
Abstract
A wealth of data has been amassed that details a complex, yet accessible, series of pathways by which the immune system, notably inflammation, can influence the brain and behavior. These data have opened the window to a diverse array of novel targets whose potential efficacy is tied to specific neurotransmitters and neurocircuits as well as specific behaviors. What is clear is that the impact of inflammation on the brain cuts across psychiatric disorders and engages dopaminergic and glutamatergic pathways that regulate motivation and motor activity as well as the sensitivity to threat. Given the ability to identify patient populations with increased inflammation, the precision of interventions can be further tuned, in conjunction with the ability to establish target engagement in the brain through the use of multiple neuroimaging strategies. After a brief overview of the mechanisms by which inflammation affects the brain and behavior, this review examines the extant literature on the efficacy of anti-inflammatory treatments, while forging guidelines for future intelligent clinical trial design. An examination of the most promising therapeutic strategies is also provided, along with some of the most exciting clinical trials that are currently being planned or underway.
Similar content being viewed by others
INTRODUCTION
Based on increasing data regarding the impact of immunologic processes on both neuronal integrity and neuropathology, there has been tremendous interest in the notion that therapies targeting the immune system or its downstream consequences on the brain may have significant promise for psychiatric disorders. Indeed, data suggest that alterations in immune system function, notably increased inflammation, can negatively impact virtually every aspect of central nervous system (CNS) function including neurotransmitter metabolism, neuroendocrine function, synaptic plasticity, and regional brain activity (Miller and Raison, 2015). These effects of the immune system and inflammation on the brain have been implicated in a number of psychiatric diseases ranging from mood and anxiety disorders to schizophrenia as well as neurodegenerative diseases. A growing literature also indicates that the immune system plays a pivotal role in fundamental aspects of normal nervous system function including neurogenesis, synaptic remodeling, neuronal excitability, metabolic support, and even the response to medication. Therefore, therapies that promote or regulate certain elements of immune system function may provide resilience against disease. Taken together, the picture that has emerged is an immune system that plays a complex and often Janus-faced role in brain health and illness that undoubtedly demands greater understanding from both an immunological and a neurobiological perspective. Nevertheless, the question has arisen of whether there is enough knowledge at this point to be applied for intervention and determination of therapeutic benefit. The purpose of this review is to address this question and consider what strategies and targets have the greatest likelihood of success. Moreover, how immune-targeted therapies might be applied to specific patient populations to address specific symptoms within the context of psychiatric disease will be explored. It is the opinion of the authors that the time is ripe to apply our knowledge and to begin to test the hypothesis that the immune system plays an important pathophysiologic role in a significant subset of individuals with psychiatric disease. Moreover, it is suggested that the current knowledge state can be applied to the benefit of the field as well as to patients suffering from psychiatric disorders. However, without taking advantage of what is currently known, studies targeting the immune system or its effects on the brain equally run the risk of generating data that are difficult to interpret, potentially harming the field and leading to premature conclusions regarding the possibilities and the pitfalls of immune-based therapies.
In this review, we will first briefly address the evidence that the immune system plays a role in psychiatric diseases with an emphasis on the role of inflammation in mood disorders. We will then discuss what is known regarding the mechanisms by which the immune system affects the brain and behavior, and how these mechanisms can inform the development of novel targets for therapeutic intervention. Finally, we will conclude with some practical guidelines regarding intelligent clinical trial design and therapeutic and preventative strategies that may be ready for immediate application.
FOUNDATIONS FOR THE HYPOTHESIS THAT THE IMMUNE SYSTEM PLAYS A ROLE IN PSYCHIATRIC DISEASE
Probably the best studied of all the psychiatric disorders regarding immune system function are the mood disorders, with the predominance of studies focusing on patients with major depression. Early studies primarily examined adaptive immune responses and indicated that T and B cells from patients with major depression exhibited decreased proliferation following a variety of stimuli including the mitogens phytohemagglutinin and pokeweed mitogen (Schleifer et al, 1984; Stein et al, 1991; Zorrilla et al, 2001). Decreases in the number and activity of NK cells, which are believed to straddle adaptive and innate divisions of the immune system, were also described (Stein et al, 1991; Zorrilla et al, 2001). In contradistinction to the initial findings, subsequent findings suggested that activation of innate immune responses were also involved (Maes, 1995). Indeed, over the past several decades, data demonstrating increased innate immune responses and inflammation in patients with major depression have dominated the literature (Miller and Raison, 2015). For example, a multitude of studies have described increases in inflammatory cytokines in the peripheral blood and cerebrospinal fluid (CSF) of depressed patients (Howren et al, 2009; Dowlati et al, 2010; Haapakoski et al, 2015). In addition, numerous studies have documented increases in peripheral blood acute-phase reactants, chemokines, and adhesion molecules (Howren et al, 2009; Dowlati et al, 2010; Haapakoski et al, 2015). In several meta-analyses of this literature, the most well-documented and reproducible findings have included increases in peripheral blood concentrations of the cytokines interleukin-6 (IL-6) and tumor necrosis factor (TNF) and the acute-phase reactant C-reactive protein (CRP) (Howren et al, 2009; Dowlati et al, 2010; Haapakoski et al, 2015). In addition to increases in inflammatory markers in depressed subjects, a number of polymorphisms in genes associated with inflammation, including IL-1β, IL-6, and TNF, as well as CRP, have been linked to the development of mood and anxiety disorders as well as treatment response (Bufalino et al, 2012; Michopoulos et al, 2015). Epigenetic changes in genes involved in the regulation of immune responses including FK506 binding protein 5 (FKBP5) have also been implicated in anxiety disorders and the response to stress. FKBP5 is a co-chaperone of heat shock protein 90 that regulates glucocorticoid receptor sensitivity. When FKBP5 is bound to the glucocorticoid receptor complex, cortisol binds with lower affinity, and nuclear translocation of the glucocorticoid receptor is less efficient, leading to glucocorticoid resistance (Klengel et al, 2013). The impact of epigenetic changes in FKBP5 are in part related to the induction of glucocorticoid resistance and reduced hormonal (glucocorticoid) control of inflammatory responses as reflected by decreased sensitivity to the synthetic glucocorticoid dexamethasone in vitro following LPS stimulation (Klengel et al, 2013). Relative to treatment response, it should also be noted that increased inflammatory markers have been associated with treatment nonresponse, such that patients with evidence of increased inflammation before antidepressant treatment exhibit reduced treatment responsiveness, and patients who are defined as treatment resistant exhibit increased inflammatory markers (Sluzewska et al, 1997; Lanquillon et al, 2000; Raison et al, 2013a, b; Strawbridge et al, 2015).
Regarding the mechanisms that underlie the increases in inflammatory responses in patients with depression or other psychiatric disorders, data indicate that there is an increased activation of fundamental inflammatory signaling molecules and pathways including increased activation of nuclear factor-κB (NF-κB) and the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome, a complex of proteins that leads to the activation of caspase 1 that in turn cleaves pro-IL-1β and pro-IL-18 into their active forms (Alcocer-Gomez et al, 2014; Elovainio et al, 2015; Miller and Raison, 2015). Moreover, decreases in counter-regulatory immunologic functions including T regulatory cells (Tregs) and their production of the anti-inflammatory cytokines TGF-β and IL-10 have been described (Sutcigil et al, 2007; Li et al, 2010; Chen et al, 2011). Of note, depletion of Tregs leads to anxiety and depressive-like behavior in mice (Kim et al, 2012). Increases in the highly proinflammatory T helper type 17 (Th17) cells in conjunction with increased peripheral blood concentrations of IL-17 have also been found in patients with major depression (Chen et al, 2011).
Although much of the attention of the literature has been focused on immunologic parameters in the peripheral blood, it should be noted that although the literature is not entirely consistent (Clark et al, 2016), increased activation of microglial cells as well as increased expression of inflammatory cytokines and Toll-like receptors have been described in postmortem brain samples of presumably depressed suicide victims (Steiner et al, 2008, 2011; Pandey et al, 2012, 2014; Torres-Platas et al, 2014). Inconsistencies in the literature have been attributed to diagnostic considerations, use of medications, brain regions sampled, and small sample sizes. In addition, recent data suggest that microglia may exhibit dynamic alterations over time as a function of duration of illness. For example, in a recent study in laboratory animals exposed to chronic unpredictable stress, microglia were found to be activated within days following stressor exposure, whereas at the end of 5 weeks of stress, microglial numbers were decreased (Kreisel et al, 2014). Depressive-like behavior was associated with both of these microglial changes, and additional data indicated that inhibiting microglia had therapeutic efficacy early in disease, whereas stimulating microglia had antidepressant effects after prolonged stress (Kreisel et al, 2014).
Consistent with immune cell trafficking to the brain, which has been described in animal models of stress-induced depression (see below), evidence of increased perivascular macrophages expressing monocyte chemoattractant-1 (MCP-1) has been found in postmortem tissue of depressed suicide victims (Torres-Platas et al, 2014). These data indicate that inflammatory responses are found not only in the periphery but also in the brain of depressed patients, suggesting a more generalized inflammatory process. What has remained unclear however has been the relationship between peripheral and central immune compartments. The recent description of a CNS lymphatic drainage system suggests that CSF may provide more clues to CNS immune processes than was previously appreciated (Louveau et al, 2015). Thus, the presence of increased inflammatory markers in the CSF of patients with depression and their relationship with the periphery may hold additional promise in documenting and monitoring CNS inflammatory processes in the brain of living subjects.
It should be noted that similar changes in immune parameters including increases in inflammatory markers have also been described in patients with a variety of psychiatric disorders other than depression including bipolar mood disorders and anxiety disorders as well as schizophrenia (Konuk et al, 2007; Hoge et al, 2009; Pace and Heim, 2011; Goldsmith et al, 2016). Moreover, what has become increasingly clear is that none of these disorders, including notably major depression, are fundamentally inflammatory disorders, but rather in each case, there is a subgroup of patients who exhibit evidence of increased inflammation (Raison and Miller, 2011). Factors that have been associated with a greater likelihood of increased inflammation in these patient populations include treatment resistance as well as childhood maltreatment, obesity, increasing age, medical illness, and genetic predisposition (Iosifescu et al, 2003; Miller et al, 2009; Oskooilar et al, 2009; Nanni et al, 2012). Taken together, these data suggest that the various neurobiological pathways and resultant symptoms secondary to inflammation (discussed in detail below) occur across disorders (are transdiagnostic) in subgroups of patients with increased inflammation. It is these subgroups of patients with increased inflammation that bespeak specificity regarding pathogenesis, especially as it relates to the interaction of inflammatory cytokines and their signaling pathways with specific neurotransmitters in brain regions that lead to specific symptoms. This will be discussed in greater detail below.
A second major body of evidence that indicates that the immune system may play a role in depression and other psychiatric disorders are studies that have administered inflammatory cytokines (eg, interferon-α (IFN-α)) or inflammatory stimuli (eg, typhoid vaccination and endotoxin) to otherwise nondepressed individuals. These studies have reliably shown that increasing inflammation leads to symptoms that are typically seen in depressed patients including depressed mood, anhedonia, anorexia, insomnia, cognitive impairment, sleep disturbances, and suicidal ideation, as well as anxiety and in some cases irritability, mania, and psychosis (Capuron et al, 2002; Constant et al, 2005; Harrison et al, 2009a, b; Eisenberger et al, 2010a, b). Interestingly, in a direct comparison of symptoms in patients with idiopathic major depression versus patients with IFN-α-induced depression, there was almost a complete overlap of symptoms excluding an overrepresentation of psychomotor retardation and anorexia in the IFN-α-treated depressed individuals (Capuron et al, 2009). As described below, it should also be noted that the neural circuitry that is engaged by inflammatory stimuli is also consistent across experimental paradigms (eg, IFN-α, typhoid vaccination, acute endotoxin administration) and neuroimaging platforms (Miller et al, 2013).
A third body of evidence that supports the notion that inflammation may play a role in symptoms common to psychiatric disorders are a multitude of studies indicating high rates of mood and anxiety disorders in patients with a variety of medical illnesses that are associated with increased inflammation including autoimmune and inflammatory disorders, cardiovascular disease, and cancer (Evans et al, 2005). In addition, psychosocial stimuli that are associated with high rates of psychiatric sequela are linked with increased inflammation including childhood maltreatment, loneliness, and psychological (as well as physical) trauma (Danese et al, 2008; Pace and Heim, 2011; Torres et al, 2013; Cole et al, 2015).
Finally, as will be discussed in greater detail below, there is growing evidence that blockade of inflammation can improve depressive symptoms, not only in patients with autoimmune and inflammatory disorders and cancer, but also in those with depression.
MECHANISMS BY WHICH THE IMMUNE SYSTEM AFFECTS THE BRAIN AND BEHAVIOR
Providing potential clues as to what might be the most viable targets for reversing the effects of inflammation on the brain and behavior are data derived from laboratory animal and human studies that address the immunological and neurobiological mechanisms by which the immune system influences the brain.
Immunological Mechanisms
Translating peripheral immune signals to the brain
It remains unclear whether immune alterations in stress-related psychiatric disorders including major depression and anxiety disorders primarily derive from changes in immune regulation in the periphery or in the CNS. Nevertheless, as indicated above, it is apparent that evidence of immune dysregulation is present in both compartments. Moreover, studies in laboratory animals have shown that blockade of inflammatory cytokines or their signaling pathways exclusively in the periphery or exclusively in the brain can block the development of stress-induced depressive-like behavior. For example, administration of the peripherally active TNF antagonist etanercept, which does not cross the blood brain barrier (BBB), eliminated depressive-like behavior following chronic mild stress in mice (Krugel et al, 2013). In addition, studies have shown that individual differences in peripheral immune responses (IL-6 responses, in particular) are associated with the susceptibility of laboratory animals to social avoidance after repeated social defeat stress. This susceptibility to social avoidance can be reversed following peripheral administration of a monoclonal antibody to IL-6 that also does not cross the BBB (Hodes et al, 2014, 2015). Conversely, overexpression of IL-1 receptor antagonist in the brain blocked chronic stress-induced anhedonia as well as impairment in hippocampal neurogenesis, a primary mechanism through which chronic stress is believed to induce depression (Ben Menachem-Zidon et al, 2008; Goshen et al, 2008). Taken together, these data suggest that peripheral immune responses are an essential element of the induction of stress-induced depressive-like behavior in laboratory animals. However, the data also indicate that intervention can occur in the periphery or the brain to reverse the effects of the immune system on behavior.
Given the data that peripheral immune responses can drive central inflammatory responses and the related changes in behavior in laboratory animals, there has been significant interest in how peripheral inflammatory signals can reach the brain. Understanding this process may have great therapeutic promise in limiting the impact of peripheral inflammatory responses on central immune and nervous system function. A primary hurdle for cytokines to directly enter the brain involves the fact that these molecules are too large to freely pass through the BBB. A number of mechanisms that circumvent this roadblock have been described, including a ‘neural’ route and a ‘humoral’ route (Quan and Banks, 2007). The neural route involves activation of peripheral afferent nerve fibers including the vagus nerve by peripheral cytokines and has been shown to lead to the induction of cytokine signals directly in the brain (Hansen et al, 1998). Blockade of the neural route through procedures such as vagotomy have been shown to inhibit many aspects of the response to peripheral inflammatory stimuli including activation of the HPA axis, alterations in catecholamine and serotonin metabolism, depressive-like behavior such as decreased social exploration and decreased sucrose preference, and, in some studies, the fever response (Fleshner et al, 1998; Luheshi et al, 2000; Wieczorek et al, 2005). The humoral route involves the access of cytokines to the brain through leaky regions in the BBB or through the binding of cytokines to saturable transport molecules on endothelial cells that comprise the BBB (Banks, 2015). Transport molecules for a number of inflammatory factors have been described, including the cytokines IL-1α, IL-1β, TNF, and IL-6, as well as the chemokine MCP-1 also known as chemokine (C-C motif) ligand 2.
Monocytes on the move. More recently, a third pathway for inflammatory signals to reach the brain has been described. This pathway involves the migration of peripheral immune cells, primarily monocytes, to the meninges, brain vasculature, and in some cases the brain parenchyma. This ‘cellular’ pathway has received considerable recent attention and has been elaborated in exquisite detail in laboratory animal models (D'Mello et al, 2009; Wohleb et al, 2014). Based on a series of studies conducted by several investigators, in the context of stress or peripheral inflammation, catecholaminergic activation of microglia or stimulation of microglia by TNF via TNF receptor 1 in the brain leads to the release of MCP-1 into the periphery (D'Mello et al, 2009; Wohleb et al, 2014). At the same time, stress-induced release of catecholamines facilitates the production and release of monocytes from the bone marrow. Once in the peripheral circulation, these monocytes can become activated by stress-associated danger signals called damage-associated molecular patterns (DAMPs) and bacteria and bacterial products such as microbe-associated molecular patterns (MAMPs) leaked from the gut (Fleshner, 2011, 2013; Wong et al, 2016). DAMPs include a number of endogenous molecules produced as a function of cellular stress and accelerated metabolism including uric acid, adenosine triphosphate (ATP), glucose, and heat shock proteins, whereas MAMPS represent bacterial products including flagellin and LPS released from the gut (Maslanik et al, 2012a, b; Fleshner, 2013). DAMPS and MAMPS can then lead to stimulation of the NLRP3 inflammasome and NF-κB, respectively, within monocytes, and ultimately lead to the production of inflammatory cytokines including IL-1, IL-18, IL-6, and TNF (Maslanik et al, 2012a, b). These activated monocytes can then be attracted to the brain vasculature or the meninges and also traffic through the lymphatic system within the brain (D'Mello et al, 2009; Wohleb et al, 2014; Louveau et al, 2015). In these locations, activated mononuclear cells can facilitate central inflammatory responses. Taken together, the emerging importance of the cellular route in transmitting inflammatory immune signals to the brain lends itself and the chemokines and adhesion molecules involved as attractive targets for drug development.
Given the relevance of microglial activation not only for the release of inflammatory mediators within the brain but also for the production of chemokines that attract monocytes to the brain, there has been considerable interest in measuring microglial activation using neuroimaging strategies including positron emission tomography (PET). The ability to image microglial activation in the brain has multiple advantages including the ability to prospectively track the impact of inflammation on brain morphology over time as well as the ability to determine whether an anti-inflammatory strategy has an impact on inflammatory responses in the brain. In addition, as discussed in greater detail below, identifying patients with evidence of increased CNS inflammatory responses can help further target therapies and thereby personalized care. One strategy to identify activated microglia has been to use PET ligands that bind to the translocator protein (TSPO), a protein found on the outer mitochondrial membrane that exhibits increased expression following microglial activation. Several newer generation TSPO ligands have been developed and demonstrate good brain penetrance and favorable binding characteristics. Two structurally similar ligands, [11C]PBR28 and [18F]FEPPA, have been examined in both depression and schizophrenia (Vivash and O'Brien, 2016). Of note, the assessment of TSPO binding is complicated by the presence of a TSPO polymorphism (rs6971) that is known to influence the TSPO binding, leading to high-affinity, mixed-affinity, and low-affinity binders (Vivash and O'Brien, 2016). Low-affinity binders are typically excluded from study analyses, although they represent only ∼10% of the population in North America. Proof-of-concept studies have demonstrated increased TSPO binding throughout the brain following administration of endotoxin to both humans and nonhuman primates (Hannestad et al, 2012; Sandiego et al, 2015). Increases in TSPO binding in the brain of nonhuman primates were correlated with increased peripheral blood concentrations of IL-1β, IL-6, and TNF depending on the time point examined after endotoxin administration. Moreover, histological examination confirmed that TSPO binding following endotoxin was primarily increased in microglia as opposed astrocytes that also express TSPO (Hannestad et al, 2012). Interestingly, no correlations between increased peripheral blood cytokine concentrations or increased symptoms of depression and increased TSPO binding in the brain were found after endotoxin in humans, although marked increases in TSPO binding were observed (Sandiego et al, 2015). In terms of studies in patient populations, mixed results have been found regarding TSPO binding in depression and schizophrenia with both positive and negative results (Hannestad et al, 2013; Kenk et al, 2015; Bloomfield et al, 2016; Coughlin et al, 2016). Nevertheless, at least one study in patients with depression has provided promising evidence of increased TSPO binding in the prefrontal cortex, insula, and anterior cingulate cortex that correlated with depression symptom severity (Setiawan et al, 2015). Unfortunately, in this study, no significant relationship was found between peripheral inflammatory markers and central TSPO binding. These data raise questions regarding the relationship between peripheral and central inflammatory markers and are not consistent with studies in laboratory animals where peripheral inflammatory responses have been shown to drive central inflammation. Moreover, as described below, several studies have demonstrated relationships between neurotransmitter and neurocircuitry changes in the brain that correlate with peripheral immune responses. Thus, it is unclear what TSPO binding is measuring in humans, and more studies are needed to fully appreciate the information that is provided, possibly through CSF sampling of immune variables including cytokines and chemokines and/or other microglial products including quinolinic acid, glutamate, nitric oxide, or complement factors that may more directly reflect CNS microglial activity. Finally, increased attention is being paid to the nuances of the analysis of TSPO data (Turkheimer et al, 2015).
T cells to the rescue. Some of the most intriguing data that have emerged from the field of neuroimmunology indicate that in the context of CNS injury, T effector cells and even monocytes can traffic to the brain and induce repair processes that have a significant impact on the extent of tissue damage and destruction (Schwartz and Shechter, 2010; Wattananit et al, 2016). For example, after partial crush injury of the optic nerve, T cells immunized to specific CNS antigens (ie, myelin basic protein) were found to protect against secondary nerve degeneration (Moalem et al, 1999). Interestingly, a similar process has been demonstrated in the context of stress, whereby T cells were found to traffic to the CNS via a glucocorticoid-mediated pathway that involved induction of adhesion molecules including intracellular adhesion molecule-1 (ICAM-1) in the meninges (Lewitus et al, 2008). T-cell trafficking to the brain during stress was shown to reduce stress-induced anxiety-like behavior and reverse stress-induced decreases in brain-derived neurotrophic factor (BDNF) that is known to stimulate neurogenesis and possess antidepressant effects (Lewitus et al, 2008). From a therapeutic standpoint, urging T cells to traffic to the brain via administration of a CNS-related antigen has been shown to exhibit both an antianxiety and antidepressant effect depending on the animal model (Lewitus and Schwartz, 2009; Lewitus et al, 2009). Along similar lines, T cells have been shown to be essential for normal cognitive function such that T cell-deficient animals exhibit impaired learning and memory as revealed by Morris water maze, Barnes maze, and radial arm maze that can be reversed by the adoptive transfer of T cells (Kipnis et al, 2004; Ziv et al, 2006). Moreover, experiments using RAG-1 knockout mice (that are deficient in mature B and T cells) have revealed that long-term deficiencies in CD4+ T cells are associated with impaired behavioral responses including increased digging and marble-burying activities compared with wild-type mice (Rattazzi et al, 2013). Mechanistic studies have revealed that T cells trafficking to the brain (primarily the meningeal space) can produce IL-4 that stimulates astrocytes to produce growth factors including BDNF, while also leading to the skewing of meningeal macrophages from a proinflammatory M1 phenotype to an anti-inflammatory, neuroprotective M2 phenotype (Derecki et al, 2010). Interestingly in this regard, the offspring of rats that were administered helminths during pregnancy, which stimulates a potent IL-4 response, were found to exhibited markedly attenuated CNS inflammatory responses to bacterial infection (Williamson et al, 2015). Taken together, these data indicate that T cells not only play a fundamental role in neuronal integrity especially as it relates to the processes of learning and memory, but also can support resilience in the context of stress-induced behavioral changes, especially through the release of neuroprotective and anti-inflammatory factors such as IL-4.
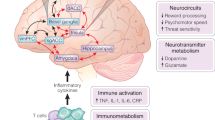
Cytokines, synaptic plasticity, and the response to antidepressants. Although much of the focus of this review has been on the potentially detrimental impact of inflammatory cytokines on behavior, a rich literature has described the impact of a variety of cytokines including inflammatory cytokines such as TNF and IL-1 on a number of CNS processes including synaptic plasticity and learning and memory as well as the response to antidepressant medications (del Rey et al, 2013). For example, mice depleted of the IL-1 type 1 receptor exhibit significant decreases in learning and memory and impaired long-term potentiation (LTP) (Yirmiya and Goshen, 2011). Interestingly, transplantation of IL-1 receptor knockout mice with wild-type neural precursor cells (expressing the IL-1 receptor), which preferentially differentiate into astrocytes, demonstrated not only complete restoration of memory function as measured by fear conditioning and water maze studies but also exhibited normal LTP in perforant path dentate gyrus synapses (Ben Menachem-Zidon et al, 2011). Similar to IL-1, deletion of TNF receptors 1 and 2 have been associated with cognitive deficits in mice including impaired maze performance and novel object recognition (Naude et al, 2014). Mechanisms that have been identified by which cytokines like TNF may influence learning and memory include the demonstration that TNF plays an important role in the regulation of synaptic efficacy through the induction of surface expression of AMPA receptors that support the preservation of synaptic strength at excitatory synapses (Beattie et al, 2002; Stellwagen and Malenka, 2006). TNF has also been implicated in the maintenance of synaptic plasticity by playing a permissive role in synaptic scaling (Stellwagen and Malenka, 2006). Of considerable interest are data suggesting that cytokines, in particular inflammatory cytokines, may play a role in the response to antidepressants. In a series of in vitro and in vivo studies, the conventional antidepressant citalopram was found to induce several inflammatory cytokines including IL-1β, IL-6, TNF, and IFN-γ, an effect that could be blocked by the nonsteroidal anti-inflammatory agent ibuprofen (Warner-Schmidt et al, 2011). In addition, administration of citalopram to TNF or IFN-γ receptor knockout mice failed to induce the expression of p11, a protein that is upregulated by BDNF and is increased in the mouse frontal cortex by multiple classes of antidepressants. Coexpression of p11 and the receptors for both TNF and IFN-γ in cortical neurons also support a role of TNF and IFN-γ in p11 expression (Warner-Schmidt et al, 2011; Svenningsson et al, 2013). Finally, administration of ibuprofen was found to block the antidepressant response to serotonin reuptake inhibitors (SSRIs) and to a lesser extent tricyclic antidepressants (TCAs) in the forced swim test and tail suspension test, but had no effect on the antidepressant efficacy of tranylcypromine and bupropion in these paradigms (Warner-Schmidt et al, 2011). These data indicate that response to certain classes of antidepressants including SSRIs and TCAs may in part be dependent on cytokine induction of key proteins that are involved in the antidepressant response. Taken together, these data emphasize the importance of hitting the ‘sweet spot,’ whereby the negative effects of inflammatory cytokines are abrogated while inhibition of the neuroregulatory functions of cytokines are avoided (Figure 1).

Hitting the sweet spot—above all do no harm. Cytokines play pivotal roles in multiple aspects of neuronal integrity including long-term potentiation, synaptic remodeling, neurogenesis, learning, memory, and possibly the response to treatments such as antidepressants. Thus, dose–response studies using anticytokine therapies must focus on those patients with evidence of excessive cytokine activation to find the optimal dose of drug that limits the detrimental effects of cytokines on brain function, while leaving their indispensible activities intact. As noted, there may be issues of the timing of these interventions as well.
NEUROBIOLOGICAL MECHANISMS: NEUROTRANSMITTERS AND NEUROCIRCUITS
Some of the most revealing work regarding promising targets to reverse the behavioral effects of the immune system and especially inflammation on the brain are studies that have examined the impact of immune and inflammatory mediators and their signaling pathways on neurotransmitter metabolism and relevant neural circuits in the brain (Miller et al, 2013; Miller and Raison, 2015). These downstream targets of inflammation may be the most ripe for clinical testing in subgroups of patients who exhibit increased inflammatory markers. Moreover, the identification of neural circuits in the brain that are affected by immune and inflammatory processes provide an outstanding opportunity to establish target engagement in the brain for drugs that either block inflammation directly or reverse the effects of inflammation on neurotransmitter systems that regulate these circuits.
Inflammation Effects on Neurotransmitter Metabolism
Primary neurotransmitter systems implicated in neuropsychiatric disorders that have been extensively studied in the context of inflammation and inflammatory cytokines include serotonin, dopamine, and glutamate. Indeed, a host of early studies demonstrated that the application of a variety of immune and inflammatory stimuli altered the concentrations of all the monoamines throughout the brain (Dunn et al, 1999; Anisman et al, 2008), with some of the earliest studies demonstrating the effect of the immune response to sheep red blood cells on norepinephrine metabolism in the hypothalamus (reviewed in (Besedovsky and del Rey, 1996)).
Serotonin
Although a number of studies have examined the impact of cytokines on serotonin metabolism, more recent work has focused on the expression and function of the serotonin transporter, a primary target of conventional antidepressant medications. Initial in vitro studies demonstrated that both IL-1β and TNF could increase the expression and function of the serotonin transporter, an effect mediated by activation of mitogen-activated protein kinase (MAPK) pathways, specifically p38 MAPK (Zhu et al, 2005, 2006). Follow-up in vivo studies confirmed the relevance of these effects by demonstrating that LPS-induced depressive-like behavior was associated with increased serotonin transporter function and could be reversed by p38 MAPK inhibitors (Zhu et al, 2010). Interestingly, in vivo studies in humans are also consistent with the effects of TNF on the serotonin transporter with increased peripheral blood TNF concentrations being associated with increased brainstem serotonin transporter binding (Krishnadas et al, 2016). Cytokine-induced activation of indoleamine 2,3 dioxygenase (IDO) has also been implicated in the effects of the immune system on serotonin. IDO activity can be stimulated by a number of inflammatory cytokines and their signaling pathways including NF-κB. When activated, IDO converts tryptophan, the primary precursor of serotonin, into kynurenine, thus reducing serotonin synthesis and availability (Robinson et al, 2006; Muller and Schwarz, 2007). Given the importance of serotonin in T-cell activation, the reduction of serotonin availability by IDO is a major pathway by which IDO regulates the immune system and T-cell function (Mellor et al, 2003). Of note, IDO inhibitors are actively being developed as anticancer agents, and are therefore currently available for testing (Lob et al, 2009). Because inhibition of serotonin transporters and increasing serotonin availability is a primary mechanism of action of conventional antidepressants, the capacity of inflammatory cytokines to increase serotonin transporter expression and function, while also reducing serotonin synthesis, may serve to undermine the ability of SSRIs to treat mood and anxiety disorders, consistent with the observation that inflammation is associated with treatment nonresponse (Raison et al, 2013a; Strawbridge et al, 2015).
Dopamine
Much attention has been paid to the impact of inflammatory cytokines on dopamine metabolism. Dopamine plays a fundamental role in the regulation of motivation and motor activity (see below) (Cousins and Salamone, 1994; Haber, 2014), and some of the hallmark clinical symptoms associated with the administration of inflammatory cytokines and cytokine inducers are anhedonia, fatigue, and psychomotor retardation (Capuron et al, 2002; Capuron and Miller, 2004). For example, the majority of patients administered IFN-α experience these neurovegetative symptoms within several weeks of beginning treatment. In contrast, the development of IFN-α-induced mood and cognitive symptoms occurs later in treatment and in a smaller subset of individuals (Capuron et al, 2002; Capuron and Miller, 2004). These mood and cognitive symptoms are responsive to SSRIs, whereas the neurovegetative symptoms seem to be relatively unresponsive to these medications (Capuron et al, 2002; Capuron and Miller, 2004).
Studies in laboratory animals and humans indicate that inflammatory cytokines appear to decrease dopamine availability and ultimately decrease dopamine release (Felger and Miller, 2012). For example, using PET, administration of radiolabeled L-DOPA to humans treated with IFN-α was found to lead to increased L-DOPA uptake and decreased L-DOPA release consistent with dopamine depletion (Capuron et al, 2012). Decreased dopamine release was also demonstrated in IFN-α-treated rhesus monkeys that, like humans, exhibit a depressive-like huddling behavior after chronic IFN-α exposure (Felger et al, 2007, 2013b). Indeed, after 4 weeks of IFN-α administration at doses similar to those given to humans for cancer treatment, rhesus monkeys exhibited decreased CSF dopamine metabolites (Felger et al, 2007). Moreover, using in vivo microdialysis of basal ganglia nuclei, these laboratory animals demonstrated decreased extracellular dopamine release following administration (by reverse microdialysis) of potassium, which leads to a voltage-dependent dopamine release, as well as amphetamine, which stimulates dopamine release and blocks dopamine reuptake (Felger et al, 2013b). These deficits in dopamine release were recovered by administration of the dopamine precursor L-DOPA via reverse microdialysis, indicating that dopamine synthesis was impaired, whereas vesicular packaging (by the vesicular monoamine transporter) and release were intact (Felger et al, 2015). Of note, decreased dopamine release during in vivo microdialysis was highly correlated with decreased effort-based consumption of sucrose pellets using a puzzle feeder (Felger et al, 2013b). No effects of IFN-α were found on consumption of sucrose pellets that were freely available in a feeding tray. Of note, mice administered LPS similarly exhibit decreased willingness to expend effort for reward, whereas they exhibit no reduction in the sensitivity to reward (Vichaya et al, 2014). The impact of inflammation on effort-based motivational behavior has also been explored in rats using a model of food deprivation (Nunes et al, 2014). In this model, under usual circumstances, food-deprived rats will choose to work (press a bar) for high carbohydrate pellets, while ignoring standard rat chow. However, administration of IL-1β or IL-6 to food-deprived animals reduces high effort-based choices for high carbohydrate pellets and biases choices for standard rat chow (Nunes et al, 2014; Yohn et al, 2016). Interestingly, administration of dopamine-active drugs including stimulants and adenosine A2A receptor antagonists (that facilitate dopamine receptor signaling) reverses the effects of IL-1β or IL-6 on effort-based choices (Nunes et al, 2014; Yohn et al, 2016).
Several studies have examined mechanisms by which dopamine synthesis might be affected by inflammation. One of the primary hypotheses is that inflammation decreases the availability of tetrahydrobiopterin (BH4). In the context of inflammation, BH4 is rapidly usurped in its role as an essential enzyme cofactor for nitric oxide synthase in the conversion of arginine to nitric oxide, an important contributor to the inflammatory response (Neurauter et al, 2008). Moreover, in the context of inflammation-induced oxidative stress, BH4 is very sensitive to being oxidized to the inactive compound dihydroxyanthopterin (Neurauter et al, 2008). Of relevance to dopamine as well as other monoamines, BH4 also serves as an essential cofactor for the rate-limiting enzymes that synthesize dopamine and serotonin including tyrosine hydroxylase and tryptophan hydroxylase, respectively (Neurauter et al, 2008; Felger and Miller, 2012; Haroon et al, 2012). BH4 is also a cofactor for phenylalanine hydroxylase that converts phenylalanine to tyrosine, the primary precursor for L-DOPA that is converted to dopamine. Studies in patients treated with IFN-α have demonstrated a decreased phenylalanine-to-tyrosine ratio consistent with decreased BH4 activity (Felger et al, 2013a). In addition, the decreased phenylalanine to tyrosine ratio in IFN-α-treated patients was correlated with increased fatigue scores and decreased CSF dopamine (Felger et al, 2013a). Decreased CSF BH4 was also associated with increased CSF IL-6 in these patients. A decreased phenylalanine-to-tyrosine ratio has also been linked with symptoms of depression in other patient populations associated with increased inflammation including patients with cancer (Hufner et al, 2015). Taken together, these data support the use of dopamine-active medications for the treatment of behavioral changes in the context of inflammation. In addition, these data suggest that drugs that activate dopamine receptors (dopamine agonists) or support dopamine synthesis may ultimately be more effective than drugs that simply block dopamine reuptake or block dopamine breakdown (eg, monoamine oxidase inhibitors) that rely on the availability of dopamine to be effective. However, given the current availability of pharmacologic agents with multiple mechanisms of action on dopamine neurotransmission, it is most likely that a combination of these medications may have the greatest likelihood of success.
Glutamate
Another neurotransmitter system that has received recent attention in terms of the effects of inflammation on the brain is glutamate. There is an extensive literature demonstrating that a variety of inflammatory cytokines including TNF, IL-1β, and IFN-γ can reduce the expression of glutamate transporters (excitatory amino acid transporters (EAATs)) on astrocytes and induce astrocytic glutamate release (Tilleux and Hermans, 2007; Ida et al, 2008; Miller et al, 2009). Indeed, TNF in vitro has been shown to activate NF-κB that binds to the EAAT2 promoter, thereby regulating its expression (Sitcheran et al, 2005). Of note, glutamate release from astrocytes has access to extrasynaptic neuronal glutamate receptors that when activated can lead to decreased BDNF and increased excitotoxicity (Hardingham et al, 2002; Hardingham and Bading, 2010). In addition, inflammatory cytokine-induced activation of IDO can lead to the production of quinolinic acid by microglia that can bind to glutamate (N-methyl-D-aspartate (NMDA)) receptors while also stimulating glutamate release from astrocytes (Tavares et al, 2002, 2005; Schwarcz et al, 2012). Indeed, in studies from postmortem tissue of presumably depressed suicide victims, activated microglia expressing quinolinic acid have been described in the anterior cingulate cortex (Steiner et al, 2011). Interestingly, studies in laboratory animals administered LPS have shown that pretreatment with the glutamate antagonist ketamine can reverse LPS-induced depressive-like behavior while having no effect on LPS-induced inflammation in the brain (Walker et al, 2013). It should be noted that hypotheses related to the role of glutamate and the role of inflammation in mood disorders have commanded much attention in the literature. However, increasing data suggest that these two pathophysiologic pathways may be intimately interrelated. For example, patients treated with IFN-α exhibit significant increases in basal ganglia and dorsal anterior cingulate cortex (dACC) glutamate concentrations as measured by magnetic resonance spectroscopy (MRS) (Haroon et al, 2014). Increases in glutamate as measured by MRS were in turn associated with IFN-α-induced depressive symptoms. These findings also appear to apply to patients with major depressive disorder where increases in peripheral blood CRP were associated with a linear and stepwise increase in basal ganglia glutamate (Haroon et al, 2016). Basal ganglia glutamate in depressed patients was in turn associated with measures of anhedonia and psychomotor retardation. Taken together, these data suggest that glutamate may represent an important final common pathway through which inflammation leads to depression. These data also allude to the notion that combinations of drugs that inhibit both inflammation and glutamate may have special relevance to treating mood and other disorders where both pathophysiologic pathways appear to be involved.
Inflammation Effects on Neurocircuitry
Through their effects on neurotransmitter metabolism, it is not surprising that inflammatory stimuli including inflammatory cytokines have been found to have an impact on circuits in the brain that regulate behavior. Much of the attention has been paid to the effects of inflammatory stimuli on the basal ganglia and its role in motivation and motor activity as well as the dACC and its role in arousal, anxiety, and alarm (Miller et al, 2013).
Inflammation effects on reward circuitry
Early studies on the effects of inflammatory stimuli on the brain included data from patients receiving the inflammatory cytokine IFN-α for malignant melanoma and hepatitis C. Initial PET studies revealed decreased metabolic activity in the prefrontal cortex consistent with the reduced glucose metabolism in this brain region commonly seen in patients with major depression (Juengling et al, 2000; Capuron et al, 2007). However, increased glucose metabolic activity was also observed in basal ganglia nuclei including the globus pallidus and putamen. Interestingly, these same areas exhibit increased metabolic activity in Parkinson’s disease, where it is believed to be due to increased oscillatory burst activity in neurons that are normally under tonic inhibition by dopamine (Wichmann and DeLong, 1999). Indeed, administration of L-DOPA has been shown to reduce glucose metabolism in Parkinson’s disease, notably in the putamen, in association with improvement in symptoms (Feigin et al, 2001). Of relevance in this regard, there is a case report of an IFN-α-treated patient who developed Parkinson-like symptoms that were relieved by L-DOPA (Bersano et al, 2008). These data suggest that administration of inflammatory stimuli like IFN-α leads to a state of decreased dopamine function that in turn results in changes in metabolic activity in relevant basal ganglia nuclei similar to that seen in Parkinson’s disease. Of note, increased metabolic activity in the basal ganglia of IFN-α-treated patients was correlated with fatigue (Capuron et al, 2007). More recent studies have used functional magnetic resonance imaging (fMRI) to further characterize the effects of inflammatory stimuli on the basal ganglia. The data indicate that administration of inflammatory cytokines (ie, IFN-α) or inducers of the inflammatory response (eg, typhoid vaccination or endotoxin) lead to decreased neural activity in basal ganglia nuclei, specifically the ventral striatum (nucleus accumbens), in response to reward anticipation or feedback (Brydon et al, 2008; Harrison et al, 2015a, b; Eisenberger et al, 2010a; Capuron et al, 2012; Dowell et al, 2015). Studies have also indicated that not only is neural activity reduced in response to these reward-based tasks, but also there is an increased response to aversive stimuli (ie, loss) (Harrison et al, 2015b). The reduced ventral striatal response to both IFN-α and endotoxin were correlated with depressive symptoms including anhedonia and fatigue as well as acute alterations in striatal microstructure as assessed by quantitative magnetization transfer (qMT) imaging (Eisenberger et al, 2010a; Capuron et al, 2012; Dowell et al, 2015). Of note, administration of typhoid vaccination has also been associated with alterations in neural activity in the substantia nigra (another basal ganglia nuclei) that was correlated with IL-6 and psychomotor slowing (Brydon et al, 2008). Taken together, these data indicate that using a host of neuroimaging paradigms, administration of a variety of inflammatory stimuli lead to consistent changes in brain regions responsible for motivation and motor activity. These findings indicate that these changes are not an idiosyncratic response to one or another inflammatory stimulus (or are a function of a given neuroimaging paradigm) but represent a reliable and reproducible effect of inflammation on the brain. It should also be noted that all of these stimuli were administered peripherally, suggesting that peripheral inflammatory responses have access to the brain and its function.
Further supporting the effects of inflammation on motivation and motor activity is a recent report on patients with major depressive disorder. In this study, increased peripheral blood concentrations of CRP were found to predict decreased functional connectivity between the striatum and the ventromedial prefrontal cortex (Felger et al, 2016). Decreased functional connectivity between ventral regions of the striatum and the ventromedial prefrontal cortex were associated with anhedonia, whereas decreased functional connectivity between dorsal regions of the striatum and the ventromedial prefrontal cortex as well as the supplementary motor area were associated with reduced psychomotor speed. Mediation analysis indicated that increased inflammation as measured by CRP led to anhedonia and psychomotor slowing through its effects on functional connectivity. These data are the first to extend the findings from studies on reward-related circuits in patients administered exogenous inflammatory stimuli to patients with endogenous increases in inflammation, thereby instantiating the relevance of increased inflammation regardless of its origin to changes in motivation and motor activity in patient populations. Finally, it should be noted that the impact of inflammation on motivation and motor activity is believed to fit into a larger evolutionary context whereby reduced motivation and motor activity would subserve the evolutionary priorities of a sick or wounded animal to retreat and withdraw, allowing energy resources to be shunted away from exploratory activities toward fighting infection and wound healing (Raison and Miller, 2013).
Inflammation effects on arousal, anxiety, and alarm
Another brain region that has been consistently associated with the effects of inflammation on behavior is the dACC, a brain region that has been referred to as a ‘neural alarm system’ based on its role in threat detection and activation of downstream arousal pathways. Early studies in IFN-α-treated patients demonstrated increased activation of the dACC during a visuospatial attention task using fMRI that highly correlated with the number of errors made on the task (Capuron et al, 2005). Activation of the dACC during a social rejection task has also been correlated with the degree of stress-induced activation of oral sTNFR2 during a public speaking stressor (Slavich et al, 2010). In addition, increased stress-induced peripheral blood IL-6 was associated with activation of the amygdala, with subjects who exhibited the highest IL-6 responses to the public speaking stressor demonstrating the greatest connectivity within threat circuitry including the amygdala (Muscatell et al, 2015). Of note, in laboratory animals exposed to social defeat stress, the amygdala is a primary site of monocyte trafficking to the brain, indicating a potential mechanism for the impact of the immune system on fear circuits (Wohleb et al, 2013). Performance of a color word Stroop task has also been associated with increased dACC activation following typhoid vaccination compared with placebo (Harrison et al, 2009a, b). Similar to the effects of inflammation on the basal ganglia, the impact of inflammation on circuits in the brain that mediate anxiety, arousal, and alarm fit into an evolutionary framework whereby the vulnerability of a wounded or sick animal is best served by hypervigilance against attack (Raison and Miller, 2013).
GUIDELINES FOR AN INFORMED APPROACH TO CLINICAL TRIALS
A rich database has been elaborated regarding the sources and consequences of immune dysregulation and inflammation and their impact on the brain and behavior as well as neuroendocrine and immune systems in neuropsychiatric diseases especially mood and anxiety disorders (Figure 2). Thus, the field is well positioned to make informed decisions regarding the development and testing of strategies to reverse the impact of the immune system and inflammation on the brain and behavior. Based on this knowledge, a series of principles can be put forward to guide interventional studies as well as drug development (Table 1) (Miller and Raison, 2015). Moreover, these principles can be used as a yardstick to measure the existing data that have examined various ‘anti-inflammatory’ treatments for psychiatric disorders. Unfortunately, as noted below, few studies have taken advantage of these principles, making the extant literature and the many associated meta-analyses almost impossible to interpret.

Causes and consequences of chronic inflammation. A wide array of genetic, developmental, lifestyle, and disease factors conspire to lead to chronic, nonresolving inflammation that has been shown to impact fundamental pathways to psychiatric pathology including the metabolism of neurotransmitters and other neuromodulators as well as neuroendocrine function, ultimately leading to distinct changes in behavior that are rooted in evolutionarily derived responses to illness. Dysregulation of immune (eg, T cell) responses in turn can exacerbate inflammation as well as affect the brain and behavior.
Enrich for Patients Based on Peripheral Immune Markers
Based on the examination of the many studies that have measured inflammatory markers in various populations of psychiatric patients, it is clear that not all patients exhibit evidence of increased inflammation. Moreover, as indicated above, there appears to be a precarious balance that must be maintained between the role of inflammatory cytokines in neuronal integrity and neuropathology (Figure 1). If the purpose of a study is to use an anti-inflammatory agent for example or a drug that targets downstream CNS pathways that are influenced by inflammation, then it would make most sense to enrich the population of patients to be examined with individuals who exhibit increased inflammatory markers. Probably the best marker for increased inflammation at this time is peripheral blood CRP. CRP is a relatively stable protein that shows little, if any, circadian variation and is not immediately influenced by acute stress (Meier-Ewert et al, 2001). Moreover, peripheral blood CRP is an easily obtainable blood test in clinics and hospitals throughout the world, and, as noted above, is reliably increased in depressed and other patient populations, has been associated with functional connectivity between the ventral and dorsal striatum and the medial prefrontal cortex, and has been found to correlate with basal ganglia concentrations of glutamate. In addition, clinical guidelines have established levels of inflammation based on peripheral blood CRP (Ridker, 2003). These guidelines are derived from the relative risk for the development of cardiovascular disease and its complications as well as diabetes. CRP concentrations of <1 mg/l are considered low inflammation, whereas concentrations of 1–3 mg/l are considered moderate inflammation. Peripheral blood CRP concentrations of >3 mg/l are considered high inflammation. Finally, it should be noted that CRP may also be used in the context of disease prevention. Indeed, an increased CRP (>3 mg/l) was found to predict the development of significant depressive symptoms in a cohort of 3397 older adults in the English Longitudinal Study of Aging (Au et al, 2015). Depressive symptoms did not predict increased CRP in this study.
Although it would be most desirable to have a CNS marker of inflammation, the currently available, second-generation TSPO PET ligands have not been studied enough to determine their utility at this time (Vivash and O'Brien, 2016). It should also be noted that at least in studies in depression, there has been no relationship found between peripheral markers of inflammation and degree of TSPO binding (Setiawan et al, 2015). This lack of association of the TSPO brain biomarker with peripheral inflammatory markers is in contradistinction with the relationship of CRP and other cytokines both peripherally and centrally (CSF) with reward circuitry or basal ganglia glutamate in depression (Haroon et al, 2016; Felger et al, 2016). More research is clearly needed in this area.
It is interesting to note that only one study published to date has stratified patients based on high or low inflammation as part of the study design, and no study has exclusively enrolled patients with high inflammation. Of the two studies that have considered baseline inflammation status, both studies used baseline inflammation as a predictor of response in patients with major depressive disorder in post hoc analysis (Raison et al, 2013b; Rapaport et al, 2015). In each case, increased peripheral blood inflammatory markers including CRP at baseline were predictors of response. One study examined the antidepressant effects of the TNF antagonist infliximab, whereas the other study examined the antidepressant effects of omega-3 fatty acid supplements. There are at least four studies on mood disorders underway or in preparation that will consider baseline inflammation in the randomization scheme, and all four studies will only enroll patients with high inflammation (NCT02363738, NCT02553915, NCT02675556, and NCT02473289). Of note, although this strategy will clearly determine whether the noted therapies are effective in patients with high inflammation, unless a ‘match/mismatch’ design is incorporated (including patients with both high and low inflammation), it will remain difficult to discern whether these treatments are broadly effective or only effective in individuals with high inflammation. Such match/mismatch designs are ideal especially for drugs with multiple off-target effects, and require stratification on the basis of high and low inflammation with the hypothesis that only patients with high inflammation will respond. Nevertheless, given the literature as it currently stands, there is limited evidence that blocking inflammation in a psychiatric patient population with increased inflammation leads to statistically significant improvement versus a patient population with low inflammation.
Establish Target Engagement in the Periphery and/or the Brain
Given the extensive literature on the impact of inflammation on various neural circuits and neurotransmitter systems in the brain, there appears to be ample opportunity to use fMRI or MRS to determine whether a given treatment reverses inflammation-induced alterations in reward or threat circuitry or glutamate metabolism as a primary outcome variable. Such a strategy would help establish target engagement in the brain for a variety of treatment approaches (eg, those targeting dopamine or glutamate pathways). As for more direct measures of inflammation in the brain, studies will have to await further development of TSPO ligands, although CSF inflammatory markers may gain wider acceptance as we better understand the lymphatic system of the CNS in humans (Louveau et al, 2015). In any case, peripheral inflammatory markers are well suited for providing evidence that an anti-inflammatory treatment is indeed ‘hitting the target.’ Of relevance in this regard, only two of the many published studies on the use of anti-inflammatory drugs to treat mood disorders or schizophrenia have established that the anti-inflammatory strategy actually reduced inflammation, and both of these found that the decrease in peripheral blood biomarkers (IL-6 in one study and inflammatory gene expression in the other) were associated with treatment response (Abbasi et al, 2012; Mehta et al, 2013). This limited use of peripheral inflammatory markers to confirm that the anti-inflammatory intervention was indeed effective further challenges meaningful interpretation of the literature using anti-inflammatory agents to treat psychiatric disorders.
Pharmacologic Specificity Is a Necessity for Hypothesis Testing
One of the major advantages of identifying the immune system and inflammation as a pathophysiologic pathway leading to neuropsychiatric disease is that there are drugs available that specifically target relevant molecules that mediate the immune response. In the case of inflammation, there are a host of monoclonal antibodies that preferentially bind to single immune molecules including inflammatory cytokines and their receptors as well as adhesion molecules (see below). These biologic agents have revolutionized the treatment of a number of disorders including autoimmune and inflammatory disorders as well as a variety of cancers. Unfortunately, only one study to date has used a biologic agent in patients with a primary psychiatric disorder, although there are at least two studies currently underway or planned as indicated below. In order to truly establish the role of inflammatory molecules in psychiatric disease, it is incumbent on the field to use these biologic agents as part of the effort to establish the hypothesis that the immune system plays a relevant role in certain symptoms and psychopathology. Nevertheless, the majority of studies to date that have examined anti-inflammatory agents in various psychiatric disorders have used drugs that have multiple off-target effects in nonselective patient populations without establishing target engagement even at the level of peripheral inflammatory markers (Kohler et al, 2014). Thus, the multitude of studies using minocycline (an inhibitor of microglial activation that is also an antibiotic with behaviorally relevant microbiome effects (Wong et al, 2016)), cyclooxygenase-2 (COX-2) inhibitors (inhibitors of prostaglandins that also bind to cadherin 11, a modulator of synaptic efficacy (Manabe et al, 2000)), aspirin, or salsalate (both inhibitors of prostaglandins that bind to cadherin 11 have antithrombotic properties and also inhibit reverse efflux of glutamate through the EAAT (De Cristobal et al, 2002)) are difficult if not impossible to interpret. Moreover, drugs that are only partially related to the immune system and inflammation, including N-acetyl-D-cysteine, an antioxidant, omega-3 fatty acids (whose anti-inflammatory status has been only marginally established (Rangel-Huerta et al, 2012)), and pioglitazone, an antidiabetic drug, have been included in meta-analyses of the efficacy of anti-inflammatory drugs for no apparent reason, further complicating the literature in this area (Rosenblat et al, 2016).
Symptom Specificity for Outcome Variables
Based on cytokine effects on the brain and specific behaviors, there are several symptom domains that warrant special focus as outcome variables in studies examining the impact of the immune system and inflammation on the brain. Studies using IFN-α, typhoid vaccination, and endotoxin in humans have all identified motivational circuits and symptoms of anhedonia as being primary targets of inflammatory stimuli on the brain and behavior (Brydon et al, 2008; Harrison et al, 2009a, b, 2015a, b; Eisenberger et al, 2010a; Capuron et al, 2012; Dowell et al, 2015). In addition, studies in patients with depression have also indicated strong relationships between peripheral inflammatory markers (eg, CRP) and changes in reward circuitry as well as alterations in glutamate metabolism in the basal ganglia, both of which were associated with symptoms of anhedonia (Haroon et al, 2016; Felger et al, 2016). These findings in humans are consistent with work in laboratory animals that has shown that motivational processes are affected by the administration of cytokines or cytokine inducers. For example, administration of IL-1β was found to block effort-based reward seeking in food-deprived rats (Nunes et al, 2014). Similar decreases in incentive-based behavior were seen following LPS administration, although in this study, sensitivity to reward remained intact (Vichaya et al, 2014). Taken together, these findings suggest that clinical trials focused on reversing inflammation or its effects on the brain and behavior should include specific assessments of motivation including both objective assessments such as the Effort Expenditure for Rewards Task (EEfRT) and clinical assessments such as the Snaith–Hamilton Pleasure Scale (SHAPS). Of note, these assessments fall under research domain criteria (RDoC) related to positive valence systems (Morris and Cuthbert, 2012; Swardfager et al, 2016).
Another symptom domain closely related to the effects of inflammation on ventral striatal circuits are those that relate to dorsal striatal circuits. More specifically, objective and clinical signs and symptoms related to psychomotor speed have been associated with exposure to inflammatory stimuli and increased inflammatory markers in psychiatric disorders such as depression (Brydon et al, 2008; Majer et al, 2008; Goldsmith et al, 2016). Moreover, psychomotor retardation as measured by the Hamilton Depression Rating Scale (HAMD) was a symptom that was found to be especially responsive to cytokine antagonism in treatment-resistant depressed patients with high inflammation (Raison et al, 2013b). Therefore, neurocognitive assessments of psychomotor performance including the finger tapping task, movement and reaction time tasks, Trails A, and the Digit Symbol Task may serve as additional relevant outcome variables (Goldsmith et al, 2016).
A third symptom domain that may have special relevance to specific outcome variables in treatment trials related to the immune system and inflammation and its effects on the brain falls into the category of negative valence systems (RDoC) involving threat sensitivity and anxiety. For example, stress-induced activation of inflammation and administration of inflammatory cytokines have been found to activate neural circuits relative to the processing of threatening stimuli including the dACC, amygdala, and insula (Capuron et al, 2005; Slavich et al, 2010; Muscatell et al, 2015). Moreover, in patient populations such as women with breast cancer, increased peripheral blood inflammatory markers (eg, CRP) have been correlated with increased activation of the amygdala during a threat sensitivity task (Muscatell et al, 2016). In addition, psychic anxiety on the Hamilton Depression rating scale exhibited a greater decrease in treatment-resistant depressed patients with high inflammation who received the TNF antagonist infliximab versus placebo (Raison et al, 2013b). The importance of anxiety as an outcome variable is also underlined by the multitude of studies demonstrating increased peripheral blood inflammatory markers in patients with a wide variety of anxiety disorders including posttraumatic stress disorder, panic disorder, generalized anxiety disorder, and obsessive–compulsive disorder (see above).
A final symptom domain that has emerged as a potential outcome variable for treatment strategies targeting the immune system and its downstream effects on the brain is sleep. There is a rich literature that has defined a reciprocal relationship between sleep and inflammation with increased inflammatory markers and inflammatory signaling pathways including NF-κB being induced by various forms of sleep deprivation, and administration of inflammatory stimuli being associated with altered sleep parameters (Redwine et al, 2000; Irwin et al, 2008; Raison et al, 2010). For example, in patients administered IFN-α, polysomnographic recordings revealed significant increases in wake after sleep onset and spontaneous arousals as well as decreased sleep efficiency (Raison et al, 2010). Interestingly, although IFN-α-treated patients exhibited marked fatigue, mean sleep latency tests during daytime hours revealed that IFN-α-treated patients took significantly longer to fall asleep during the day than control subjects. Of note, inhibition of TNF by infliximab was associated with decreased wake after sleep onset and spontaneous arousals and increased sleep efficiency in infliximab-treated depressed patients with high (CRP >5 mg/l) versus low inflammation (CRP ⩽5 mg/l), controlling for changes in scores of depression (Weinberger et al, 2015).
In sum, objective and clinical outcome variables including neuroimaging of neurocircuits focused on motivation and motor activity as well as anxiety (eg, RDoC positive and negative valence systems) and sleep will provide the highest probability of detecting a signal in clinical trials focused on targeting the immune system and its downstream effects on the brain. These outcome variables will very likely outperform the more traditional assessments focused on broad spectrum, nonspecific measures of depressive or anxiety symptoms as represented by scales such as the HAMD, the Montgomery Asberg Depression Rating Scale, the Beck Depression Inventory, the Inventory of Depressive Symptoms, the Beck Anxiety Inventory, and others. Of note, no study to date has focused primary outcomes on the above symptom constructs, although studies in schizophrenia, focusing on negative symptoms, have come the closest in this regard. For example, both a meta-analysis and two recent randomized clinical trials have demonstrated that minocycline (see caveats above) was superior to placebo on the PANSS negative subscale scores and the SANS, particularly the avolition measure (Chaudhry et al, 2012; Ghanizadeh et al, 2014; Oya et al, 2014).
TARGETS FOR TRANSLATING BEHAVIORAL IMMUNOLOGY INTO TREATMENT
Given the extensive amount of data that have been collected relevant to the effects of the immune system and its activation on the brain and behavior, there are a number of opportunities for the elaboration of clinical trials using currently available pharmacologic and behavioral strategies that can satisfy many of the guidelines recommended above. One important caveat, however, is that interventions that target the impact of the immune system on the brain cannot be viewed in the same light as conventional antidepressants, antianxiety agents, or anti-schizophrenia agents. The immune system is a pathophysiologic pathway that leads to a series of symptoms described above that cut across diagnoses (transdiagnostic). Therefore, although populations of patients in clinical studies may be recruited based on conventional diagnostic nosology, there should be no misapprehension that the proposed treatments are specific for any disorder, just as inflammation is not specific to any disorder. Indeed, the proposed treatments are relevant for psychiatric or medical disorders where the above-noted symptoms and inflammation coexist. Interestingly, few of the most promising targets have yet to be explored (Table 2 and Figures 3 and 4), and those that have been explored have yielded mixed results, largely because of the failure to use the informed clinical trial guidelines indicated above.

Neurotransmitter targets for therapeutic development in psychiatric disorders. Aside from the more obvious immune targets of therapeutic interventions (eg, cytokines and their receptors, adhesion molecules, and chemokines), there are a host of metabolic and immunologic signaling pathways that mediate the downstream effects of inflammation on neurotransmitter systems including the synthesis, reuptake and release of dopamine, glutamate, and serotonin. Moreover, there is increasing interest in the role of the kynurenine pathway in multiple aspects of the impact of inflammation on excitotoxicity and neurotransmitter metabolism. 5HT: serotonin; 5HTT: serotonin transporter; BH4: tetrahydrobiopterin; DA: dopamine; EAAT: excitatory amino acid transporter; Glu: glutamate; IDO: indoleamine 2,3 dioxygenase; IL: interleukin; KA: kynurenic acid; KAT: kynurenine aminotransferase; KMO: kynurenine 3-monooxygenase; KYN: kynurenine; MAPK: mitogen-activated protein kinase; nAChR: nicotinic acetylcholine receptor; NF-κB: nuclear factor-κB; NMDAR: N-methyl-D-aspartate receptor; Phe: phenylalanine; QA: quinolinic acid; TH: tyrosine hydroxylase; TNF: tumor necrosis factor; TRP: tryptophan; Tyr: tyrosine.

Neuroendocrine and immunomodulatory targets for therapeutic development in psychiatric disorders. Neuroendocrine and immune pathways can serve as additional targets to modulate the immune system leading to neuroprotective and immunoregulatory responses involving the elaboration of growth factors such as BDNF and T-cell subtypes (eg, Tregs and ChAT+ T cells) with anti-inflammatory properties. These targets include administration of glucocorticoids that support trafficking of immunomodulatory, IL-4-producing T cells to the brain as well as electroceutical interventions involving stimulation of the efferent vagus to induce Ach-producing T cells that, via the α7 nAChR, can inhibit NF-κB and TLR-4 expression. Finally, infectious agents and manipulations of the microbiome can foster the development of Tregs that have a multiplicity of anti-inflammatory effects. ACh: acetylcholine; BDNF: brain-derived neurotrophic factor; CD: cluster of differentiation; ChAT: choline acetyltransferase; CTLA: cytotoxic T-lymphocyte-associated protein; GR: glucocorticoid receptor; IFN: interferon; IL: interleukin; M: macrophage; M. vaccae: Mycobacterium vaccae; nAChR: nicotinic acetylcholine receptor; NF-κB: nuclear factor-κB; TGF: transforming growth factor; TLR: Toll-like receptor; Tregs: T regulatory cells.
Ready for Prime Time?
Immunological targets
Monoclonal antibodies. The most obvious prime time immunologic targets for clinical trials are the mediators of inflammation themselves including inflammatory cytokines and adhesion molecules as well as cellular components of the inflammatory response (Table 2). FDA-approved biologic agents include monoclonal antibodies (mAbs) to the cytokines and their receptors TNF, IL-1, IL-6R, and IL-12/23 and the cell adhesion molecule α4-integrin. The mAbs to IL-17 have also been recently been approved for use in the United States. FDA-approved biologic agents are also available that block B-cell activation. These drugs are generally approved for use in autoimmune and inflammatory disorders including rheumatoid arthritis, ulcerative colitis and Crohn’s disease, psoriasis, multiple sclerosis, and ankylosing spondylitis. Biologic agents have a substantial advantage over the majority of anti-inflammatory agents because of their potent anti-inflammatory activity, specificity, and lack of off-target effects as noted above. Based on these qualities, mAbs to inflammatory mediators represent a high-value pharmacologic strategy for future studies investigating and treating the impact of the immune system and inflammation on the brain and behavior.
Given data demonstrating the reliable increases in TNF, IL-1β, and IL-6 in depression and other psychiatric disorders, of the currently available monoclonal antibodies, those targeting these cytokines are the best positioned for application in hypothesis-testing studies and in clinical trials. Moreover, the recent and pending availability of biosimilars (biologic generics) in the United States will reduce the cost of these agents. As revealed in a recent meta-analysis of the literature, mAbs against TNF as well as IL-12/23 have already shown antidepressant activity in nondepressed individuals with psoriasis in several large studies (Kohler et al, 2014). One clinical trial in treatment-resistant depression has also revealed that mAbs to TNF (infliximab) appear to be effective in treatment-resistant depressed patients but only in those with increased inflammation (Raison et al, 2013b). Following up on this study are clinical trials examining the efficacy of infliximab in patients with bipolar depression and a CRP of >5 mg/l (NCT02363738) and a phase II trial of an anti-IL-6 mAb (sirukumab) in patients with depression and a CRP of >3 mg/l (NCT02473289). These trials will likely yield important data, given that they will be the first anticytokine treatment studies solely focused on depressed patients with increased inflammation. Nevertheless, the primary end point of these studies (ie, decrease in MADRS or HAMD17, respectively) is nonspecific relative to what is known about the effects of the immune system on the brain, and therefore these studies run the risk of having the primary behavioral targets of the treatment being overshadowed by nonspecific symptoms that may improve equally well with placebo. Moreover, there is no measure of whether the treatment has engaged relevant neurocircuits, neurotransmitter systems, or cell types in the brain (ie, target engagement), and hence a failed trial may be difficult to interpret.
Of note, there are many caveats with mAb treatments, notably the risk of infection and reactivation of tuberculosis as well as risk for certain neoplastic diseases and demyelinating conditions (Kopylov et al, 2015; Singh et al, 2015). Moreover, for some biologics, neutralizing antibodies can undermine successful therapy over time. One issue that stands out is the tremendous lack of data investigating the immunologic processes that may drive psychiatric disorders. Whether the cytokines of interest are primarily derived by T or B cells or monocytes is unknown, and the relative distribution of cytokines (skewed to Th17, Th1, Th2, M1 or M2) across patient groups or longitudinally in a given group has not been established. Moreover, although animal studies have emphasized the importance of immune cells trafficking to the brain, it remains unclear what roles adhesion molecules (for which there are mAbs) are playing in this process. Meta-analyses of immune markers have been conducted in many patient populations, but these studies do not provide systematic, mechanistic immunologic data within any one group, and therefore which cytokine targets are of most relevance awaits further study. Finally, it should be noted that aside from inhibiting inflammatory cytokines using mAbs, there has been recent development of technology to administer anti-inflammatory cytokines. For example, XT-101 is a plasmid DNA encoding human IL-10 that is being developed in clinical trials for the treatment of pain (Kwilasz et al, 2015).
Microglia. There has been increasing interest in the use of various pharmacologic agents that can reduce microglial activation in the CNS. Drugs such as minocycline, a broad-spectrum tetracycline antibiotic that effectively crosses the BBB, have been shown to have potent anti-inflammatory and neuroprotective effects in laboratory animal models of neurodegenerative disorders as well as following stress or LPS (Henry et al, 2008; Kreisel et al, 2014). Unfortunately, unlike the biologic agents that target peripheral inflammatory responses (where target engagement can be established in the periphery), microglial activation has proven difficult to assess in humans, even with currently available second-generation TSPO ligands, the exception being following administration of endotoxin (see above). Another challenge for minocycline is its effects on the microbiome that has been shown to affect mood regulation (Wong et al, 2016; Zheng et al, 2016). Being an antibiotic, minocycline will have significant effects on the relative distribution of bacteria in many areas of the body. How these ‘indirect’ effects on the microbiome might influence the immune system remains unclear and will undoubtedly complicate interpretation of results. Indeed, recent data suggest that treatment of mice with an antibiotic cocktail that disrupts the gut microflora leads to reduced neurogenesis and reduced performance on the novel object recognition task (Mohle et al, 2016). These effects were mediated by antibiotic-induced loss of interactions between monocytes and the gut microflora and could be restored by adoptive transfer of monocytes after antibiotic treatment. These data emphasize the importance of the gut microbiome to normal CNS function via its effects on circulating immune cells, and lend caution to the use of antibiotics for the treatment of neuropsychiatric disorders (Mohle et al, 2016). Finally, it should be noted that data from laboratory animals exposed to 5 weeks of chronic unpredictable stress exhibited decreased microglia in the hippocampus and prefrontal cortex in association with decreased sucrose preference and social exploration (Kreisel et al, 2014). Interestingly, microglial activation in these chronically stressed animals using LPS was found to reverse these depressive-like behaviors, suggesting that microglial changes during stress are dynamic (see above) and activating microglia in the context of chronic stress may actually have antidepressant effects. Based on these considerations, until more work has established the validity of the current strategies for measuring microglial activation in the brain, the utility and interpretability of studies using minocycline and other potential therapeutic agents that act primarily through reducing (or increasing) microglial activation is limited.
Inflammatory signaling pathways. Inhibitors of COX-2 have been some of the most commonly used anti-inflammatory agents in clinical trials of psychiatric disorders, primarily depression, and schizophrenia. COX-2 inhibitors block the production of prostaglandins that are primary downstream mediators of the inflammatory response and have been shown to be increased in the peripheral blood of patients with depression (Lieb et al, 1983). The number of studies of COX-2 inhibitors in depression is relatively small (n=5, excluding acetylsalicylic acid), despite the fact that no less than 3 meta-analyses of these studies have appeared in the literature (threatening to outnumber the number of primary studies themselves; Faridhosseini et al, 2014; Kohler et al, 2014; Na et al, 2014). The largest of these studies was a randomized, placebo-controlled preventative trial that included over 4000 patients and showed no advantage of the COX-2 inhibitor celecoxib over placebo in preventing the development of depression in an elderly cohort of subjects (Fields et al, 2012). Of the 4 remaining trials, all were small (~n=20 per group) and all used celecoxib versus placebo as an add-on to a conventional antidepressant in patients with major depression. The 4 studies showed a positive benefit for celecoxib at some point in the trial, although the one study conducted in Germany was plagued by a dropout rate of 50% in the celecoxib group and 60% in the placebo group (Muller et al, 2006), and of the remaining 3 studies, all of which were conducted in Iran, one showed no effect at 8 weeks (Hashemian et al, 2011); yet, for unclear reasons, in each meta-analysis, the more positive 4-week data from this study (that appear in the literature as an abstract) are included in the efficacy evaluation (Muller et al, 2006; Akhondzadeh et al, 2009; Hashemian et al, 2011; Abbasi et al, 2012). Of note, the Iranian group has also published positive data for celecoxib in OCD, acute bipolar mania, and depression in breast cancer, indicating an extraordinary efficacy that warrants further evaluation (Arabzadeh et al, 2015; Mohammadinejad et al, 2015; Shalbafan et al, 2015). Finally, there are some data to support that acetylsalicylic acid may augment the response to conventional antidepressant therapy, although this is a small literature that is in need of larger studies (Mendlewicz et al, 2006). Of note, a larger study has been completed with results pending (NCT01429272). Taken together, the findings with COX-2 inhibitors are far from convincing, and further trials from a more diverse group of investigators are needed. Moreover, COX-2 inhibitors present more challenges with off-target effects than mAbs as noted above. Nevertheless, given the option to measure target engagement in the periphery with COX-2 inhibitors as well as acetylsalicylic acid (and salsalate), further data from these anti-inflammatory strategies may yield more clearcut data if informed designs are employed.
Although the evidence presented above indicates that drugs that target p38 MAPK, NF-κB, and the inflammasome might be useful for the inhibition of inflammatory responses and their effects on behavior, these drugs are not at a sufficient stage of development to warrant consideration as ready for application to psychiatric disorders now or in the near future. p38 MAPK inhibitors are the closest in this regard with multiple ongoing trials for asthma, rheumatoid arthritis, and cardiovascular disease, and one negative study in depression. The same could be said of drugs that block oxidative stress (eg, N-acetyl cysteine). Given that there are many sources of oxidative stress (other than inflammation), it seems somewhat overinclusive to consider drugs that target oxidative stress in the absence of inflammation as relevant to the effects of inflammation on the brain and behavior. Therefore, oxidative stress should be considered a downstream target of inflammatory processes that warrants consideration only in concert with inflammation. Of interest however is the ability to measure oxidative balance in the CNS using MRS assessments of glutathione (GSH). Although there are data suggesting an inverse relationship between GSH and anhedonia in depressed patients and controls, no data on GSH levels in the brain as they relate to markers of inflammation are available (Lapidus et al, 2014). Further evaluation of the utility of GSH as a marker of the downstream effects of inflammation on the brain await further development, and will facilitate the testing of drugs that target oxidative stress as viable treatments for the effects of inflammation on the brain.
Neurotransmitter targets
As indicated in Figure 3, there are a number of therapeutic targets that address the downstream effects of the immune system and inflammation on neurotransmitter metabolism.
Dopamine. As described above, data in laboratory animals and humans indicate that dopamine is a primary target of the effects of inflammation on the brain. These findings have been manifested by alterations in dopamine metabolism following the administration of inflammatory stimuli as well as the presence of decreased effort-based motivation for reward (Kitagami et al, 2003; Felger et al, 2013b, 2015). In addition, increased peripheral CRP in patients with major depression has been associated with symptoms of anhedonia as well as psychomotor retardation that is mediated by inflammation-induced alterations in functional connectivity between ventral and dorsal striatum and ventromedial prefrontal cortex (Felger et al, 2016). Reversal of cytokine-induced alterations in effort-based motivation in laboratory animals by pharmacologic agents that increase dopaminergic signaling further support the role of dopamine in mediating the impact of inflammation on reward-related behaviors (Nunes et al, 2014). Taken together, these data make a strong case for pharmacologic agents that increase dopamine function through increased dopamine synthesis or release, decreased dopamine reuptake or breakdown, and increased dopamine receptor signaling. Relevance to target engagement in the brain, these pharmacologic strategies alone or in combination would ostensibly reverse inflammation-induced decreases in functional connectivity between striatum and prefrontal cortex as well as reverse objective measures of effort-based reward and measures of psychomotor slowing. Relevant pharmacologic agents include dopamine agonists, stimulants, bupropion, monoamine oxidase-B inhibitors, adenosine A2A receptor antagonists, and drugs that support dopamine synthesis through supplementation of BH4 including saproterin and folic acid, L-methylfolate, and S-adenosylmethionine (SAMe) that help convert the inactive BH2 to BH4. Clinical trials have been conducted using L-methylfolate and SAMe in depression with mixed results (Papakostas et al, 2012; Sarris et al, 2015). Interestingly, however, a recent study using L-methylfolate in patients with an inadequate response to antidepressants demonstrated in a post hoc analysis that combinations of elevated CRP, TNF, IL-6, and IL-8 along with BMI predicted an improved antidepressant response to L-methylfolate treatment (Shelton et al, 2015). Stratified analyses based on inflammatory biomarkers have not been conducted with SAMe. These data support the value of enriching patient populations for targeted analyses, while also indicating that treatments that can increase BH4 (and potentially dopamine synthesis) may have efficacy in the treatment of depression in patients with increased biomarkers of inflammation. Nevertheless, although promising, adding measures of target engagement in the brain as well as outcome variables related to motivation and motor activity will significantly strengthen the ultimate interpretation of these data.
Glutamate. Another emerging neurotransmitter target relative to the effects of inflammation on the brain is glutamate. As noted previously, inflammatory cytokines can have marked effects on glutamate metabolism, especially in the basal ganglia as documented by MRS (Haroon et al, 2014, 2016). Nevertheless, although there are drugs currently available and in development that block glutamate receptors (eg, memantine) or influence glutamate reuptake (eg, riluzole), there have been no studies examining the ability of glutamate antagonists to reverse inflammation-induced alterations in the CNS and the associated changes in motivation and motor activity in patients with depression or other psychiatric disorders. One study in rodents demonstrated that the NMDA antagonist ketamine was able to reverse LPS-induced depressive-like behavior including sucrose preference, a measure of anhedonia (Walker et al, 2013). Ketamine had no effect on LPS-induced inflammation in the brain or CNS activation of IDO or decrease in BDNF expression. Moreover, blockade of AMPA receptors was able to reverse ketamine’s effects on LPS-induced depressive-like behavior, indicating that the effects of ketamine were specific to its impact on glutamate signaling. Interestingly, a recent study demonstrated that serum concentrations of TNF, IL-1β, and IL-6 were significantly higher in treatment-resistant depressed patients who responded to ketamine versus nonresponders (Fan et al, 2015). Similarly, in a laboratory animal model of treatment-resistant depression, response to ketamine was also associated with increased measures of inflammation (Walker et al, 2015). These findings are consistent with the notion that drugs that impact glutamate signaling may be especially relevant in patients with increased inflammatory markers. Taken together, these data indicate that glutamate is a high-value target for reversing the effects of inflammation on the brain and behavior.
IDO and the kynurenine pathway. Laboratory animal studies have provided a rich array of experiments that demonstrate the role of IDO and its activation of the kynurenine pathway (KP) in the development of depressive symptoms in the context of a variety of immune stimuli. Studies in mice exposed to LPS as well as Bacille Calmette–Guerin (BCG) have demonstrated that inhibition of IDO by 1 methyltryptophan (1 MT) completely reversed increases in the kynurenine/tryptophan ratio and reversed depressive- and anxiety-like behaviors including sucrose preference, whereas it had no effect on inflammatory markers in the brain (O'Connor et al, 2009; Salazar et al, 2012). Although much attention has been paid to the two primary pathways downstream of IDO leading to quinolinic acid (the ‘neurotoxic pathway’) and kynurenic acid (the ‘neuroprotective pathway’), data suggest that this distinction may be too simplistic, and it is very apparent that both pathways can contribute to pathology. In support of this notion, it should be mentioned that both quinolinic acid and kynurenic acid were significantly elevated in the CSF of IFN-α-treated individuals (Raison et al, 2010). Moreover, in an elegant series of studies using animals with the neurotoxic pathway disabled through genetic deletion of kynurenine 3-monooyxgenase or 3-hydroxyanthranilic acid dioxygenase, decreased sucrose preference following LPS persisted and was accompanied by significant increases of kynurenic acid in the nucleus accumbens (Parrott et al, 2016). These data are consistent with previous studies demonstrating that direct administration of kynurenic acid into the rat striatum was associated with substantial reductions in striatal dopamine (Wu et al, 2007). These effects of kynurenic acid have been shown to be mediated by antagonism of the α7 nicotinic receptor that ultimately leads to reduced glutamate-stimulated dopamine release (Wu et al, 2007). Given the association of sucrose preference with motivational pathways that involve dopamine and the nucleus accumbens, these data suggest that although the neurotoxic pathway may have effects on some inflammation-related behaviors and contribute to neurodegeneration through glutamate signaling, the effects of inflammation on motivation (and potentially other behaviors related to frontostriatal activity including cognitive function and psychomotor performance) may be mediated by kynurenic acid and the so-called ‘neuroprotective pathway’ and its effects on dopamine. Consistent with this notion, data in bipolar patients have indicated that increased CSF kynurenic acid is associated with a genetic variant that is linked to cognitive dysfunction as well as IL-1β induction and subsequent activation of the KP in postmortem brain tissue (Sellgren et al, 2015). Given recent interest in the development of α7 receptor nicotinic receptor agonists for treatment of cognitive deficits and negative symptoms in schizophrenia, it is intriguing to note that early data suggested positive effects of the α7 receptor nicotinic receptor agonist TC-5619 on avolition and apathy as well as cognition in schizophrenia (Lieberman et al, 2013). However, these effects were not replicated in a subsequent large multisite clinical trial, although no attempt was made to consider responders versus nonresponders as a function of inflammation or other mechanistic pathways (eg, the KP) (Walling et al, 2016). Whether α7 receptor nicotinic receptor agonists should be considered exclusively in patients with high inflammation and anhedonia and/or cognitive dysfunction remains an open question worthy of further exploration. It should also be noted that the neuroprotective effects of kynurenic acid have been well established in neurodegenerative disorders (O'Farrell and Harkin, 2015), and therefore drugs that block kynurenic acid (as opposed to stimulating the α7 receptor nicotinic receptor) should be considered with caution.
Neuroendocrine targets
Based on the role of stress-induced catecholamines in activating microglia (and other cell types), while also mobilizing immune cells from the bone marrow to the peripheral circulation, adrenergic antagonists may play an important role early in the stress response by reducing inflammatory responses (Wohleb et al, 2011, 2014). In addition, given the potent anti-inflammatory effects of glucocorticoids coupled with their role in supporting the trafficking of potentially protective T cells to the brain during acute stress, glucocorticoid agents may have a place in inhibiting stress-induced inflammatory responses before they evolve (Rhen and Cidlowski, 2005; Lewitus et al, 2008) (Figure 4). As noted previously, individuals with a genetic predisposition (eg, via FKBP5 polymorphisms) to the consequences of stressful life events including both early-life stress and adult trauma may be especially vulnerable to the inflammatory effects of acute stress by virtue of dysfunction of the glucocorticoid receptor (GR) (Klengel et al, 2013). Of note, there is a rich literature demonstrating the negative impact of cytokines on GR function both in vitro and in vivo, including disrupting the translocation of the GR from the cytoplasm to nucleus as well as through inhibitory protein–protein interactions in the nucleus (Pace et al, 2007). These effects of cytokines on GR function have been shown to be mediated by p38 MAPK that, as noted above, can also affect serotonin transporter expression (Wang et al, 2004). Studies have been conducted and are underway using both adrenergic antagonists and synthetic glucocorticoids in the context of acute trauma to prevent development of PTSD (Amos et al, 2014). Nevertheless, although increased inflammatory markers including CRP have been shown to predict the subsequent development of PTSD (Eraly et al, 2014; Michopoulos et al, 2015), no studies have determined whether those individuals with increased inflammatory markers are the ones who might be most likely to respond to adrenergic or glucocorticoid agonists. Moreover, whether the success of these interventions is in part due to inhibition of stress-induced inflammatory responses or even through glucocorticoid-mediated trafficking of neuroprotective T cells to the brain has yet to be established. Interestingly, recent laboratory animal data suggest that blockade of IL-1 can reduce stress-enhanced fear learning in an animal model of PTSD (Jones et al, 2015). Clearly, more work on the role of inflammatory pathways in the development and treatment of fear and anxiety-related disorders in the context of trauma is warranted.
Another neuroendocrine pathway that has received increasing attention, especially as it relates to drug development, is the cholinergic anti-inflammatory pathway (Figure 4) (Tracey, 2009). As part of the autonomic nervous system, it is known that the parasympathetic nervous system and related vagal tone play an important role in the maintenance of a variety of homeostatic functions that occur largely at rest including digestion, urination, and salivation, as well as sexual arousal. Interestingly, recent data indicate that vagal tone also regulates innate immune system responses through the activation of CD4+ T cells that have the capacity to produce and release acetylcholine (Rosas-Ballina et al, 2011). Relevant to innate immune regulation and inflammation, acetylcholine is capable of binding to the α7 nicotinic acetylcholine receptor on macrophages and other cell types leading to inhibition of NF-κB, a process that involves microRNA-124 (Sun et al, 2013). In addition, α7 nicotinic receptor signaling can inhibit inflammasome activation in macrophages by preventing the release of mitochondrial DNA, a DAMP and thus a NLRP3 ligand (Lu et al, 2014). Of note, adoptive transfer of vagally activated CD4+ T cells to nude mice (that are devoid of T cells) led to reduced endotoxin-induced TNF responses in these animals (Rosas-Ballina et al, 2011). In terms of the translational implications of these findings, recent work indicates that galantamine, an allosteric potentiating ligand of the α7 nicotinic acetylcholine receptor, can reduce the expression of diabetes through suppression of inflammatory cytokine responses (Hanes et al, 2015), and afferent stimulation of the vagus nerve reduces the inflammatory cytokine response to endotoxin (Borovikova et al, 2000). Clinical trials are currently underway to test whether afferent vagal nerve electrostimulation (in contrast to the efferent nerve stimulation currently used in depression) can treat inflammatory disorders including Crohn’s disease (NCT01569503). Given the role of α7 nicotinic acetylcholine receptor agonists in both reversing the effects of kynurenic acid in the brain (see above) and potentially inhibiting NF-κB in the periphery, such drugs might have special relevance for reversing the CNS and peripheral effects of inflammation on behavior (Figure 4).
Bolstering resilience
Although early in development, an alternative strategy to directly targeting inflammation or its downstream impact on neurotransmitter systems and neurocircuits in the brain through pharmaceutical or electroceutical means is to take advantage of the immunoregulatory and neuroregulatory properties of immune cells themselves. As described above, T cells can be attracted to the brain through immunization with CNS antigens where they can promote the release of growth factors and limit M1 inflammatory responses. Nevertheless, the risk of inducing a more fulminant immune response to CNS antigens remains a concern with this approach. Another strategy is to stimulate the production of IL-4-producing T cells as was accomplished in pregnant damns using administration of helminths (Figure 4). Helminth infection has also been shown to promote anti-inflammatory responses involving IL-4 and M2 macrophages in the gut via effects on the microbiome in a mouse model of inflammatory bowel disease (Ramanan et al, 2016). Relevant in this regard, studies are exploring this immunomodulatory strategy using both helminth ova and hookworm larvae in separate studies to treat relapsing and remitting multiple sclerosis (NCT00645749 and NCT01470521, respectively). Enhancement of Treg function is another strategy that can promote innate immune system regulation. One approach has involved the administration of certain bacteria such as Mycobacterium vaccae or parasites that stimulate Treg production and function (Figure 4) (Zuany-Amorim et al, 2002). For example, immunization with M. vaccae was found to reduce anxiety-like and fear behaviors in mice exposed to chronic psychosocial stress, an effect that could be eliminated by depletion of Tregs (Reber et al, 2016). Given that inflammatory cytokines can disrupt T-cell signaling and development, an additional benefit of certain anti-inflammatory strategies including anti-TNF drugs is an increase in the number and function of Tregs that has been seen in rheumatoid arthritis (Bayry et al, 2007). As described above, afferent vagal nerve stimulation also has been shown to promote the development of T cells that express choline acetyltransferase and produce the anti-inflammatory acetylcholine. Finally, an emerging field of immunomodulatory treatments involves stem cell therapies. For example, mesenchymal stem cells (MSCs) have been shown to promote the development and differentiation of Tregs, while also releasing a number of immunomodulatory factors including TGF-β, IL-10, and IL-1ra that can inhibit inflammation (Shi et al, 2011; Francois et al, 2012). Several clinical trials are underway using MSCs in inflammatory and autoimmune disorders, and one clinical trial is underway examining the impact of a single infusion of allogeneic human MSCs for patients with treatment-resistant depression and increased inflammation (CRP>3 mg/l) (NCT02675556).
Diet, supplements, and behavioral strategies
Although the targeting of specific immunologic mechanisms becomes more complex in the context of lifestyle factors and supplements, there are sufficient data to consider these interventions as relevant to the prevention and treatment of neuropsychiatric disorders that occur in the context of immune dysregulation including inflammation. The primary caveat however is that these interventions likely interact with the nervous and immune systems at multiple levels, therefore making it difficult to isolate the ‘active ingredient’ of their efficacy. There is a rich literature demonstrating the effects of exercise, weight reduction, certain diets, as well as supplements such as omega-3 fatty acids, yoga, massage, tai chi, cognitive behavioral therapy, and meditation in reducing psychiatric symptoms such as depression and anxiety. Moreover, many of these interventions have been shown to induce a variety of immune changes including a reduction in inflammation (Bower and Irwin, 2016). For example, mindfulness meditation has been shown to increase functional connectivity between the posterior cingulate cortex and the left dorsolateral prefrontal cortex that in turn was associated with decreases in IL-6 over a 4-month period (Creswell et al, 2016). In addition, both cognitive behavioral therapy and tai chi were associated with reduced levels of CRP, monocyte production of inflammatory cytokines, and inflammatory gene expression in elderly patients with insomnia (Irwin et al, 2015). A hatha yoga program was also shown to reduce LPS-induced peripheral blood mononuclear cell production of TNF, IL-6, and IL-1β, as well as fatigue, at 3 months in breast cancer survivors (Kiecolt-Glaser et al, 2014). Nevertheless, the data that the efficacy of these interventions is dependent on their demonstrated effects on the immune system are generally lacking.
Two of the most studied interventions include dietary weight loss and exercise. Both of these interventions have been shown to have antidepressant and antianxiety effects (Fabricatore et al, 2011; Schuch et al, 2016). Moreover, both of these interventions have been shown to reduce a variety of inflammatory markers in longitudinal studies (Forsythe et al, 2008; Woods et al, 2009). More recent research using measures of body composition have suggested that the amount of weight loss during exercise is the most significant factor in reducing inflammation, whereas other studies using BMI concluded that the effects of exercise are independent of weight reduction (Woods et al, 2009; Fisher et al, 2011). Nevertheless, the relationship among weight loss or exercise, inflammation, and depression or anxiety is only beginning to be established. For example, although a 16-week exercise program was shown to reduce peripheral blood concentrations of CRP in a sample of 319 sedentary women, changes in inflammation, which were especially pronounced in obese exercisers, were not associated with improvements in mood (Arikawa et al, 2011). Nevertheless, it should be noted that these women were not clinically depressed. In a study of depressed patients who had exhibited a partial response to a SSRI and were randomly assigned to two levels of exercise, not only did baseline TNF predict the response to the exercise intervention, but a significant correlation was also found between the decrease in IL-1β and the improvement in depression severity controlling for BMI (Rethorst et al, 2013). Body composition was not measured. Clearly, more research is needed in this area. Of interest, data indicate that exercise can activate kynurenine aminotransferase (KAT) in skeletal muscle in both laboratory animals and humans via a peroxisome proliferator-activated γ coactivator-1α1 (PGC-1α1)-dependent pathway (Agudelo et al, 2014). KAT in turn can convert kynurenine into kynurenic acid that is unable to cross the BBB. Mice genetically engineered to overexpress PGC-1α1 were found to be resistant to the depressive effects of chronic mild stress or direct kynurenine administration, effects mediated by their ability to reduce plasma kynurenine through KAT activation (Agudelo et al, 2014). These findings suggest that in addition to reducing inflammation and symptoms of depression, exercise may be able to limit the effects of inflammation-induced activation of IDO and the KP though stimulation of PGC-1α1 in muscle tissue.
The impact of the vast array of dietary supplements including curcumin, resveratrol, and ginger supplementation on the immune system and inflammation is beyond the scope of this review; however, one dietary supplement has considerably more data than the others. Omega-3 fatty acids have been extensively studied as antidepressants, with meta-analyses demonstrating significant antidepressant efficacy especially for eicosapentaenoic acid (EPA) (Mocking et al, 2016). Relevant to the immune system, in a recent study it was demonstrated that patients with a compilation of inflammatory markers including increased CRP and IL-1ra had a greater antidepressant response to EPA, whereas individuals with increased CRP and IL-6 were less responsive to placebo (Rapaport et al, 2015). These data extend the current literature on the antidepressant effects of omega-3 fatty acids to potentially include an anti-inflammatory component that is currently under investigation (see NCT02553915). Although there are some in vitro and in vivo data supporting anti-inflammatory effects of omega-3 fatty acids in humans (Rangel-Huerta et al, 2012), data linking these effects to their impact on behavior including depression are warranted.
SUMMARY
Based on the rich and rapidly developing literature on the impact of the immune system and inflammation on the brain and behavior, there are a number of high-value targets that warrant further drug development and clinical testing. Nevertheless, there are also a number of guidelines that will facilitate this research including the enriching of patient populations for study, the establishment of target engagement, the use of relevant behavioral end points, and the focus on drugs with specific mechanisms of action. A number of trials that embrace at least some of these features are underway, and should reveal more information regarding the relevance of the immune system as a novel treatment target that holds promise for patients with a wide variety of psychiatric disorders and evidence of increased inflammation.
FUNDING AND DISCLOSURE
In the past 3 years, JCF has consulted for Proctor and Gamble and Pfizer. No funding or sponsorship was provided by these companies for the current work, and all views expressed herein are solely those of the authors. The authors declare no conflict of interest.
References
Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S (2012). Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord 141: 308–314.
Agudelo LZ, Femenia T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V et al (2014). Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159: 33–45.
Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B et al (2009). Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety 26: 607–611.
Alcocer-Gomez E, de Miguel M, Casas-Barquero N, Nunez-Vasco J, Sanchez-Alcazar JA, Fernandez-Rodriguez A et al (2014). NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun 36: 111–117.
Amos T, Stein DJ, Ipser JC (2014). Pharmacological interventions for preventing post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev 7: Cd006239.
Anisman H, Merali Z, Hayley S (2008). Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol 85: 1–74.
Arabzadeh S, Ameli N, Zeinoddini A, Rezaei F, Farokhnia M, Mohammadinejad P et al (2015). Celecoxib adjunctive therapy for acute bipolar mania: a randomized, double-blind, placebo-controlled trial. Bipolar Disord 17: 606–614.
Arikawa AY, Thomas W, Schmitz KH, Kurzer MS (2011). Sixteen weeks of exercise reduces C-reactive protein levels in young women. Med Sci Sports Exerc 43: 1002–1009.
Au B, Smith KJ, Gariepy G, Schmitz N (2015). The longitudinal associations between C-reactive protein and depressive symptoms: evidence from the English Longitudinal Study of Ageing (ELSA). Int J Geriatr Psychiatry 30: 976–984.
Banks WA (2015). The blood-brain barrier in neuroimmunology: tales of separation and assimilation. Brain Behav Immun 44: 1–8.
Bayry J, Siberil S, Triebel F, Tough DF, Kaveri SV (2007). Rescuing CD4+CD25+ regulatory T-cell functions in rheumatoid arthritis by cytokine-targeted monoclonal antibody therapy. Drug Discov Today 12: 548–552.
Beattie E, Stellwagen D, Morishita W, Bresnahan J, Ha B, Von Zastrow M et al (2002). Control of synaptic strength by glial TNFalpha. Science 295: 2282–2285.
Ben Menachem-Zidon O, Avital A, Ben-Menahem Y, Goshen I, Kreisel T, Shmueli EM et al (2011). Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav Immun 25: 1008–1016.
Ben Menachem-Zidon O, Goshen I, Kreisel T, Ben Menahem Y, Reinhartz E, Ben Hur T et al (2008). Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 33: 2251–2262.
Bersano A, Aghemo A, Rumi MG, Ballabio E, Candelise L, Colombo M (2008). Recovery after L-DOPA treatment in peginterferon and ribavirin induced parkinsonism. Eur J Intern Med 19: 370–371.
Besedovsky HO, del Rey A (1996). Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev 17: 64–102.
Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR et al (2016). Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [(11)C]PBR28 PET brain imaging study. Am J Psychiatry 173: 44–52.
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR et al (2000). Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405: 458–462.
Bower JE, Irwin MR (2016). Mind-body therapies and control of inflammatory biology: a descriptive review. Brain Behav Immun 51: 1–11.
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD (2008). Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry 63: 1022–1029.
Bufalino C, Hepgul N, Aguglia E, Pariante CM (2012). The role of immune genes in the association between depression and inflammation: a review of recent clinical studies. Brain Behav Immun 31: 31–47.
Capuron L, Fornwalt FB, Knight BT, Harvey PD, Ninan PT, Miller AH (2009). Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord 119: 181–185.
Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB et al (2002). Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26: 643–652.
Capuron L, Miller AH (2004). Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry 56: 819–824.
Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS et al (2005). Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry 58: 190–196.
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B et al (2007). Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 32: 2384–2392.
Capuron L, Pagnoni G, Drake D, Woolwine B, Spivey J, Crowe R et al (2012). Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 69: 1044–1053.
Chaudhry IB, Hallak J, Husain N, Minhas F, Stirling J, Richardson P et al (2012). Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol 26: 1185–1193.
Chen Y, Jiang T, Chen P, Ouyang J, Xu G, Zeng Z et al (2011). Emerging tendency towards autoimmune process in major depressive patients: a novel insight from Th17 cells. Psychiatry Res 188: 224–230.
Clark SM, Pocivavsek A, Nicholson JD, Notarangelo FM, Langenberg P, McMahon RP et al (2016). Reduced kynurenine pathway metabolism and cytokine expression in the prefrontal cortex of depressed individuals. J Psychiatry Neurosci 41: 150226.
Cole SW, Capitanio JP, Chun K, Arevalo JM, Ma J, Cacioppo JT (2015). Myeloid differentiation architecture of leukocyte transcriptome dynamics in perceived social isolation. Proc Natl Acad Sci USA 112: 15142–15147.
Constant A, Castera L, Dantzer R, Couzigou P, de Ledinghen V, Demotes-Mainard J et al (2005). Mood alterations during interferon-alfa therapy in patients with chronic hepatitis C: evidence for an overlap between manic/hypomanic and depressive symptoms. J Clin Psychiatry 66: 1050–1057.
Coughlin JM, Wang Y, Ambinder EB, Ward RE, Minn I, Vranesic M et al (2016). In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [(11)C]DPA-713 PET and analysis of CSF and plasma. Transl Psychiatry 6: e777.
Cousins MS, Salamone JD (1994). Nucleus accumbens dopamine depletions in rats affect relative response allocation in a novel cost/benefit procedure. Pharmacol Biochem Behav 49: 85–91.
Creswell JD, Taren AA, Lindsay EK, Greco CM, Gianaros PJ, Fairgrieve A et al (2016). Alterations in resting-state functional connectivity link mindfulness meditation with reduced interleukin-6: a randomized controlled trial. Biol Psychiatry 80: 53–61.
D'Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102.
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008). Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65: 409–415.
De Cristobal J, Cardenas A, Lizasoain I, Leza JC, Fernandez-Tome P, Lorenzo P et al (2002). Inhibition of glutamate release via recovery of ATP levels accounts for a neuroprotective effect of aspirin in rat cortical neurons exposed to oxygen-glucose deprivation. Stroke 33: 261–267.
del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO (2013). A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav Immun 33: 15–23.
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR et al (2010). Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 207: 1067–1080.
Dowell NG, Cooper EA, Tibble J, Voon V, Critchley HD, Cercignani M et al (2015). Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol Psychiatry 79: 320–328.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK et al (2010). A meta-analysis of cytokines in major depression. Biol Psychiatry 67: 446–457.
Dunn AJ, Wang J, Ando T (1999). Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv Exp Med Biol 461: 117–127.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010a). Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68: 748–754.
Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR (2010b). Inflammation and social experience: an inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav Immun 24: 558–563.
Elovainio M, Taipale T, Seppala I, Mononen N, Raitoharju E, Jokela M et al (2015). Activated immune-inflammatory pathways are associated with long-standing depressive symptoms: evidence from gene-set enrichment analyses in the Young Finns Study. J Psychiatr Res 71: 120–125.
Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A et al (2014). Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry 71: 423–431.
Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KR et al (2005). Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry 58: 175–189.
Fabricatore AN, Wadden TA, Higginbotham AJ, Faulconbridge LF, Nguyen AM, Heymsfield SB et al (2011). Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond) 35: 1363–1376.
Fan N, Luo Y, Xu K, Zhang M, Ke X, Huang X et al (2015). Relationship of serum levels of TNF-alpha, IL-6 and IL-18 and schizophrenia-like symptoms in chronic ketamine abusers. Schizophr Res 169: 10–15.
Faridhosseini F, Sadeghi R, Farid L, Pourgholami M (2014). Celecoxib: a new augmentation strategy for depressive mood episodes. A systematic review and meta-analysis of randomized placebo-controlled trials. Hum Psychopharmacol 29: 216–223.
Feigin A, Fukuda M, Dhawan V, Przedborski S, Jackson-Lewis V, Mentis MJ et al (2001). Metabolic correlates of levodopa response in Parkinson's disease. Neurology 57: 2083–2088.
Felger J, Miller A (2012). Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol 33: 315–327.
Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM et al (2007). Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry 62: 1324–1333.
Felger JC, Hernandez CR, Miller AH (2015). Levodopa reverses cytokine-induced reductions in striatal dopamine release. Int J Neuropsychopharmacol 18.
Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL et al (2013a). Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun 31: 153–160.
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X et al (2016). Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry (in press).
Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR et al (2013b). Chronic interferon-alpha decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 38: 2179–2178.
Fields C, Drye L, Vaidya V, Lyketsos C (2012). Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: findings from a randomized controlled trial. Am J Geriatr Psychiatry 20: 505–513.
Fisher G, Hyatt TC, Hunter GR, Oster RA, Desmond RA, Gower BA (2011). Effect of diet with and without exercise training on markers of inflammation and fat distribution in overweight women. Obesity (Silver Spring) 19: 1131–1136.
Fleshner M (2011). The gut microbiota: a new player in the innate immune stress response? Brain Behav Immun 25: 395–396.
Fleshner M (2013). Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun 27: 1–7.
Fleshner M, Goehler LE, Schwartz BA, McGorry M, Martin D, Maier SF et al (1998). Thermogenic and corticosterone responses to intravenous cytokines (IL-1beta and TNF-alpha) are attenuated by subdiaphragmatic vagotomy. J Neuroimmunol 86: 134–141.
Forsythe LK, Wallace JM, Livingstone MB (2008). Obesity and inflammation: the effects of weight loss. Nutr Res Rev 21: 117–133.
Francois M, Romieu-Mourez R, Li M, Galipeau J (2012). Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther 20: 187–195.
Ghanizadeh A, Dehbozorgi S, OmraniSigaroodi M, Rezaei Z (2014). Minocycline as add-on treatment decreases the negative symptoms of schizophrenia; a randomized placebo-controlled clinical trial. Recent Pat Inflamm Allergy Drug Discov 8: 211–215.
Goldsmith DR, Haroon E, Woolwine BJ, Jung MY, Wommack EC, Harvey PD et al (2016). Inflammatory markers are associated with decreased psychomotor speed in patients with major depressive disorder. Brain Behav Immun 56: 281–288.
Goldsmith DR, Rapaport MH, Miller BJ (2016). A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. (in press).
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T et al (2008). Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13: 717–728.
Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimaki M (2015). Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav Immun 49: 206–215.
Haber SN (2014). The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 282: 248–257.
Hanes WM, Olofsson PS, Kwan K, Hudson LK, Chavan SS, Pavlov VA et al (2015). Galantamine attenuates type 1 diabetes and inhibits anti-insulin antibodies in non-obese diabetic mice. Mol Med 21: 702–708.
Hannestad J, DellaGioia N, Gallezot JD, Lim K, Nabulsi N, Esterlis I et al (2013). The neuroinflammation marker translocator protein is not elevated in individuals with mild-to-moderate depression: a [(1)(1)C]PBR28 PET study. Brain Behav Immun 33: 131–138.
Hannestad J, Gallezot JD, Schafbauer T, Lim K, Kloczynski T, Morris ED et al (2012). Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. Neuroimage 63: 232–239.
Hansen MK, Taishi P, Chen Z, Krueger JM (1998). Vagotomy blocks the induction of interleukin-1beta (IL-1beta) mRNA in the brain of rats in response to systemic IL-1beta. J Neurosci 18: 2247–2253.
Hardingham GE, Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11: 682–696.
Hardingham GE, Fukunaga Y, Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414.
Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T et al (2016). Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry (in press).
Haroon E, Raison CL, Miller AH (2012). Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37: 137–162.
Haroon E, Woolwine BJ, Chen X, Pace TW, Parekh S, Spivey JR et al (2014). IFN-alpha-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology 39: 1777–1785.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009a). Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66: 407–414.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ et al (2009b). Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry 66: 415–422.
Harrison NA, Cercignani M, Voon V, Critchley HD (2015a). Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology 40: 831–838.
Harrison NA, Voon V, Cercignani M, Cooper EA, Pessiglione M, Critchley HD (2015b). A neurocomputational account of how inflammation enhances sensitivity to punishments versus rewards. Biol Psychiatry 80: 73–81.
Hashemian F, Majd M, Hosseini SM, Sharifi A, Panahi MVS, Bigdeli O (2011). A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in the treatment of a drug-naive women with major depression. Klin Psikofarmakol Bul 14: S183–S184.
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT et al (2008). Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation 5: 15.
Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015). Neuroimmune mechanisms of depression. Nat Neurosci 18: 1386–1393.
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D et al (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA 111: 16136–16141.
Hoge EA, Brandstetter K, Moshier S, Pollack MH, Wong KK, Simon NM (2009). Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress Anxiety 26: 447–455.
Howren MB, Lamkin DM, Suls J (2009). Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71: 171–186.
Hufner K, Oberguggenberger A, Kohl C, Geisler S, Gamper E, Meraner V et al (2015). Levels in neurotransmitter precursor amino acids correlate with mental health in patients with breast cancer. Psychoneuroendocrinology 60: 28–38.
Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H (2008). Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett 432: 232–236.
Iosifescu DV, Nierenberg AA, Alpert JE, Smith M, Bitran S, Dording C et al (2003). The impact of medical comorbidity on acute treatment in major depressive disorder. Am J Psychiatry 160: 2122–2127.
Irwin MR, Olmstead R, Breen EC, Witarama T, Carrillo C, Sadeghi N et al (2015). Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol Psychiatry 78: 721–729.
Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC et al (2008). Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 64: 538–540.
Jones ME, Lebonville CL, Barrus D, Lysle DT (2015). The role of brain interleukin-1 in stress-enhanced fear learning. Neuropsychopharmacology 40: 1289–1296.
Juengling FD, Ebert D, Gut O, Engelbrecht MA, Rasenack J, Nitzsche EU et al (2000). Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology (Berl) 152: 383–389.
Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G et al (2015). Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull 41: 85–93.
Kiecolt-Glaser JK, Bennett JM, Andridge R, Peng J, Shapiro CL, Malarkey WB et al (2014). Yoga's impact on inflammation, mood, and fatigue in breast cancer survivors: a randomized controlled trial. J Clin Oncol 32: 1040–1049.
Kim SJ, Lee H, Lee G, Oh SJ, Shin MK, Shim I et al (2012). CD4+CD25+ regulatory T cell depletion modulates anxiety and depression-like behaviors in mice. PLoS One 7: e42054.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004). T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA 101: 8180–8185.
Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T (2003). Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res 978: 104–114.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Kohler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O et al (2014). Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71: 1381–1391.
Konuk N, Tekin IO, Ozturk U, Atik L, Atasoy N, Bektas S et al (2007). Plasma levels of tumor necrosis factor-alpha and interleukin-6 in obsessive compulsive disorder. Mediators Inflamm 2007: 65704.
Kopylov U, Vutcovici M, Kezouh A, Seidman E, Bitton A, Afif W (2015). Risk of lymphoma, colorectal and skin cancer in patients with IBD treated with immunomodulators and biologics: a Quebec claims database study. Inflamm Bowel Dis 21: 1847–1853.
Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV et al (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 19: 699–709.
Krishnadas R, Nicol A, Sassarini J, Puri N, Burden AD, Leman J et al (2016). Circulating tumour necrosis factor is highly correlated with brainstem serotonin transporter availability in humans. Brain Behav Immun 51: 29–38.
Krugel U, Fischer J, Radicke S, Sack U, Himmerich H (2013). Antidepressant effects of TNF-alpha blockade in an animal model of depression. J Psychiatr Res 47: 611–616.
Kwilasz AJ, Grace PM, Serbedzija P, Maier SF, Watkins LR (2015). The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology 96: 55–69.
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H (2000). Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology 22: 370–379.
Lapidus KA, Gabbay V, Mao X, Johnson A, Murrough JW, Mathew SJ et al (2014). In vivo (1)H MRS study of potential associations between glutathione, oxidative stress and anhedonia in major depressive disorder. Neurosci Lett 569: 74–79.
Lewitus GM, Cohen H, Schwartz M (2008). Reducing post-traumatic anxiety by immunization. Brain Behav Immun 22: 1108–1114.
Lewitus GM, Schwartz M (2009). Behavioral immunization: immunity to self-antigens contributes to psychological stress resilience. Mol Psychiatry 14: 532–536.
Lewitus GM, Wilf-Yarkoni A, Ziv Y, Shabat-Simon M, Gersner R, Zangen A et al (2009). Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry 65: 283–288.
Li Y, Xiao B, Qiu W, Yang L, Hu B, Tian X et al (2010). Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J Affect Disord 124: 68–75.
Lieb J, Karmali R, Horrobin D (1983). Elevated levels of prostaglandin E2 and thromboxane B2 in depression. Prostaglandins Leukot Med 10: 361–367.
Lieberman JA, Dunbar G, Segreti AC, Girgis RR, Seoane F, Beaver JS et al (2013). qA randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology 38: 968–975.
Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P (2009). Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer 9: 445–452.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523: 337–341.
Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J et al (2014). alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med 20: 350–358.
Luheshi GN, Bluthe RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M et al (2000). Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Auton Neurosci 85: 127–132.
Maes M (1995). Evidence for an immune response in major depression: a review and hypothesis. Prog Neuropsychopharmacol Biol Psychiatry 19: 11–38.
Majer M, Welberg LA, Capuron L, Pagnoni G, Raison CL, Miller AH (2008). IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav Immun 22: 870–880.
Manabe T, Togashi H, Uchida N, Suzuki SC, Hayakawa Y, Yamamoto M et al (2000). Loss of cadherin-11 adhesion receptor enhances plastic changes in hippocampal synapses and modifies behavioral responses. Mol Cell Neurosci 15: 534–546.
Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M (2012a). The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun 28: 54–62.
Maslanik T, Tannura K, Mahaffey L, Loughridge AB, Beninson L, Ursell L et al (2012b). Commensal bacteria and MAMPs are necessary for stress-induced increases in IL-1beta and IL-18 but not IL-6, IL-10 or MCP-1. PLoS One 7: e50636.
Mehta D, Raison CL, Woolwine BJ, Haroon E, Binder EB, Miller AH et al (2013). Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav Immun 31: 205–215.
Meier-Ewert HK, Ridker PM, Rifai N, Price N, Dinges DF, Mullington JM (2001). Absence of diurnal variation of C-reactive protein concentrations in healthy human subjects. Clin Chem 47: 426–430.
Mellor AL, Munn D, Chandler P, Keskin D, Johnson T, Marshall B et al (2003). Tryptophan catabolism and T cell responses. Adv Exp Med Biol 527: 27–35.
Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N (2006). Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol 21: 227–231.
Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO et al (2015). Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry 172: 353–362.
Miller AH, Haroon E, Raison CL, Felger JC (2013). Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety 30: 297–306.
Miller AH, Maletic V, Raison CL (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65: 732–741.
Miller AH, Raison CL (2015). Are anti-inflammatory therapies viable treatments for psychiatric disorders?: where the rubber meets the road. JAMA Psychiatry 72: 527–528.
Miller AH, Raison CL (2015). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16: 22–34.
Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999). Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5: 49–55.
Mocking RJ, Harmsen I, Assies J, Koeter MW, Ruhe HG, Schene AH (2016). Meta-analysis and meta-regression of omega-3 polyunsaturated fatty acid supplementation for major depressive disorder. Transl Psychiatry 6: e756.
Mohammadinejad P, Arya P, Esfandbod M, Kaviani A, Najafi M, Kashani L et al (2015). Celecoxib versus diclofenac in mild to moderate depression management among breast cancer patients: a double-blind, placebo-controlled, randomized trial. Ann Pharmacother 49: 953–961.
Mohle L, Mattei D, Heimesaat MM, Bereswill S, Fischer A, Alutis M et al (2016). Ly6C(hi) monocytes provide a link between antibiotic-induced changes in gut microbiota and adult hippocampal neurogenesis. Cell Rep 15: 1945–1956.
Morris SE, Cuthbert BN (2012). Research domain criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci 14: 29–37.
Muller N, Schwarz MJ (2007). The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry 12: 988–1000.
Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B et al (2006). The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 11: 680–684.
Muscatell KA, Dedovic K, Slavich GM, Jarcho MR, Breen EC, Bower JE et al (2015). Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav Immun 43: 46–53.
Muscatell KA, Eisenberger NI, Dutcher JM, Cole SW, Bower JE (2016). Links between inflammation, amygdala reactivity, and social support in breast cancer survivors. Brain Behav Immun 53: 34–38.
Na KS, Lee KJ, Lee JS, Cho YS, Jung HY (2014). Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: a meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry 48: 79–85.
Nanni V, Uher R, Danese A (2012). Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am J Psychiatry 169: 141–151.
Naude PJ, Dobos N, van der Meer D, Mulder C, Pawironadi KG, den Boer JA et al (2014). Analysis of cognition, motor performance and anxiety in young and aged tumor necrosis factor alpha receptor 1 and 2 deficient mice. Behav Brain Res 258: 43–51.
Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M et al (2008). Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab 9: 622–627.
Nunes EJ, Randall PA, Estrada A, Epling B, Hart EE, Lee CA et al (2014). Effort-related motivational effects of the pro-inflammatory cytokine interleukin 1-beta: studies with the concurrent fixed ratio 5/ chow feeding choice task. Psychopharmacology (Berl) 231: 727–736.
O'Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J et al (2009). Induction of IDO by Bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol 182: 3202–3212.
O'Farrell K, Harkin A (2015). Stress-related regulation of the kynurenine pathway: relevance to neuropsychiatric and degenerative disorders. Neuropharmacology.
Oskooilar N, Wilcox CS, Tong ML, Grosz DE (2009). Body mass index and response to antidepressants in depressed research subjects. J Clin Psychiatry 70: 1609–1610.
Oya K, Kishi T, Iwata N (2014). Efficacy and tolerability of minocycline augmentation therapy in schizophrenia: a systematic review and meta-analysis of randomized controlled trials. Hum Psychopharmacol 29: 483–491.
Pace TW, Heim CM (2011). A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav Immun 25: 6–13.
Pace TW, Hu F, Miller AH (2007). Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21: 9–19.
Pandey GN, Rizavi HS, Ren X, Bhaumik R, Dwivedi Y (2014). Toll-like receptors in the depressed and suicide brain. J Psychiatr Res 53: 62–68.
Pandey GN, Rizavi HS, Ren X, Fareed J, Hoppensteadt DA, Roberts RC et al (2012). Proinflammatory cytokines in the prefrontal cortex of teenage suicide victims. J Psychiatr Res 46: 57–63.
Papakostas GI, Shelton RC, Zajecka JM, Etemad B, Rickels K, Clain A et al (2012). L-methylfolate as adjunctive therapy for SSRI-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry 169: 1267–1274.
Parrott JM, Redus L, Santana-Coelho D, Morales J, Gao X, O’Connor JC (2016). Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl Psychiatry (in press).
Quan N, Banks WA (2007). Brain-immune communication pathways. Brain Behav Immun 21: 727–735.
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G et al (2010). CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15: 393–403.
Raison CL, Felger JC, Miller AH (2013a). Inflammation and treatment resistance in major depression: a perfect storm. Psychiatric Times.
Raison CL, Miller AH (2011). Is depression an inflammatory disorder? Curr Psychiatry Rep 13: 467–475.
Raison CL, Miller AH (2013). The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D). Mol Psychiatry 18: 15–37.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF et al (2013b). A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70: 31–41.
Raison CL, Rye DB, Woolwine BJ, Vogt GJ, Bautista BM, Spivey JR et al (2010). Chronic interferon-alpha administration disrupts sleep continuity and depth in patients with hepatitis C: association with fatigue, motor slowing, and increased evening cortisol. Biol Psychiatry 68: 942–949.
Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y et al (2016). Helminth infection promotes colonization resistance via type 2 immunity. Science 352: 608–612.
Rangel-Huerta OD, Aguilera CM, Mesa MD, Gil A (2012). Omega-3 long-chain polyunsaturated fatty acids supplementation on inflammatory biomarkers: a systematic review of randomised clinical trials. Br J Nutr 107 (Suppl 2): S159–S170.
Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R et al (2015). Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: a proof-of-concept study. Mol Psychiatry 21: 71–79.
Rattazzi L, Piras G, Ono M, Deacon R, Pariante CM, D'Acquisto F (2013). CD4(+) but not CD8(+) T cells revert the impaired emotional behavior of immunocompromised RAG-1-deficient mice. Transl Psychiatry 3: e280.
Reber SO, Siebler PH, Donner NC, Morton JT, Smith DG, Kopelman JM et al (2016). Immunization with a heat-killed preparation of the environmental bacterium Mycobacterium vaccae promotes stress resilience in mice. Proc Natl Acad Sci USA 113: E3130–E3139.
Redwine L, Hauger RL, Gillin JC, Irwin M (2000). Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J Clin Endocrinol Metab 85: 3597–3603.
Rethorst CD, Toups MS, Greer TL, Nakonezny PA, Carmody TJ, Grannemann BD et al (2013). Pro-inflammatory cytokines as predictors of antidepressant effects of exercise in major depressive disorder. Mol Psychiatry 18: 1119–1124.
Rhen T, Cidlowski JA (2005). Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353: 1711–1723.
Ridker PM (2003). Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 107: 363–369.
Robinson CM, Hale PT, Carlin JM (2006). NF-kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine 35: 53–61.
Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C et al (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334: 98–101.
Rosenblat JD, Kakar R, Berk M, Kessing LV, Vinberg M, Baune BT et al (2016). Anti-inflammatory agents in the treatment of bipolar depression: a systematic review and meta-analysis. Bipolar Disord 18: 89–101.
Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O'Connor JC (2012). Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav 62: 202–209.
Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, Lin SF et al (2015). Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci USA 112: 12468–12473.
Sarris J, Price LH, Carpenter LL, Tyrka AR, Ng CH, Papakostas GI et al (2015). Is S-adenosyl methionine (SAMe) for depression only effective in males? A re-analysis of data from a randomized clinical trial. Pharmacopsychiatry 48: 141–144.
Schleifer SJ, Keller SE, Meyerson AT, Raskin MJ, Davis KL, Stein M (1984). Lymphocyte function in major depressive disorder. Arch Gen Psychiatry 41: 484–486.
Schuch FB, Vancampfort D, Richards J, Rosenbaum S, Ward PB, Stubbs B (2016). Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res 77: 42–51.
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13: 465–477.
Schwartz M, Shechter R (2010). Protective autoimmunity functions by intracranial immunosurveillance to support the mind: the missing link between health and disease. Mol Psychiatry 15: 342–354.
Sellgren CM, Kegel ME, Bergen SE, Ekman CJ, Olsson S, Larsson M et al (2015). A genome-wide association study of kynurenic acid in cerebrospinal fluid: implications for psychosis and cognitive impairment in bipolar disorder. Mol Psychiatry (in press).
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G et al (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72: 268–275.
Shalbafan M, Mohammadinejad P, Shariat SV, Alavi K, Zeinoddini A, Salehi M et al (2015). Celecoxib as an adjuvant to fluvoxamine in moderate to severe obsessive-compulsive disorder: a double-blind, placebo-controlled, randomized trial. Pharmacopsychiatry 48: 136–140.
Shelton RC, Pencina MJ, Barrentine LW, Ruiz JA, Fava M, Zajecka JM et al (2015). Association of obesity and inflammatory marker levels on treatment outcome: results from a double-blind, randomized study of adjunctive L-methylfolate calcium in patients with MDD who are inadequate responders to SSRIs. J Clin Psychiatry 76: 1635–1641.
Shi M, Liu ZW, Wang FS (2011). Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol 164: 1–8.
Singh JA, Cameron C, Noorbaloochi S, Cullis T, Tucker M, Christensen R et al (2015). Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta-analysis. Lancet 386: 258–265.
Sitcheran R, Gupta P, Fisher PB, Baldwin AS (2005). Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. EMBO J 24: 510–520.
Slavich GM, Way BM, Eisenberger NI, Taylor SE (2010). Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci USA 107: 14817–14822.
Sluzewska A, Sobieska M, Rybakowski JK (1997). Changes in acute-phase proteins during lithium potentiation of antidepressants in refractory depression. Neuropsychobiology 35: 123–127.
Stein M, Miller AH, Trestman RL (1991). Depression, the immune system, and health and illness. Findings in search of meaning. Arch Gen Psychiatry 48: 171–177.
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C et al (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42: 151–157.
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z et al (2011). Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8: 94.
Stellwagen D, Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440: 1054–1059.
Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ (2015). Inflammation and clinical response to treatment in depression: a meta-analysis. Eur Neuropsychopharmacol 25: 1532–1543.
Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF et al (2013). MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res 23: 1270–1283.
Sutcigil L, Oktenli C, Musabak U, Bozkurt A, Cansever A, Uzun O et al (2007). Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin Dev Immunol 2007: 76396.
Svenningsson P, Kim Y, Warner-Schmidt J, Oh YS, Greengard P (2013). p11 and its role in depression and therapeutic responses to antidepressants. Nat Rev Neurosci 14: 673–680.
Swardfager W, Rosenblat JD, Benlamri M, McIntyre RS (2016). Mapping inflammation onto mood: Inflammatory mediators of anhedonia. Neurosci Biobehav Rev 64: 148–166.
Tavares RG, Schmidt AP, Abud J, Tasca CI, Souza DO (2005). In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem Res 30: 439–444.
Tavares RG, Tasca CI, Santos CES, Alves LB, Porciuncula LO, Emanuelli T et al (2002). Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int 40: 621–627.
Tilleux S, Hermans E (2007). Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85: 2059–2070.
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N (2014). Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun 42: 50–59.
Torres MA, Pace TW, Liu T, Felger JC, Mister D, Doho GH et al (2013). Predictors of depression in breast cancer patients treated with radiation: role of prior chemotherapy and nuclear factor kappa B. Cancer 119: 1951–1959.
Tracey KJ (2009). Reflex control of immunity. Nat Rev Immunol 9: 418–428.
Turkheimer FE, Rizzo G, Bloomfield PS, Howes O, Zanotti-Fregonara P, Bertoldo A et al (2015). The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans 43: 586–592.
Vichaya EG, Hunt SC, Dantzer R (2014). Lipopolysaccharide reduces incentive motivation while boosting preference for high reward in mice. Neuropsychopharmacology 39: 2884–2890.
Vivash L, O'Brien TJ (2016). Imaging microglial activation with TSPO PET: lighting up neurologic diseases? J Nucl Med 57: 165–168.
Walker AJ, Foley BM, Sutor SL, McGillivray JA, Frye MA, Tye SJ (2015). Peripheral proinflammatory markers associated with ketamine response in a preclinical model of antidepressant-resistance. Behav Brain Res 293: 198–202.
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B et al (2013). NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38: 1609–1616.
Walling D, Marder SR, Kane J, Fleischhacker WW, Keefe RS, Hosford DA et al (2016). Phase 2 trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull 42: 335–343.
Wang X, Wu H, Miller AH (2004). Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry 9: 65–75.
Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P (2011). Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc Natl Acad Sci USA 108: 9262–9267.
Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J et al (2016). Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36: 4182–4195.
Weinberger JF, Raison CL, Rye DB, Montague AR, Woolwine BJ, Felger JC et al (2015). Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav Immun 47: 193–200.
Wichmann T, DeLong MR (1999). Oscillations in the basal ganglia. Nature 400: 621–622.
Wieczorek M, Swiergiel AH, Pournajafi-Nazarloo H, Dunn AJ (2005). Physiological and behavioral responses to interleukin-1beta and LPS in vagotomized mice. Physiol Behav 85: 500–511.
Williamson LL, McKenney EA, Holzknecht ZE, Belliveau C, Rawls JF, Poulton S et al (2015). Got worms? Perinatal exposure to helminths prevents persistent immune sensitization and cognitive dysfunction induced by early-life infection. Brain Behav Immun 51: 14–28.
Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT et al (2011). beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci 31: 6277–6288.
Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2014). Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8: 447.
Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013). Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33: 13820–13833.
Wong ML, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J et al (2016). Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry 21: 797–805.
Woods JA, Vieira VJ, Keylock KT (2009). Exercise, inflammation, and innate immunity. Immunol Allergy Clin North Am 29: 381–393.
Wu HQ, Rassoulpour A, Schwarcz R (2007). Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain? J Neural Transm 114: 33–41.
Yirmiya R, Goshen I (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25: 181–213.
Yohn SE, Arif Y, Haley A, Tripodi G, Baqi Y, Muller CE et al (2016). Effort-related motivational effects of the pro-inflammatory cytokine interleukin-6: pharmacological and neurochemical characterization. Psychopharmacology (Berl) (in press).
Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X et al (2016). Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host's metabolism. Mol Psychiatry 21: 786–796.
Zhu CB, Blakely RD, Hewlett WA (2006). The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 31: 2121–2131.
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD (2005). p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem 280: 15649–15658.
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010). Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35: 2510–2520.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N et al (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9: 268–275.
Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A et al (2001). The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 15: 199–226.
Zuany-Amorim C, Sawicka E, Manlius C, Le Moine A, Brunet LR, Kemeny DM et al (2002). Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced allergen-specific regulatory T-cells. Nat Med 8: 625–629.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Miller, A., Haroon, E. & Felger, J. Therapeutic Implications of Brain–Immune Interactions: Treatment in Translation. Neuropsychopharmacol 42, 334–359 (2017). https://doi.org/10.1038/npp.2016.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.167
This article is cited by
-
Immunoregulatory role of the gut microbiota in inflammatory depression
Nature Communications (2024)
-
The pharmacological bases for repurposing statins in depression: a review of mechanistic studies
Translational Psychiatry (2023)
-
Efficacy of eicosapentaenoic acid in inflammatory depression: study protocol for a match-mismatch trial
BMC Psychiatry (2022)
-
Functional connectivity in reward circuitry and symptoms of anhedonia as therapeutic targets in depression with high inflammation: evidence from a dopamine challenge study
Molecular Psychiatry (2022)
-
PCOS and Depression: Common Links and Potential Targets
Reproductive Sciences (2022)
