Figure S3B). Testing 24 h later revealed that extinction was selectively impaired in animals updated to delay; animals in the trace–trace group extinguished normally despite intra-amygdala APV (Figure 4d), as previously observed (Kwapis et al, 2014a). For the group updated to delay, a mixed-model ANOVA on the test data revealed a significant main effect for drug (F(1,13)=7.38, p<0.05), period (F(1,13)=61.15, p<0.001), and a significant drug x period interaction (F(1,13)=7.56, p<0.05). For Trace, we found a significant effect for period (F(1,12)=24.03, p<0.001) but no significant effect of drug (F(1,12)=0.19, p>0.05) or interaction (F(1,12)=0.87, p>0.05). Follow-up post hoc tests revealed that APV infusion during extinction led to increased freezing during the CS period of the test session for animals updated from trace to delay (p<0.01). This manipulation had no effect on freezing levels for animals in the trace–trace (no shift) group (p>0.05).

Updating trace fear memory with a single trial of delay changes the neural circuit so that the amygdala is now required for extinction. (a) The experimental timeline. (b) Freezing during each period (Baseline, BL; CS presentation, CS; trace interval, TI, post-CS, post) of the update session. (c) Freezing during the first 8 CS presentations of the extinction session. (d) Freezing during the CS test. Intra-amygdala APV impaired extinction for animals given the shifted delay update but had no effect on animals given the non-shifted trace update. Data are presented as mean±SEM **p<0.01; n=6–8/group. ACSF, artificial cerebrospinal fluid (vehicle); AMY, amygdala; APV, aminophosphonovaleric acid (NMDAR antagonist); BL, baseline; CS, conditional stimulus.
Thus, the delay update changed the neural circuit required for extinction; whereas the amygdala was not required for extinction in the trace–trace group, extinction engaged the amygdala in the trace–delay update group, even though these animals were initially trained with trace fear conditioning. Updating procedures can therefore force a complex trace fear memory to recruit brain structures that are unique to the better-defined basic delay fear circuit for extinction.
Experiment 4: Updating Procedures Can Make Trace Fear Memory No Longer Require Cortical Structures Unique to Trace Conditioning
Experiment 3 demonstrated that the updating process can change the neural circuit supporting memory by recruiting new structures that are typically not involved in extinguishing the original association. We next tested whether the opposite is also true, if structures that typically support the original memory are no longer required after the update session. To this end, we tested whether the RSC would be required to retrieve the trace-to-delay updated memory. As the RSC is required to retrieve trace, but not delay fear memory (Kwapis et al, 2014a), if the memory trace reorganizes to the delay fear circuit, the RSC should no longer be required for retrieval.
To test this, we trained (Supplementary Figure S2D) and updated animals as before and allowed the memory to restabilize for 24 h (Figure 5a and b). We then infused APV into the RSC before testing (‘Retrieval’ test; Figure 5c; delay ACSF, n=9; APV, n=8; trace ACSF, n=7; APV, n=6). Intra-RSC NMDAR blockade disrupted memory retrieval for trace–trace animals, as we have previously demonstrated (Kwapis et al, 2014a; Kwapis et al, 2015b). Animals updated from trace to delay, however, no longer required the RSC for retrieval, as APV had no effect on memory retrieval in this group. For animals updated from trace to delay, a mixed-model ANOVA on the ‘retrieval’ test revealed a significant effect of period (F(1,15)=46.84, p<0.001) but no main effect of drug (F(1,15)=0.008, p>0.05) and no significant interaction (F(1,15)=0.16, p>0.05). For animals in the trace–trace group, we found a significant main effect of period (F(1,11)=48.22, p<0.0001) and a significant period x drug interaction (F(1,11)=7.03, p<0.05), although there was no significant main effect of drug (F(1,11)=4.35, p=0.06). Follow-up post hoc comparisons revealed that APV disrupted freezing during the CS period only in animals in the trace–trace group (p<0.01). No difference in CS freezing was observed for animals in the shifted trace–delay group (p>0.05). Thus, intra-RSC infusion of APV no longer affected memory retrieval following an update from trace to delay.

Updating trace memory with a single trial of delay changes the neural circuit so that the RSC is no longer necessary for memory retrieval. (a) The experimental timeline. (b) Freezing during each period (Baseline, BL; CS presentation, CS; trace interval, TI, post-CS, post) of the update session. (c) Freezing during the ‘Retrieval’ CS test following APV infusion. NMDAR blockade in the RSC impaired memory retrieval for animals give the trace update (n=6–7/group) but had no effect on memory retrieval in the delay update group (n=8–9/group). (d) Mean time spent freezing during the ‘Recovery’ test. Freezing fully recovered in the trace–trace group after elimination of APV. Data are presented as mean±SEM **p<0.01. Note: Supplementary Figure captions supplied with the figures. APV, aminophosphonovaleric acid (NMDAR antagonist); BL, baseline; CS, conditional stimulus; RSC, retrosplenial cortex.
This demonstrates that cortical structures involved in the trace circuit are no longer required after updating to delay. Notably, the trace–trace animals’ memory recovered when retested 24 h later (Figure 5d; post hoc comparisons for delay and trace p>0.05), indicating that NMDAR blockade in the RSC only transiently impairs trace memory. This demonstrates that updating procedures can change the neural circuit supporting a memory, so that certain structures required for retrieval are no longer necessary.
Taken together, our results demonstrate that reconsolidation-dependent updating can alter the neural circuitry supporting memory, so that a relatively complex trace fear memory relies on structures unique to the delay fear circuit and no longer requires participation of structures unique to the trace fear circuit.
DISCUSSION
In the present study, we tested whether the reconsolidation-dependent updating process could be leveraged to reorganize the neural circuit supporting a memory in addition to revising the content of that memory. We found that trace fear memory undergoes a protein synthesis-dependent updating process following exposure to a single trial of delay conditioning (Figure 2) but consistent with previous reports, this process is not initiated when new information is eliminated from the retrieval trial (Diaz-Mataix et al, 2013; Jarome et al, 2015; Sevenster et al, 2012). Further, this updating process reorganizes the neural circuit supporting the memory, so that after restabilization, complex trace fear memory relies on the better-defined basic delay circuit, characterized by amygdala-dependent extinction (Figure 4) and RSC-independent memory retrieval (Figure 5). These results demonstrate for the first time that reconsolidation-dependent updating can reorganize the neural circuit supporting memory.
Post-traumatic stress disorder and other anxiety disorders associated with traumatic memories are typically treated with extinction-based approaches (Kwapis and Wood, 2014b; Parsons and Ressler, 2013; Rothbaum and Davis, 2003). Unfortunately, extinction does not result in permanent reduction in fear responding, as observed by the phenomena of spontaneous recovery, reinstatement, and renewal (Davis, 2011; Parsons and Ressler, 2013; Pavlov, 1927). Further, a vast majority of the research on fear extinction comes from rodent studies that use delay fear conditioning to model anxiety, with far less work focusing on the circuit-level mechanisms supporting more complex associations that may better relate to explicit memory in humans (Kwapis et al, 2015a; Kwapis et al, 2014a). Thus, understanding how the reconsolidation-dependent updating process functions to modify the content and location of memory is critical for developing effective strategies to attenuate traumatic memories, especially those complex associations that may not rely on a well-defined neural circuit. Here, we demonstrate that a poorly defined, complex trace memory circuit can be fundamentally reorganized so that it relies on a better-defined neural circuit that may be more amenable to behavioral or pharmacological manipulation.
While our results suggest that the fear memory actually reorganizes in response to the update session (changing from the trace circuit to circuitry that better resembles the delay circuit) we cannot conclusively determine that the memory relies exclusively on the basic delay fear circuit and not some hybrid of the trace and delay circuits. Although this is possible, our data suggest that the memory is reorganized to the delay circuit, rather than simply recruiting parts of the delay circuit in addition to the established trace circuit. In particular, because the RSC was no longer required for memory retrieval after the update, our results suggest that the memory is reorganized to the delay circuit and no longer requires participation of some of the distributed cortical structures necessary for trace memory retrieval.
Alternatively, one could argue that the update session is encoded as a new, separate memory that simply outcompetes the original memory, which remains intact but dormant. While we cannot conclusively rule this out, we do not think this is the case for two reasons. First, the original trace fear memory is made labile following standard retrieval (Figure 1) or a delay update (Figure 2), as ANI administered after the update session impaired fear expression in both cases. If the original memory is unaffected by the update session, which is encoded as a new memory, freezing in response to the original memory would be expected to remain intact. This observed impairment is consistent with numerous studies reporting disruption of the original memory when an amnesic agent is applied following a standard CS retrieval trial (Jarome et al, 2012; Lee, 2008, 2010; Nader et al, 2000; Parsons et al, 2006b) or following an updating trial in other paradigms (Morris et al, 2006; Rossato et al, 2007; Winters et al, 2011). Second, as the update session needs to share a high degree of similarity to the original training session to drive protein synthesis-dependent reconsolidation (Besnard, 2012; Lee, 2008, 2010; Rossato et al, 2007; Winters et al, 2011), it seems that the update session interacts with the original memory, rather than encoding a new, separate association. The update session therefore appears to be changing the original association, rather than being encoded as a separate, unrelated memory.
It is also possible that the update session interrupted the consolidation process for the original memory, which may not be complete by 24 h after training. The timecourse of memory consolidation is not entirely clear, with cellular consolidation believed to last on the scale of hours followed by a much longer process termed ‘systems consolidation’ believed to last days to weeks (Kim and Fanselow, 1992; Rudy and Sutherland, 2008; Squire, 1992; Tayler et al, 2013). We do not think that our results are due to an interruption of consolidation, however, for two main reasons. First, cellular consolidation is generally agreed to be complete by 24 h, as ANI and other amnesic agents do not disrupt memory at this timepoint in the absence of retrieval (Duvarci et al, 2005; Misanin et al, 1968; Nader et al, 2000). Although systems consolidation may be ongoing at 24 h, the memory most likely still relies on the acquisition circuit, as the memory has not had sufficient time to be consolidated to cortical circuits, even on the shortest timeframe (Tse et al, 2011). Second, as both groups of animals have an identical training session (all of the animals get TFC), if the update session interrupts the consolidation process, it would be expected to affect both groups in a similar manner. Nonetheless, it would be interesting to determine whether this type of updating-based reorganization works on older, remote fear memories, which are sometimes resistant to reconsolidation (Frankland et al, 2006; Graff et al, 2014; Milekic and Alberini, 2002; Suzuki et al, 2004). It is possible that the updating session would need to occur within a time window after acquisition (possibly during the systems consolidation timeframe) to drive reorganization of the circuitry. It would also be interesting to determine how broadly these findings apply to other forms of memory. For example, one could test whether the opposite update (delay-to-trace) could also reorganize the neural circuit of a delay-trained memory to more closely reflect the circuit supporting trace fear. Finally, it would also be valuable to determine whether this type of updating-based reorganization occurs for other forms of memory (eg, appetitive memory). Identifying these boundary conditions will be a critical step towards applying reconsolidation-based strategies to attenuate traumatic memory in a clinical setting.
One potential criticism of our study is that ANI (Figures 1 and 2) can have unintended side effects (Flexner and Goodman, 1975; Iordanov et al, 1997; Radulovic and Tronson, 2008; Rudy, 2008) and APV may not block retrieval in some brain structures critical for memory formation (Campeau et al, 1992; Falls et al, 1992; Kwapis et al, 2015a). Regardless of how ANI is acting, it is clear that its application in the amygdala affects both delay and trace memory following a standard CS-only retrieval trial (Figure 1) and that its application only affects memory following presentation of new information (Figure 2). Similarly, although blocking NMDA receptors may not affect memory retrieval in some brain structures (Mei et al, 2011), numerous studies have shown that APV-mediated NMDAR inhibition in the amygdala can disrupt delay fear extinction (Falls et al, 1992; Kwapis et al, 2014a; Laurent et al, 2008) and blocking NMDA receptors in the RSC effectively disrupts memory retrieval for trace and context (but not delay) fear (Corcoran et al, 2011; Kwapis et al, 2014a; Kwapis et al, 2015b). These drugs, therefore, while not perfect, are effective tools to interrogate the neural circuit supporting the updated memory.
In conclusion, we found that reconsolidation-dependent updating procedures can be leverage to reorganize a relatively complex trace fear memory so that it relies on the more basic delay fear circuit. This is the first demonstration that the reconsolidation-dependent updating process can reorganize the neural circuit supporting a memory in addition to revising that memory’s content. Further, these results suggest that reconsolidation-dependent updating could be used to force a complex traumatic memory to rely on a better-defined neural circuit that may be more amenable to behavioral or pharmacological manipulation.
FUNDING AND DISCLOSURE
This research was supported by the National Institute of Mental Health (NIMH) grant R01MH069558 to FJH and NIMH grant F31MH090685 to JLK. The authors declare no conflict of interest.
References
Alberini CM, Ledoux JE (2013). Memory reconsolidation. Curr Biol 23: R746–R750.
Besnard A (2012). A model of hippocampal competition between new learning and memory updating. J Neurosci 32: 3281–3283.
Campeau S, Miserendino MJ, Davis M (1992). Intra-amygdala infusion of the N-methyl-D-aspartate receptor antagonist AP5 blocks acquisition but not expression of fear-potentiated startle to an auditory conditioned stimulus. Behav Neurosci 106: 569–574.
Clem RL, Huganir RL (2010). Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330: 1108–1112.
Corcoran KA, Donnan MD, Tronson NC, Guzman YF, Gao C, Jovasevic V et al (2011). NMDA receptors in retrosplenial cortex are necessary for retrieval of recent and remote context fear memory. J Neurosci 31: 11655–11659.
Davis M (2011). NMDA receptors and fear extinction: implications for cognitive behavioral therapy. Dialogues Clin Neurosci 13: 463–474.
Diaz-Mataix L, Ruiz Martinez RC, Schafe GE, LeDoux JE, Doyere V (2013). Detection of a temporal error triggers reconsolidation of amygdala-dependent memories. Curr Biol 23: 467–472.
Duvarci S, Nader K, LeDoux JE (2005). Activation of extracellular signal-regulated kinase- mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci 21: 283–289.
Falls WA, Miserendino MJD, Davis M (1992). Extinction of fear-potentiated startle–blockade by infusion of an nmda antagonist into the amygdala. J Neurosci 12: 854–863.
Flexner LB, Goodman RH (1975). Studies on memory: inhibitors of protein synthesis also inhibit catecholamine synthesis. Proc Natl Acad Sci USA 72: 4660–4663.
Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, Silva AJ (2006). Stability of recent and remote contextual fear memory. Learn Mem 13: 451–457.
Gilmartin MR, Helmstetter FJ (2010). Trace and contextual fear conditioning require neural activity and NMDA receptor-dependent transmission in the medial prefrontal cortex. Learn Mem 17: 289–296.
Gilmartin MR, Kwapis JL, Helmstetter FJ (2012). Trace and contextual fear conditioning are impaired following unilateral microinjection of muscimol in the ventral hippocampus or amygdala, but not the medial prefrontal cortex. Neurobiol Learn Mem 97: 452–464.
Gilmartin MR, Miyawaki H, Helmstetter FJ, Diba K (2013). Prefrontal activity links nonoverlapping events in memory. J Neurosci 33: 10910–10914.
Goltseker K, Levi L, Barak S (2016). Counterconditioning during reconsolidation prevents relapse of cocaine memories. Neuropsychopharmacology 42: 716–726.
Graff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J et al (2014). Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156: 261–276.
Guimarais M, Gregorio A, Cruz A, Guyon N, Moita MA (2011). Time determines the neural circuit underlying associative fear learning. Front Behav Neurosci 5: 89.
Haubrich J, Crestani AP, Cassini LF, Santana F, Sierra RO, Alvares Lde O et al (2015). Reconsolidation allows fear memory to be updated to a less aversive level through the incorporation of appetitive information. Neuropsychopharmacology 40: 315–326.
Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL et al (1997). Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 17: 3373–3381.
Jarome TJ, Ferrara NC, Kwapis JL, Helmstetter FJ (2015). Contextual information drives the reconsolidation-dependent updating of retrieved fear memories. Neuropsychopharmacology 40: 3044–3052.
Jarome TJ, Kwapis JL, Werner CT, Parsons RG, Gafford GM, Helmstetter FJ (2012). The timing of multiple retrieval events can alter GluR1 phosphorylation and the requirement for protein synthesis in fear memory reconsolidation. Learn Mem 19: 300–306.
Jarome TJ, Werner CT, Kwapis JL, Helmstetter FJ (2011). Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS ONE 6: e24349.
Kim JJ, Fanselow MS (1992). Modality-specific retrograde amnesia of fear. Science 256: 675–677.
Kochli DE, Thompson EC, Fricke EA, Postle AF, Quinn JJ (2015). The amygdala is critical for trace, delay, and contextual fear conditioning. Learn Mem 22: 92–100.
Kwapis JL, Jarome TJ, Helmstetter FJ (2015a). The role of the medial prefrontal cortex in trace fear extinction. Learn Mem 22: 39–46.
Kwapis JL, Jarome TJ, Lee JL, Gilmartin MR, Helmstetter FJ (2014a). Extinguishing trace fear engages the retrosplenial cortex rather than the amygdala. Neurobiol Learn Mem 113: 41–54.
Kwapis JL, Jarome TJ, Lee JL, Helmstetter FJ (2015b). The retrosplenial cortex is involved in the formation of memory for context and trace fear conditioning. Neurobiol Learn Mem 123: 110–116.
Kwapis JL, Jarome TJ, Lonergan ME, Helmstetter FJ (2009). Protein kinase Mzeta maintains fear memory in the amygdala but not in the hippocampus. Behav Neurosci 123: 844–850.
Kwapis JL, Jarome TJ, Schiff JC, Helmstetter FJ (2011). Memory consolidation in both trace and delay fear conditioning is disrupted by intra-amygdala infusion of the protein synthesis inhibitor anisomycin. Learn Mem 18: 728–732.
Kwapis JL, Wood MA (2014b). Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci 37: 706–720.
Laurent V, Marchand AR, Westbrook RF (2008). The basolateral amygdala is necessary for learning but not relearning extinction of context conditioned fear. Learn Mem 15: 304–314.
Lee JL (2008). Memory reconsolidation mediates the strengthening of memories by additional learning. Nat Neurosci 11: 1264–1266.
Lee JL (2010). Memory reconsolidation mediates the updating of hippocampal memory content. Front. Behav. Neurosci. 4: 168.
Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK et al (2008). Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319: 1253–1256.
Mei B, Li F, Gu Y, Cui Z, Tsien JZ (2011). NMDA receptors are not required for pattern completion during associative memory recall. PLoS ONE 6: e19326.
Milekic MH, Alberini CM (2002). Temporally graded requirement for protein synthesis following memory reactivation. Neuron 36: 521–525.
Misanin JR, Miller RR, Lewis DJ (1968). Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace. Science 160: 554–555.
Monfils MH, Cowansage KK, Klann E, LeDoux JE (2009). Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 324: 951–955.
Morris RG, Inglis J, Ainge JA, Olverman HJ, Tulloch J, Dudai Y et al (2006). Memory reconsolidation: sensitivity of spatial memory to inhibition of protein synthesis in dorsal hippocampus during encoding and retrieval. Neuron 50: 479–489.
Nader K, Hardt O (2009). A single standard for memory: the case for reconsolidation. Nat. Rev. Neurosci. 10: 224–234.
Nader K, Schafe GE, Le Doux JE (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722–726.
Parsons RG, Gafford GM, Baruch DE, Riedner BA, Helmstetter FJ (2006a). Long-term stability of fear memory depends on the synthesis of protein but not mRNA in the amygdala. Eur J Neurosci 23: 1853–1859.
Parsons RG, Gafford GM, Helmstetter FJ (2006b). Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci 26: 12977–12983.
Parsons RG, Ressler KJ (2013). Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci 16: 146–153.
Pavlov IP (1927) Conditioned Reflexes: An Investigation of the Physiological Activity of the Cerebral Cortex. Oxford University Press: London.
Radulovic J, Tronson NC (2008). Protein synthesis inhibitors, gene superinduction and memory: too little or too much protein? Neurobiol Learn Mem 89: 212–218.
Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB et al (2011). Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci 14: 1302–1308.
Rossato JI, Bevilaqua LR, Myskiw JC, Medina JH, Izquierdo I, Cammarota M (2007). On the role of hippocampal protein synthesis in the consolidation and reconsolidation of object recognition memory. Learn Mem 14: 36–46.
Rothbaum BO, Davis M (2003). Applying learning principles to the treatment of post-trauma reactions. Ann N Y Acad Sci 1008: 112–121.
Rudy JW (2008). Is there a baby in the bathwater? Maybe: some methodological issues for the de novo protein synthesis hypothesis. Neurobiol Learn Mem 89: 219–224.
Rudy JW, Sutherland RJ (2008). Is it systems or cellular consolidation? Time will tell. An alternative interpretation of the Morris group's recent science paper. Neurobiol Learn Mem 89: 366–369.
Sara SJ (2000). Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 7: 73–84.
Schiller D, Monfils MH, Raio CM, Johnson DC, Ledoux JE, Phelps EA (2010). Preventing the return of fear in humans using reconsolidation update mechanisms. Nature 463: 49–53.
Sevenster D, Beckers T, Kindt M (2012). Retrieval per se is not sufficient to trigger reconsolidation of human fear memory. Neurobiol Learn Mem 97: 338–345.
Squire LR (1992). Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 99: 195–231.
Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004). Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24: 4787–4795.
Tayler KK, Tanaka KZ, Reijmers LG, Wiltgen BJ (2013). Reactivation of neural ensembles during the retrieval of recent and remote memory. Curr Biol 23: 99–106.
Tse D, Takeuchi T, Kakeyama M, Kajii Y, Okuno H, Tohyama C et al (2011). Schema-dependent gene activation and memory encoding in neocortex. Science 333: 891–895.
Winters BD, Tucci MC, Jacklin DL, Reid JM, Newsome J (2011). On the dynamic nature of the engram: evidence for circuit-level reorganization of object memory traces following reactivation. J Neurosci 31: 17719–17728.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website .
Supplementary information
Rights and permissions
About this article

Cite this article
Kwapis, J., Jarome, T., Ferrara, N. et al. Updating Procedures Can Reorganize the Neural Circuit Supporting a Fear Memory. Neuropsychopharmacol 42, 1688–1697 (2017). https://doi.org/10.1038/npp.2017.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.23
This article is cited by
-
Aging mice show impaired memory updating in the novel OUL updating paradigm
Neuropsychopharmacology (2020)
