
Abstract
Stress exposure is associated with individual differences in corticolimbic structure and function that often mirror patterns observed in psychopathology. Gene x environment interaction research suggests that genetic variation moderates the impact of stress on risk for psychopathology. On the basis of these findings, imaging genetics, which attempts to link variability in DNA sequence and structure to neural phenotypes, has begun to incorporate measures of the environment. This research paradigm, known as imaging gene x environment interaction (iGxE), is beginning to contribute to our understanding of the neural mechanisms through which genetic variation and stress increase psychopathology risk. Although awaiting replication, evidence suggests that genetic variation within the canonical neuroendocrine stress hormone system, the hypothalamic-pituitary-adrenal axis, contributes to variability in stress-related corticolimbic structure and function, which, in turn, confers risk for psychopathology. For iGxE research to reach its full potential it will have to address many challenges, of which we discuss: (i) small effects, (ii) measuring the environment and neural phenotypes, (iii) the absence of detailed mechanisms, and (iv) incorporating development. By actively addressing these challenges, iGxE research is poised to help identify the neural mechanisms underlying genetic and environmental associations with psychopathology.
Similar content being viewed by others
INTRODUCTION
All organisms strive to maintain homeostasis by regulating physiology and behavior within a dynamic equilibrium. Stress, the perception of inadequate resources in the context of environmental pressures appraised as threatening, disrupts homeostasis by triggering physiologic and behavioral responses to meet the immediate demands on an individual (Ganzel et al, 2010; Selye, 1936). Although stress promotes adaptive responses to challenge when motivation is high and resources available, the relationship between stress, especially that which is chronic, unpredictable, and uncontrollable, and the experience of psychopathology is unequivocal (McEwen and Gianaros, 2010). Nearly 40% of individuals report experiencing adversity during childhood, which predicts 30% of adult-onset and 45% of childhood-onset psychiatric disorders (Green et al, 2010; Kessler et al, 2010). Similarly, evidence suggests that ubiquitous adult-onset stressful experiences, as well as perceived stress increase risk for the emergence and relapse of various forms of psychopathology (Dohrenwend, 2000; Monroe and Reid, 2009). These robust transdiagnostic associations have propelled research across species to investigate the neurobiology of stress responsiveness and adaptation, as well as how individual differences in this neurobiology develop and confer relative vulnerability or resiliency to the pathogenic effects of stress (de Kloet et al, 2005; Hill et al, 2012; Lucassen et al, 2014; Lupien et al, 2009; McCrory et al, 2011; McEwen and Morrison, 2013; Teicher and Samson, 2013).
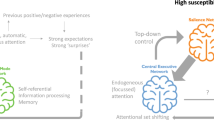
The neurobiology of stress responsiveness is well understood due in large part to its conservation across species. When a threat is detected coordinated autonomic, neuroendocrine, metabolic and immune system responses are initiated by governing interconnected corticolimbic circuitry, which also functions to return the body to homeostasis following stressor removal (de Kloet et al, 2005; Ulrich-Lai and Herman, 2009; Whalen and Phelps, 2009). The central structure, or hub, within this network is the amygdala, which has extensive afferent and efferent connections with other corticolimbic circuitry nodes including the thalamus, sensory cortex, autonomic and neuromodulatory brainstem nuclei, hypothalamus, insula, hippocampal formation, and prefrontal cortex (Figure 1; for more detailed reviews of this circuitry see (Duvarci and Pare, 2014; Hariri, 2015; Janak and Tye, 2015; Kim et al, 2011; Price and Drevets, 2012; Whalen and Phelps, 2009). Broadly, the amygdala and its connections to these regions are necessary for first recognizing possible threat in the environment and then generating and regulating appropriate reactions in physiology and behavior.

The amygdala functions as the hub of the corticolimbic circuit. The basolateral complex of the amygdala (BLA) receives low-resolution sensory information from the thalamus and olfactory tract, as well as high-resolution sensory information from unimodal sensory cortex and multimodal association areas. The BLA relays this information to the central nucleus of the amygdala (CeA), which further projects to the sublenticular extended amygdala (SLEA) composed of the substantia innominata (SI) and the bed nucleus of the stria terminalis (BNST). The CeA and SLEA drive physiobehavioral output through projections to corticolimbic nodes (brainstem, hypothalamus, insula, hippocampal formation, prefrontal cortex). CeA and SLEA projections to brainstem nuclei facilitate sympathetic arousal while relays to the hypothalamus excite the hypothalamic-pituitary-adrenal (HPA) axis producing a stress hormone response. Diffuse cholinergic projections from the nucleus basalis of Meynert within the SI to the cortex increase neuronal sensitivity to input, including sensory and interoceptive information, to facilitate acuity and awareness/alertness. BLA and SLEA projections to the insula facilitate interoceptive awareness in conjunction with convergent somatosensory and interoceptive projections from the body, allowing for a representation of one's bodily states (eg, heart rate). Amygdala and EA projections to the hippocampus potentiate encoding and recall of the context in which a stimulus triggered an amygdala response. A secondary role of the hippocampus within the corticolimbic system is to provide negative feedback inhibition of the HPA axis. Corticolimbic nodes further project back to the amygdala to further regulate its function. For instance, projections from the dorsomedial PFC can serve to inhibit amygdala response during emotion regulation and fear extinction. (For more detailed reviews of this circuitry see Duvarci and Pare, 2014; Hariri, 2015; Janak and Tye, 2015; Kim et al, 2011; Price and Drevets, 2012; Whalen and Phelps, 2009.)
Given the quintessential role of corticolimbic circuitry in stress responsiveness, it is not surprising to find consistent differences in corticolimbic function and structure across various forms of psychopathology and associated behavioral risk factors, such as negative emotionality or neuroticism (Hariri, 2015). In particular, relative hyperactivity of the amygdala to threatening contexts and stimuli has emerged as a core feature of stress-related disorders such as anxiety, PTSD, and depression (Bruhl et al, 2014; Groenewold et al, 2013; Hayes et al, 2012; Swartz and Monk, 2014a). Highly convergent findings from preclinical models of human psychopathology, especially anxiety disorders, further underscore the importance of corticolimbic circuitry broadly and the amygdala specifically in the emergence of stress-related disorders (Bukalo et al, 2014; Duvarci and Pare, 2014). Relative to the consistent and convergent observations of amygdala hyperactivity in stress-related disorders, differences in amygdala structure have been mixed. For instance, first-episode depression is associated with larger amygdala volume, whereas smaller volumes or no differences have been observed in patients with recurrent depression (Frodl et al, 2002, 2003). Mixed volumetric findings have also been reported among individuals with PTSD and anxiety disorders as well as individuals stratified according to the experience of early life stress (Hanson et al, 2015; Hilbert et al, 2014; Luby et al, 2013; Sheridan et al, 2012; Tottenham et al, 2010). What is clear, however, is that there are vast individual differences in stress-related corticolimbic structure and function, with increasing evidence that these differences may mediate associations between stress and later psychopathology (Burghy et al, 2012; Gee et al, 2013; Gorka et al, 2014; Swartz et al, 2014b; 2015; Tottenham et al, 2011).
Imaging genetics is an emerging research strategy poised to identify mediating mechanisms through which variability in the genome and epigenome shape individual differences in corticolimbic function and structure associated with risk for psychopathology (Bogdan et al, 2013a; Hariri, 2009). Inspired by: (i) non-human animal models and neuroimaging research documenting effects of the environment, and in particular stress, on brain function and structure (Gee et al, 2013; Lupien et al, 2009; Tottenham and Sheridan, 2009), and (ii) models of gene × environment interaction influencing psychopathology (Caspi et al, 2010; Karg et al, 2011; Zannas and Binder, 2014, but see also Duncan and Keller, 2011), imaging genetics studies have begun to examine how assessed and manipulated environments moderate associations between genetic variation and the brain.
Our review highlights emerging imaging gene × environment interaction (iGxE) research that is beginning to inform our understanding of individual differences in corticolimbic circuitry and how these differences may confer psychopathology risk (Caspi and Moffitt, 2006; Hyde et al, 2011a). We begin by providing a brief introduction of the basic structure and regulation of the hypothalamic-pituitary-adrenal (HPA) axis, as well as its associations with psychopathology and corticolimbic structure and function to help orient the reader to the processes subsequently considered within the iGxE framework. Next, we review iGxE research of the HPA axis and corticolimbic circuitry, as well as several iGxE studies that have been conducted with variants outside of the canonical HPA axis. Lastly, we address several key challenges that confront iGxE and are familiar to traditional genetic, neuroimaging, psychiatric, and environmental research, including: (i) small effects of common genetic variation further constrained by the frequency of environmental events (Duncan and Keller, 2011), (ii) how best to assay and/or manipulate the environment and neural phenotypes (Monroe, 2008, iii) limited understanding of detailed mechanisms (Bogdan et al, 2013a), and (iv) the importance of considering developmental timing (Lupien et al, 2009). By actively confronting these challenges, iGxE, in concert with traditional neuroimaging, environmental, and molecular and behavioral genetic research can uniquely inform who is at risk for psychopathology and through what specific neurogenetic mechanisms this risk emerges. A deeper understanding of risk mechanisms can subsequently inform ongoing efforts to refine psychiatric nosology and identify novel therapeutic targets to combat the development of stress-related psychopathology.
HPA AXIS
Within corticolimbic circuitry, the neuroendocrine HPA axis is a central regulator of stress responsiveness and adaptation (for reviews see de Kloet et al, 2005; Lupien et al, 2009; Ulrich-Lai and Herman, 2009). HPA axis activity follows a daily oscillation governed by the circadian system and is provoked by stress. The HPA axis is controlled by the paraventricular nucleus (PVN) of the hypothalamus, which receives afferent innervation from the central amygdala (CeA) and sublenticular extended amygdala (SLEA), as well as brainstem nuclei, other hypothalamic nuclei, the hippocampus, and prefrontal cortex (Ulrich-Lai and Herman, 2009). These projections convey a wide array of sensory, emotional, contextual, and perceptual information and serve to activate (and inhibit) a three-step hormonal cascade.
PVN activation stimulates the release of corticotropin-releasing hormone (CRH), which triggers adrenocorticotropic hormone (ACTH) secretion from the pituitary. ACTH stimulates release of cortisol (corticosterone in rodents), which operates on multiple targets through a binary corticosteroid receptor system consisting of mineralocorticoid (MR) and glucocorticoid (GR) receptors. Owing to their high affinity for cortisol, MRs are typically occupied throughout the circadian cycle allowing cortisol to provide a stable excitatory tone in the hippocampus that inhibits the HPA axis under basal and stressful circumstances. In contrast, GRs, which have a low affinity for cortisol, only become occupied following large spikes in cortisol, such as circadian rhythm peaks (eg, the awakening response) or following stress. Within the hippocampus, GRs inhibit continued HPA axis activity and facilitate a return to homeostasis after a stressor has passed (though cortisol-GR binding in other regions, such as the amygdala, can potentiate the HPA axis response; Kolber and Muglia, 2009). Generally, CRH and ACTH signaling stimulate HPA axis activity while cortisol-MR binding constrains the initial HPA axis response, and cortisol-GR binding returns the body to homeostasis after a stress-precipitated response (de Kloet et al, 2005; Lupien et al, 2009).
The influence of the HPA axis extends directly to other brain regions and the transcriptome. HPA axis hormone receptors are widespread throughout the brain with high concentrations in corticolimbic structures. For instance, CRH binding within the amygdala has been tightly linked to models of stress-related psychopathology in rodents, including anxiety and depression (Binder and Nemeroff, 2010; Hauger et al, 2009; Kehne and Cain, 2010). The HPA axis gains access to the genome through intracellular corticosteroid receptors. When bound with cortisol, these receptors can translocate to the nucleus where they bind to regulatory elements of DNA, known as GR response elements (GREs), to enhance or suppress the transcription of a wide range of genes (de Kloet et al, 2005, 2008; Menke et al, 2012). Access to the transcriptome allows the HPA axis to extend its reach to a diverse array of proteins and may contribute to the pleiotropic effects of stress, including subsequent vulnerability to psychopathology (Arloth et al, 2015). Interestingly, individual differences in cortisol-stimulated gene expression are more robust predictors of psychopathology than baseline differences (Menke et al, 2012), emphasizing the importance of considering the transcriptome in the context of stress and related psychopathology (Frodl et al, 2012, 2014a). Here, it is important to note that there are many additional factors that interact with the HPA axis (eg, urocortins, vasopressin, neuropeptide Y, inflammation) that are appealing candidates for further study in the context of stress-related disorders; however, with few exceptions these additional factors have yet to be extensively investigated in the iGxE literature and hence are not further considered in our review (Hauger et al, 2006; Horowitz and Zunszain, 2015).
Genetic and Environmental Origins of HPA Axis Variability
There are vast individual differences in diurnal HPA axis rhythms and responsiveness to challenge that are generally stable over time and moderately to largely heritable (Federenko et al, 2004; Franz et al, 2010; Gustafsson et al, 2011; Van Hulle et al, 2012; Wust et al, 2000). Candidate gene and genome-wide association studies (GWAS) have begun to link common genetic variation within the HPA axis cascade to individual differences in diurnal and stress-evoked HPA axis function (Bolton et al, 2014; DeRijk, 2008, 2009). Furthermore, many of these variants have been shown to interact with stress exposure to convey vulnerability to psychopathology and other stress-related diseases (for reviews see: DeRijk et al, 2008; Zannas and Binder, 2014).
In addition to genetic factors, animal models have consistently documented that stress, particularly when chronic and early in life, results in long-lasting changes to the HPA axis (for review see: Lupien et al, 2009). For example, maternal separation in rodents increases CRH receptor expression in the pituitary and reduces hippocampal GR binding sites, resulting in heightened basal HPA axis output and an atypical diurnal pattern (Anisman et al, 1998; McEwen, 2000). Similar associations (ie, elevated ACTH and cortisol and reduced GR expression in the hippocampus) have been found in non-human primates and humans exposed to adversity in early life suggesting that these effects are conserved across species (Miller et al, 2007; Tyrka et al, 2013).
Rodent models suggest that early life stress-related HPA axis differences may emerge from epigenetic modifications. Most notably, Meaney and colleagues have shown that rat maternal care affects the later adult behavior of offspring through epigenetic regulation of the HPA axis (Turecki and Meaney, 2014; Weaver et al, 2004, 2005). Briefly, rats raised by a low-caring mother (ie, one providing little licking and grooming, as well as arched back nursing) show behavioral susceptibility to later stress (eg, anxiety-like behavior), which is mediated by a cascade of cellular changes that persist throughout the rat’s lifespan. Offspring of low-caring mothers have reduced nerve growth factor-inducible protein A expression, which leads to increased methylation of the GR gene (NR3C2) in the hippocampus, and thus less GR expression. As GR-cortisol binding within the hippocampus provides negative feedback regulation of the HPA axis, this epigenetic change results in poor stress recovery, much like what is observed in humans with depression. Remarkably consistent findings in a human post mortem study suggest a striking conservation of this epigenetic pathway across species (McGowan et al, 2009). Perhaps most interestingly, in cross-fostering experiments, this epigenetic change was transmitted inter-generationally whereby low-caring mothers raised offspring, who became low-caring mothers themselves transmitting this epigenetic signature across generations creating a heritable behavioral pattern uncoupled from DNA sequence.
HPA Axis Variability, Psychopathology, and Corticolimbic Structure and Function
Decades of research have shown that the HPA axis is disrupted across psychopathologies (de Kloet et al, 2005; Doom and Gunnar, 2013; Faravelli et al, 2012; Marques et al, 2009). The most consistent evidence is found regarding depression, which is often characterized by elevations in CRH, ACTH, and cortisol, as well as impaired negative feedback of the HPA axis (Pariante and Lightman, 2008). Alongside non-human animal models demonstrating that HPA axis manipulation (eg, knockout, pharmacologic challenge) produces anxiety- and depressive-like behaviors (Kolber and Muglia, 2009; Lupien et al, 2009), human studies indicate that chronic HPA axis stimulation (eg, for the treatment of medical conditions such as asthma or rheumatoid arthritis) can induce severe psychiatric symptoms that remit following treatment cessation (Fardet et al, 2012; Patten and Neutel, 2000; Wada et al, 2000). Moreover, successful treatment of stress-related disorders normalizes HPA axis function, prompting hope that novel treatments targeting this system may be more broadly therapeutically efficacious (de Kloet et al, 2008; Ising and Holsboer, 2007; Otte et al, 2010; Thomson and Craighead, 2008).
In contrast to many studies linking HPA axis function to psychopathology, far less research has examined HPA axis correlates of corticolimbic function and structure in humans. A few notable exceptions complement a rich foundation of non-human animal data (Dedovic et al, 2009; Lupien et al, 2009; McEwen and Gianaros, 2011). For instance, individuals with Cushing’s syndrome (ie, a tumor leading to large amounts of ACTH and cortisol release) have increased threat-related activation of the amygdala and hippocampus, as well as diminished hippocampal gray matter volume (Andela et al, 2015; Maheu et al, 2007, 2008). Human pharmacologic challenge studies document that cortisol signaling increases amygdala activation and functional connectivity with other corticolimbic nodes in response to threat-related stimuli (Henckens et al, 2010, 2012; Vogel et al, 2015).
Similarly, circulating concentrations of endogenous cortisol, correlate with differential activity and connectivity within corticolimbic circuitry. Specifically, elevated cortisol is associated with increased amygdala and decreased ventromedial prefrontal cortex (vmPFC) activation during regulation of negative affect, providing an intriguing putative mechanism (ie, deficient emotion regulation) through which HPA axis dysfunction may contribute to psychopathology (Urry et al, 2006). Recent evidence further suggests that early life stress exposure leads to early ‘maturation’ of amygdala-prefrontal connectivity during adolescence (ie, negative coupling comparable to that in adults) that is mediated by circulating concentrations of cortisol, and that individuals exposed to early life stress exhibiting relatively immature amygdala-prefrontal functional connectivity experience more anxiety (Gee et al, 2013). Lastly, in girls, greater early life adversity has been associated with heightened cortisol during childhood, which negatively predicts amygdala-vmPFC resting-state intrinsic coupling and greater subsequent risk for stress-related disorders in young adulthood (Bogdan and Hariri, 2012a; Burghy et al, 2012). Collectively, these studies suggest that dysfunction of the HPA axis may play a causal role in upregulating amygdala activity and altering its functional connectivity with the prefrontal cortex, which may weaken the capacity to effectively regulate stress responses and hence, contribute to the emergence of psychopathology.
iGxE OF CORTICOLIMBIC CIRCUITRY
Given clear associations between exposure to stress and expression of psychopathology, as well as transdiagnostic evidence for HPA axis dysregulation, understanding how genetic variation moderates the impact of stress on corticolimbic structure and function is critical for identifying mechanisms of risk and resilience. Although in its infancy, research adopting an iGxE framework is beginning to improve our etiologic understanding of psychopathology risk by identifying putative neural mechanisms. Current iGxE research has primarily focused on candidate polymorphisms of documented functional relevance in the HPA axis that are also predictive of psychopathology risk. More recently, and consistent with the polygenic architecture of HPA axis function, as well as the resolution at which neural and behavioral genetics research is conducted, additive genetic profiles have been constructed that seek to better capture the cumulative influence of multiple variants on HPA axis response and regulation, and subsequently, the impact of such variability on corticolimbic phenotypes, as well as associated risk for psychopathology. Below, we first review studies of candidate polymorphisms within the HPA axis, before turning to polygenic scores and investigations of iGxE outside of the canonical HPA axis (Table 1; Table 2).
FK506 Binding Protein 5 (FKBP5)
FK506 binding protein 5 (FKBP5) is a co-chaperone of the GR complex that diminishes GR sensitivity to cortisol, as well as its access to the transcriptome. Seminal studies by Binder, Ressler, and colleagues have identified a common functional polymorphism within FKBP5, rs1360780, associated with pleiotropic stress-related health effects (eg, depression, immune dysfunction (Zannas and Binder, 2014). The minor T allele of this C/T polymorphism is associated with greater FKBP5 expression following GR activation (Binder et al, 2004). The T allele further predicts a prolonged cortisol response to stress and impaired negative feedback of the HPA axis, as well as elevated early life stress-related depression and PTSD (Binder et al, 2008; Zannas and Binder, 2014; Zimmermann et al, 2011).
Recently, the molecular mechanisms that likely drive the functional consequences of rs1360780 genotype have been detailed (Klengel et al, 2013). This polymorphism influences the 3D structure of FKBP5 by changing the physical proximity of a long-range enhancer region in intron 2 with the transcription start site. Specifically, the greater FKBP5 expression in T allele carriers appears to result from increased physical contact between a GRE within intron 2 and the transcription start site which is likely driven by T allele conferred increased affinity for TATA-box binding protein. As such, the T allele facilitates FKBP5 expression in the context of GR activation, and hence, reduced cortisol-GR binding and impaired HPA axis negative feedback. This effect can be further compounded by T allele-specific childhood stress-related demethylation of a functional GRE within intron 7 of FKBP5, which comes into physical contact with the transcription start site. Demethylation here enhances FKBP5 expression in the context of GR stimulation, resulting in a further reduction in HPA axis negative feedback among T allele carriers. This pattern of allele-specific methylation likely originates from prolonged HPA axis response following stress, owing to the 3D conformational changes linked to rs1360780 genotype. Interestingly, the epigenetic effects on FKBP5 may only emerge during early life, which may help explain the preponderance of research suggesting that adversity in early life, but not adulthood, robustly moderates the effects of this genotype on multiple stress-related phenotypes.
On the basis of clinical associations and the functional characterization of rs1360780 genotype, several recent iGxE studies have examined the impact of this polymorphism and stress exposure on corticolimbic structure and function. For example, we have shown that rs1360780 genotype interacts with childhood adversity to predict increased threat-related reactivity of the dorsal amygdala in European-American (n=139) T allele carriers (White et al, 2012). This rs1360780 genotype x early life stress interaction has subsequently been replicated in young European (n=153) and European-American (n=308) adults in two independent studies (Di Iorio et al, submitted; Holz et al, 2014). That this finding has been most pronounced in the dorsal amygdala, which encompasses the CeA and thus the principal efferent innervation of the PVN (Figure 1), is consistent with rodent literature suggesting that stress upregulates FKBP5 in the CeA, but not the basolateral complex (Scharf et al, 2011). Thus, the interaction of FKBP5 genotype and early adversity on dorsal amygdala activity may reflect sensitized responses to threat. This interpretation is consistent with data suggesting that T allele carriers show an attention bias toward threatening stimuli, as well as increased function connectivity throughout corticolimbic circuitry (Fani et al, 2013; Holz et al, 2014).
In contrast to research on the interaction between early life stress and rs1360780 genotype, evidence for main effects with regard to corticolimbic structure and function has been less consistent. For instance, one study reported a main effect of rs1360780 genotype on threat-related amygdala reactivity, wherein the risk T allele was associated with greater reactivity (Holz et al, 2014). However, other studies have yielded null main effects on amygdala reactivity (Di Iorio et al, submitted; White et al, 2012). Hippocampal structure has been the most studied structural phenotype with null reports of rs1360780 main effects on hippocampal volume (Fani et al, 2013; Hernaus et al, 2014; Pagliaccio et al, 2014). Notably, FKBP5 methylation in intron 7 has been negatively correlated with anterior hippocampal volume, further suggesting that neural effects may only be observable in the context of adversity (Klengel et al, 2013). Moreover, an effect of rs1360780 genotype on differences in hippocampal function and morphology has been observed in a trauma-exposed sample of African-Americans (Fani et al, 2013). Specifically, the risk T allele was associated with greater threat-related hippocampal activity, as well as greater spatial displacement in hippocampal morphology in the CA1 region. This finding is particularly intriguing in light of evidence that CA1 atrophy and impaired long-term potentiation are induced by stress and chronic corticosterone administration in rodents (Alfarez et al, 2003; Sousa et al, 2000), as well as abnormalities in CA1 shape in stress-related psychopathology (Kempton et al, 2011).
Lastly, some effects of rs1360780 genotype have been reported with the structure of the anterior cingulate cortex (ACC), which contributes to monitoring and regulating behavioral responses including those to threat. In a healthy Japanese sample, the T allele has been linked to reduced dorsal ACC volume and reduced white matter integrity in the dorsal anterior cingulum as well as the posterior cingulum, two pathways linking the ACC with other brain regions. Impaired white matter integrity of the posterior cingulum has also been reported among African-American T homozygotes (Fani et al, 2014). These findings suggest that FKBP5 rs1360780 genotype may disrupt white matter integrity within the cingulum and, possibly, white matter connectivity between the ACC and corticolimbic nodes including the amygdala and hippocampus, which may impact the capacity to effectively regulate stress and threat responses. However, notably, these white matter integrity results were not consistent with methodology (ie, mean diffusivity, fractional anisotropy) across these two studies suggesting that further validation is needed.
Collectively, iGxE studies of FKBP5 rs1360780 genotype suggest that the risk T allele is associated with potentiated amygdala reactivity to threat in the context of early life adversity. Such potentiated reactivity may set the stage for the development of stress-related disorders later in life (Figure 2). That this association primarily arises in the context of early life adversity is concordant with evidence that allele-specific epigenetic signatures in intron 7 of FKBP5 are developmentally constrained (Klengel et al, 2013). Importantly, however, this does not suggest that later life stress may not be important. Indeed, given evidence that FKBP5 genotype moderates susceptibility to later PTSD (Binder et al, 2008), this interaction and the heightened amygdala reactivity it confers may predispose individuals to greater sensitivity to future stressful experiences. Although requiring larger samples, it would be interesting to test whether the early life stress × rs1360780 genotype interaction better predicts future psychopathology and related neural phenotypes in the context of recent life stress (ie, early life stress × recent life stress × genotype). It will also be important to assess whether FKBP5 methylation, HPA axis function, and related transcriptome effects mediate the relationship of the interaction between rs1360780 genotype and early life stress and corticolimbic function and structure and further, if these differences mediate the link to psychopathology (Figure 2).

Understanding neural mechanisms underlying psychopathology in iGxE research: Moderated mediational models of FKBP5. Structural equation models allow researchers to examine indirect pathways to examine theoretical mechanisms. (a) FKBP5 genotype is proposed to interact with early life stress to influence FKBP5 methylation. This may, in turn produce differences in cortisol output (and its effect on the transcriptome), and in turn, amygdala activation to threat-related stimuli. (b) Similarly, amygdala activation (in conjunction with many other phenotypes) may partially mediate the association between the interaction of FKBP5 genotype and early life stress on psychopathology. Although models a and b could be combined, it is unlikely that large enough data sets will exist in the near future that have adequately captured these phenotypes.
Not surprisingly, this research has motivated attempts to develop FKBP5 treatments. Up until recently, this work has largely been unsuccessful due to the structural similarity between FKBP5 and FKBP2 (which has opposing functional effects). Notably, however, an FKBP5 specific inhibitor was recently developed that produced antidepressant like effects in mouse models of stress (Gaali et al, 2015). These results are particularly encouraging and are suggestive that FKBP5 inhibition may, eventually, become a useful tool in the psychiatric treatment arsenal.
MR Receptor (NR3C2)
A functional missense Iso/Val polymorphism (rs5522) located in exon 2 of the MR gene (NR3C2) influences its functional efficiency. The Val allele has been associated with reduced cortisol-related function, as well as blunted diurnal cortisol variation and heightened stress reactivity as indexed by neuroendocrine, autonomic, and self-report measures in some but not all studies (DeRijk et al, 2006, 2008; van Leeuwen et al, 2010). Interestingly, reduced MR expression is found in post mortem brains of depressed individuals consistent with observations that the Val allele predicts late life depression and depression-like HPA axis dysfunction (DeRijk et al, 2008; Klok et al, 2011a; van Leeuwen et al, 2010). Moreover, there is evidence from animal models that upregulated MR expression in the basolateral amygdala, wherein primary synaptic circuits for inhibition of amygdala output are located, can have anxiolytic and antidepressant effects (Mitra et al, 2009).
We have found evidence that the val allele interacts with early life stress to influence threat-related amygdala reactivity in children (n=279) (Bogdan et al, 2012b). Specifically, there was a positive association between emotional neglect and threat-related amygdala reactivity in Iso allele homozygotes. In contrast, in the context of low childhood adversity, val-allele carriers had increased amygdala reactivity relative to iso allele homozygotes. There were no genotype differences in reactivity at high levels of maltreatment. Thus, even in the context of low prior adversity, Val allele carriers display amygdala activation similar to individuals with stress-related psychopathology. As such, Val allele carriers may be more vulnerable to the development of stress-related illness, even in the absence of significant environmental stress, perhaps due to the relative reduction in HPA axis inhibition. This interpretation is consistent with evidence that MR blockade raises basal cortisol levels but does not affect stress-induced HPA axis response (Vogel et al, 2015). Notably, this relationship has only been observed in children just entering adolescence and was not replicated in young adults (n=308; Di Iorio et al, submitted). Given evidence of continued amygdala development into adulthood, it is possible that stress-related increases may be observed in Val allele carriers later in life; however, it is also possible that this is a false positive. Clearly, additional replication attempts and extensions to different developmental periods are needed. Lastly, emerging research has shown that along with rs2070950, rs5522 forms a haplotype that is further predictive of differential MR function and risk for psychopathology (Klok et al, 2011b). This haplotype may help explain conflicting reports of the relationship between the rs5522 variant in single SNP analyses of HPA axis function and psychopathology (Klok et al, 2011a; van Leeuwen et al, 2011) and allow future iGxE research to more accurately characterize functional consequences of variation in the MR gene on stress-related risk phenotypes (Di Iorio et al, submitted).
Polygenic Profiles
The vast majority of iGxE research has examined the association between single-polymorphic loci and individual differences in neural phenotypes. In light of small effects typically conferred by single polymorphisms and the utility of multilocus approaches in traditional psychiatric research (Purcell et al, 2009), imaging genetics broadly has begun to incorporate multilocus genetic profiles to quantify aggregate genetic influence (Bogdan et al, 2013a; Holmes et al, 2012; Nikolova et al, 2011). This approach has two primary scoring schemes: (i) the summation of risk alleles or weighted effects providing a ‘risk’ score for a given condition across the genome or within a specified set of genes, or (ii) a biologically informed multilocus profile score (BIMPS) based upon previously reported associations with gene function or downstream consequences representing the function of a biological system (eg, the HPA axis).
Most polygenic research in imaging genetics has used BIMPS. In this approach, genetic polymorphisms are selected based upon known functional associations (eg, receptor expression). In the case of iGxE, this approach has been most often used with regard to measures of HPA axis function (eg, cortisol levels). Strengths of this approach include the use of a priori knowledge, which enhances the interpretation of findings and better integrates this research within the general field of neuroscience and psychiatry. A related limitation, of course is the necessity for a priori functional knowledge. With few exceptions (eg, FKBP5 rs1360780), common polymorphisms have generally been poorly characterized with little to no replication of functional associations. Moreover, additive polygenic profiles typically assume additive effects, despite the strong likelihood of epistatic interactions. Notably, when polygenic approaches are constrained to neural systems and pathways they likely bring genetic data to a resolution that is more consistent with neural and behavioral phenotypes. That is, as neuroimaging assesses higher-order circuit function reflecting the overall activity of thousands of neurons and glia, a polygenic profile assesses higher-order function of a signaling cascade in comparison with a single polymorphism with a likely small effect on only one component of a signaling cascade.
In a study of young adult college students (n=308) we have found that a BIMPS reflective of HPA axis function comprised of FKBP5 rs1360780, CRHBP rs10473984, CRHR1 rs110402; NR3C2 rs5522/rs4635799, predicts threat-related amygdala reactivity and amygdala gray matter volume in the context of early life stress (Di Iorio et al, submitted). Specifically, individuals with elevated BIMPS, reflective of relatively increased HPA axis activity and/or impaired negative feedback, also reporting greater exposure to early life stress had increased threat-related amygdala activity and reduced amygdala gray matter volume, whereas an opposite pattern of findings was observed in those with low BIMPS. In addition, we found that HPA BIMPS predicted elevated anxious arousal, though there was no evidence that the effects on the amygdala mediated this relationship. These functional findings are consistent with a wealth of literature suggesting that HPA axis hyperactivity predicts relatively exaggerated corticolimbic responses and, importantly, identifies individual polygenic liability to these effects.
That elevated exposure to early life stress was associated with smaller amygdala gray matter volume in those with high HPA axis BIMPS (ie, associated with heightened HPA axis activity), but reduced amygdala gray matter volume in those with low BIMPS provides insight into inconsistent (ie, larger, smaller, equivalent) amygdala volume reports in those exposed to early life stress (Hanson et al, 2015; Sheridan et al, 2012; Tottenham et al, 2010). Specifically, these data suggest that individual differences in HPA axis function may meaningfully contribute to variability in early stress-related brain structure. Some prior evidence is consistent with this speculation as temporally-limited ELS predicts increased amygdala volume, whereas a longer duration of exposure, which is associated with impaired HPA axis negative feedback, has been linked to reduced amygdala volume (Mehta et al, 2009). This interpretation is consistent with the hypothesis that initial stress-related hypertrophy within the amygdala may eventually lead to amygdala atrophy in the context of continued adversity and/or HPA axis activity (McEwen, 2003).
In an independent longitudinal study of children (n=120), we have found that an HPA axis genetic risk profile predicts cortisol response to an acute stressor, which mediates the relationship between the genetic profile and later childhood amygdala volume (Pagliaccio et al, 2014). Here, the risk profile was comprised of ten SNPs (CRHR1 (rs4792887, rs110402, es242941, rs242939, rs1876828), NR3C2 (rs5522), NR3C1 (rs41423247, rs10482605, rs10052957), and FKBP5 (rs1360780)) associated with HPA axis function and/or depressive phenotypes. Consistent with literature from which the profile was derived, the profile was associated with elevated cortisol response to acute stress. Most interestingly, the profile interacted with stressful life events during early childhood to predict increased amygdala and hippocampal gray matter volume measured between ages 7–12, which is consistent with a cortisol-mediated mechanism predicting later structural differences. Notably, this finding is opposite the finding we have observed in young adults mentioned above (Di Iorio et al, submitted). This lack of consistency may be the result of developmental timing wherein, in young childhood, early life stress correlates with larger amygdala volume (ie, hypertrophy) in the context of elevated HPA axis activity; however, later, in the context of continued HPA axis activation, this association may transition to atrophy as has been speculated (McEwen, 2003). It will be critically important for further longitudinal research to examine transitions between developmental windows (eg, childhood to adolescence) to directly test this speculation.
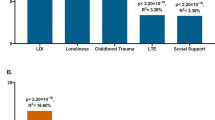
Lastly, the pleiotropic effects of stress likely emerge, at least in part, due to the influence of HPA axis function on the transcriptome. In a multisite collaborative study led by Binder’s group (Figure 3), we recently identified genetic polymorphisms that predict transcript-specific mRNA expression differences following GR agonism (Arloth et al, 2015). Interestingly, we found that polymorphisms predicting GR-driven differences in gene expression were also overrepresented in the major depression mega analysis conducted by the Psychiatric Genomics Consortium (PGC; http://www.med.unc.edu/pgc/downloads). Further, we found that an additive profile across these SNPs predicted depression in a community sample, as well as abnormal amygdala activity (specifically, increased reactivity to neutral relative to threatening facial expression) in a sample of young adult college students. Thus, these results suggest that SNPs predicting individual differences in GR-related gene transcription and depression may potentiate a non-specific amygdala response that speculatively may lead to fear overgeneralization that alters perception of otherwise neutral stimuli as threatening. We are currently collecting concurrent imaging and gene expression data in the context of a dexamethasone challenge to ascertain whether GR-related gene expression mediates associations between dexamethasone challenge and corticolimbic function.

Genetic determinants of the transcriptome response predict depression and generalized amygdala function. In a multisite study, the GR agonist dexamethasone was administered to healthy and depressed participants who were genotyped. RNA expression was measured before and after (+3 h) dexamethasone administration. First, we identified SNPs that were associated with dexamethasone-related changes in gene expression. Next, we examined whether these SNPs were significantly enriched in psychiatric disorders. (a) Evidence of significant enrichment in patients with depression from the PGC depression mega analysis. The red line indicates the number of SNPs that were predictive of GR-related gene expression and depression; the blue and gray bars represent enrichment with regard to random SNPs and SNPs associated with baseline gene expression, respectively. As can be seen, permutation analyses indicated that GR-expression related SNPs are overrepresented in their association with depression. (b–e) the results of these genetic polymorphisms on threat-related amygdala function. We first created a multilocus profile score representing SNPs associated with both GR-related gene expression and depression before regressing this score on threat-related amygdala activation. Curiously, the genetic profile was predictive of relatively reduced amygdala reactivity to threatening stimuli ((c) angry and fearful faces>neutral faces). However, post hoc tests revealed that this profile was predictive of elevated amygdala response to neutral faces ((d); ie, neutral faces>shape matching control task) while being unrelated to threatening faces ((e) angry and fearful faces>shape matching control task). These findings suggest that SNPs associated with both GR-related gene expression and depression predict an overgeneralized amygdala response, wherein neutral faces elicit relatively increased activation. Figure adapted from Arloth et al, 2015.
iGxE Outside of the HPA Axis
Several candidate polymorphisms residing outside the HPA axis have been examined in an iGxE framework with regard to corticolimbic structure and function in the context of stress (Table 2). Because the vast majority of this work has investigated the 5-HTTLPR variant of the serotonin transporter gene (SLC6A4) and the Vall66Met (rs6265) polymorphism of the brain-derived neurotrophic factor gene (BDNF) we focus predominantly on these lines of research.
The serotonin transporter-linked polymorphic region (5-HTTLPR) is an imperfect repeat polymorphism within the 5′ regulatory region of the serotonin transporter gene (SLC6A4) that, in conjunction with 5′ SNPs rs25531 and rs25532, may alter serotonin transporter expression, confer vulnerability to the depressogenic effects of stress, and be associated amygdala function. Relative to the long allele, the short allele at this locus has been linked in some, but not all, studies to reduced 5-HTT expression, increased vulnerability to the depressogenic effects of stress, and heightened amygdala reactivity to threatening stimuli (Bastiaansen et al, 2014; Duncan and Keller, 2011; Hariri et al, 2002; Karg et al, 2011; Kaufman, 2015; Lesch et al, 1996; Murphy et al, 2013; Sharpley et al, 2014). A handful of iGxE studies have examined whether this polymorphism interacts with the environment to predict variability in corticolimbic circuitry with evidence that the short allele is associated with elevated amygdala activity at rest and in response to threatening stimuli among those exposed to relatively elevated life stress (Table 1; Alexander et al, 2012; Canli et al, 2006; Williams et al, 2009). Moreover, two 5-HTTLPR iGxE studies suggest that 5-HTTLPR short allele carriers have smaller hippocampal volume in the context of early life stress (Frodl et al, 2010) and stressful life events (Rabl et al, 2014); however, these findings were not replicated with regard to ELS (Gatt et al, 2009) and were in an opposing direction when considering perceived stress in another study (Zannas et al, 2013). Lastly, one study provides evidence that, in women, 5-HTTLPR genotype moderates the association between stressful life events and resting-state functional connectivity of the parahippocampus and posterior cingulate cortex, as well as structural connectivity of the hippocampus and both the amygdala and putamen (Favaro et al, 2014). Notably, iGxE research on the 5-HTTLPR to date has been conducted in relatively small samples with no direct replications (ie, same neural phenotype and environmental assessment/manipulation). As such, although this preliminary research suggests that stress and 5-HTTLPR genotype may interact to shape corticolimbic structure and function, which may contribute to increased stress-related psychopathology in short allele carriers, validation of these results are needed, particularly in light of mixed effects reported in the literature (Bastiaansen et al, 2014; Duncan and Keller, 2011; Karg et al, 2011; Kaufman, 2015; Murphy et al, 2013). Given evidence that 5-HTTLPR × stress effects are most pronounced when considering early life adversity, as opposed to lifetime measures of stress, iGxE studies focusing on early life stress exposure may prove most fruitful (Bogdan et al, 2014; Karg et al, 2011).
The Met allele of the BDNF Vall66Met (rs6265) polymorphism has been linked to reduced BDNF function, as well as depression and reduced hippocampal volume in some, but not all studies (Egan et al, 2003; Elzinga et al, 2011; Verhagen et al, 2010). Perhaps most interestingly from a GxE perspective, there is evidence that Met allele carriers who have been exposed to childhood abuse had the lowest levels of serum BDNF (Elzinga et al, 2011). Given the key role of BDNF supporting the survival and growth of neurons, several iGxE studies have examined associations between rs6265 genotype and gray matter volume. Across these studies, reduced hippocampal volume has been reported among Met allele carriers who have been exposed to elevated levels of stress (Table 2; Carballedo et al, 2013; Frodl et al, 2014b; Gatt et al, 2009; Rabl et al, 2014; but see also Gerritsen et al, 2012). These results suggest that BDNF reductions in Met allele carriers exposed to early life stress, may potentiate stress- and cortisol-related effects on hippocampal volume, perhaps by not buffering the survival of these cells (Frodl and O’Keane, 2013). Such stress-related moderation may explain inconsistent main effect associations between rs6265 genotype and hippocampal volume (Molendijk et al, 2012).
In addition to the SLC6A4 5-HTTLPR and BDNF Val66Met polymorphisms, a handful of other iGxE studies have explored polymorphisms within other genes (ADRA2B, NPSR1, COMT, OPRK1; Table 1). Because these findings have largely emerged in single studies, we only briefly review polymorphisms for which two or more iGxE reports exist (additional single studies are noted in Table 2). First, a common deletion (−4825 indel) in the gene coding for the presynaptic noradrenergic α2B receptor (ADRA2B) is associated with blunted agonist-promoted receptor desensitization, elevated re-experiencing of traumatic memory and elevated amygdala reactivity to negative pictures, may moderate the effects of acute stress on corticolimbic circuitry (de Quervain et al, 2007; Rasch et al, 2009; Small et al, 2001). However, the two reports iGxE reports on this findings have used vastly different paradigms. In one study, acute stress blunted amygdala reactivity to emotionally morphing faces among non-deletion carriers (Cousijn et al, 2010); however, in the other, it was deletion carriers who had blunted amygdala reactivity when recognizing pictures depicting neutral facial expressions, whereas fearful faces were not associated with −4825 indel genotype (Li et al, 2015). Whether these different findings are the result of task differences or may be indicative of false positives is presently unclear.
Three studies have examined whether a common functional polymorphism (rs4680; Val158Met) within the catechol-O-methyltransferase gene (COMT), which codes for a catecholamine catabolic enzyme, moderates associations between stress exposure and corticolimbic neural phenotypes. The Val158Met polymorphism has been primarily associated with differential prefrontal dopamine function, schizophrenia, and working memory, though meta-analyses suggest these main effects may not be stable (Bilder et al, 2004; Egan et al, 2001; Munafo et al, 2005; Rabl et al, 2014; Wardle et al, 2013). Met allele homozygosity, which is associated with reduced COMT expression and hence elevated catecholamine levels in the prefrontal cortex, is predictive of a negative relationship between stressful life events and hippocampal volume; an opposing, positive pattern was observed in Val homozygotes (Rabl et al, 2014).
An independent line of research has examined associations between COMT rs4680 genotype and stress with working memory and related neural function. One such study found that among Met allele homozygotes, acute stress induced poorer working memory performance, and blunted dorsolateral prefrontal cortex (DLPFC) activation; whereas performance benefits and increased DLPFC activation was observed in Val allele carriers (Qin et al, 2012). Lastly, in one of the first neuroimaging studies incorporating measures of methylation, Ursini et al (2011) noted that the Val allele at rs4680 creates a CpG site wherein methylation can occur that is absent in the Met allele. Here, in Val homozygotes, a composite measure of lifetime stress was associated with reduced methylation in COMT, which in turn was associated with increased peripheral measures of COMT expression. Perhaps most interestingly, elevated methylation, which was associated with reduced stress exposure, was predictive of greater working memory performance and reduced prefrontal cortex activity (interpreted as greater efficiency) among Val homozygotes. Thus these results, suggest that in the absence of stress exposure, the increased methylation present among Val homozygotes may produce COMT expression more similar to the Met allele and improvements in working memory performance; whether this would then leave individuals homozygous for the Val allele with no lifetime stress exposure susceptible to acute stress-induced deficits in memory performance and blunted DLPFC activation is unclear. However, it is possible that such GxE interactions may at least partially contribute to mixed main effects observed in the literature of this candidate polymorphism (Munafo et al, 2005; Wardle et al, 2013). Collectively, these results point to the importance of considering GxE in the context of neuroimaging, particularly when there are probable molecular mechanisms that may promote such effects (eg, the creation of a methylation cite by the Val polymorphism of the Val158Met polymorphism).
CONCLUSIONS AND FUTURE DIRECTIONS
Genetic variation, particularly within the HPA axis, predicts stress-related differences in coticolimbic structure and function. The most replicated association has been an interaction between FKBP5 rs1360780 genotype and early life adversity predicting threat-related amygdala function. This finding complements psychiatric associations by providing evidence that potentiated stress-related enhancement of corticolimbic function is a plausible neurobiological mechanism underlying clinical association. Greater faith in this finding comes from convergent research in non-human animals and the molecular characterization of this polymorphism (Klengel et al, 2013; Scharf et al, 2011; Zannas and Binder, 2014). With the exception of the BDNF rs6265 Met allele conferring reduced hippocampal volume in the context of elevated stress, other investigations of single-polymorphic loci have not yet been replicated (Frodl et al, 2014b). Recently, polygenic approaches have been used which aggregate genetic variation across the HPA axis. This approach is intuitively appealing as it is likely more consistent with the resolution at which neural and behavioral genetics research is conducted and polygenic HPA axis research is already beginning to shed light on individual difference factors that may contribute to mixed findings within the psychiatric/early life stress literature (eg, amygdala structure enlargement and reductions (Di Iorio et al, submitted; Pagliaccio et al, 2014)). However, notably, such polygenic analyses have not been regularly conducted and are in need of replication and developmental extension. Like all research approaches, iGxE research is confronted by many challenges, four of which are among the most daunting: (i) small effects, (ii) measuring the environment and neural phenotypes, (iii) understanding detailed mechanisms, and (iv) incorporating development into research design. Here we review these challenges and discuss strategies to address them.
Small Effects—Increasing Sample Size and Polygenic Analyses
Much like the effects of common genetic variation on psychiatric vulnerability and behavior, evidence suggests that common polymorphisms will have, at most, only a small effect on neural measures (Hibar et al, 2015; Iacono, 2014). Such weak penetrance is difficult to detect and may account for inconsistent findings (ie, false positives and negatives) within imaging genetics, particularly in studies using small samples, such as those within the iGxE field reviewed here. Traditionally, the prohibitive monetary and resource cost to both neuroimaging and genetic research has constrained sample sizes. However, improvements in availability, affordability, and data management and analyses in both neuroimaging and genetics have allowed for the development of large-scale studies (Bogdan et al, 2012b; Holmes et al, 2015; Nikolova et al, 2012; Schumann et al, 2010), multisite data pooling protocols (Barch et al, 2013; Holmes et al, 2012), and data sharing networks (Thompson et al, 2014) in an effort to increase power to detect small effects.
Neuroimaging measures in genetics research have been lauded for their value as biological processes intermediate to genes and the expression of clinical syndromes (ie, as intermediate phenotypes; Bogdan et al, 2013b; Cannon and Keller, 2006; Gottesman and Gould, 2003; Hariri and Weinberger, 2003; Hasler et al, 2004; Meyer-Lindenberg and Weinberger, 2006). Consequently, we and others have suggested that these phenotypes should produce relatively larger effects than more distal behavioral and psychopathology phenotypes (Bogdan et al, 2013b; Hariri and Weinberger, 2003; Hasler et al, 2004; Meyer-Lindenberg and Weinberger, 2006). Although there is some evidence to support the assumption of larger effects sizes in neuroimaging relative to clinical and behavioral phenotypes, this evidence is also confounded by studies characterized by smaller samples, which generally report larger effects (Rose and Donohoe, 2013). A series of reports originating from the Minnesota Twin Family Study, the largest genetic study of EEG-related phenotypes (n=4905), suggests that neural phenotypes may be linked to common genetic variation with approximately the same strength of traditional psychiatric and behavioral phenotypes (Iacono, 2014). The largest investigation of intermediate phenotypes (discovery n=13 171; replication n=17 546), the ENIGMA consortium, recently identified genome-wide significant signals for measures of brain volume (Hibar et al, 2015). The strongest effect emerged within the putamen for an intergenic SNP, rs945270, which is associated with KTN1 expression; critically, the effect of this SNP is sobering as it explained only 0.52% of variance in putamen volume. This relatively small effect, is consistent with effects of single variants on other complex traits (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Although intermediate phenotypes have been assumed to provide better traction into genetic architecture contributing to psychopathology, this evidence challenges this assumption. Future research would ideally compare effect sizes to neural intermediate phenotypes and psychiatric and behavioral phenotypes within the same sample. If indeed, neural intermediate phenotypes are not associated with larger genetic effects, intermediate phenotype research may prove most useful for understanding the neurobiological mechanisms through which genetic variation influences behavior.
Although GxE research is intuitively appealing on a theoretical basis, it is met with unique challenges from a statistical power perspective (Dick et al, 2015; Duncan and Keller, 2011). In GxE research, both genotype frequency and variability in environmental experience constrain the sample; this is potentiated in SNPs with relatively rare minor alleles and in environments that are extreme or specific. For example, a recent report suggests that opioid-related genetic variation moderates the association between stressful life events and depression in a modestly sized sample (n=420). However, the only group that differed had a cell sample size of 6 (ie, carriers of the rare G allele at rs1799971 who were also exposed to targeted rejection events (Slavich et al, 2014)); effects driven by such small cells, in the absence of rare highly penetrant mutations, are unlikely to be stable. If iGxE research is associated with similar effects of traditional psychiatric GxE research (eg, Duncan and Keller, 2011), all iGxE studies to date have been vastly underpowered. Lastly, the statistical power of GxE is further reduced, particularly in small studies, due to the need to account for genotype × covariate and environment × covariate interactions in analyses, which limits degrees of freedom more than main effect analyses (Keller, 2014). Going forward it will be important for iGxE research to consider such interactions between predictor variables and covariates of no interest (ie, G × covariates and E × covariates), which is becoming increasingly common (Carey et al, in press; Di Iorio et al, submitted; Pagliaccio et al, 2014, 2015; Rabl et al, 2014).
In addition to increasing sample size, iGxE research may be able to increase power by using polygenic analytic techniques. Following increasing accessibility of genome-wide data, novel methods have been developed to leverage its high dimensionality. In particular, in addition to the additive profile scores discussed above, three additional approaches may prove fruitful: (i) polygenic risk scores, (ii) gene/systems-level testing, and (iii) regression trees.
The polygenic risk scoring approach generally relies upon well-powered GWAS results (though investigations have been conducted in an unweighted fashion using candidate genetic loci (Pearson et al, 2014) that are relevant to the neural phenotypes under study such as those provided by mega analyses of categorical disorder risk conducted by the PGC (http://www.med.unc.edu/pgc/downloads; Smoller et al, 2013). A benefit of this approach is that it likely better captures the polygenic nature of psychopathology. A limitation is that these genetic risk scores are based on statistical association with a phenotype and in isolation provide limited insight into potential biological mechanisms underlying observed neural associations. Moreover, like the vast majority of widely employed polygenic methods, this approach assumes an additive model, that neglects potential epistatic effects. Notably, although epistatic effects are suggested from molecular interactions in non-human animal models (eg, Gray et al, 2015), reported epistatic interactions in the imaging genetics literature have not been replicated to our knowledge (eg, Andreasen et al, 2012; Tan et al, 2007).
Perhaps the most important limitations of disorder-based polygenic risk scores when used in iGxE research, is that they were generated without consideration of the environment and are inherently constrained by the phenotype from which they were generated. Psychiatric disorders are characterized by heterogeneity in presentation, stability, and symptoms (Flint and Kendler, 2014; Hasler et al, 2004). For instance, according to DSM-5, five of a possible nine symptoms are required to be diagnosed with depression (with at least one of these symptoms being either depressed mood or anhedonia; American Psychiatric Association, 2013). This results in hundreds of possible different symptom combinations that qualify for the same diagnosis but presumably reflect distinct underlying pathophysiologies. This heterogeneity within disorders produces polygenic risk scores that may not clearly map onto discrete behaviors associated with a particular neural phenotype. For example, although anhedonia, which reflects an inability to anticipate and/or experience pleasure, is a cardinal symptom of depression, according to some studies it is only the fifth most common symptom endorsed by depressed patients with a varied expression ranging from 37–77% (Lewinsohn et al, 1998; Pelizza and Ferrari, 2009). As such, polygenic risk scores generated from a GWAS of depression diagnosis alone likely include estimates unrelated to anhedonia that may not be appropriate predictors for a neuroimaging study of reward processing.
Put more generally, it is possible that distinct underlying pathophysiologies may have a more specific polygenic basis than what is captured in polygenic disorder risk scores. If this in indeed the case, a research domain criteria approach to generating polygenic risk scores that is free from categorical diagnostic status may help catalyze research that will be more readily translated to iGxE research (Insel et al, 2010). Notably, however, given the small effects of common genetic variants, this approach is constrained by access to large data sets with measured phenotypes; as such, GWAS of individual symptoms may be a powerful starting point. In addition to their potential utility for iGxE research, such analyses could potentially inform psychiatric nosology by showing genetic consistency or divergence across specific symptoms and syndromes (Bulik-Sullivan et al, 2015).
This limitation notwithstanding, polygenic risk scores hold great promise for iGxE research. In addition to their use across the genome, polygenic scores could be calculated for sets of SNPs within distinct neural systems. Such an approach may be particularly important for providing SNP level priors, when there is evidence to support a systems-level involvement in a given neural phenotype, but limited evidence to prioritize particular SNPs (eg, the circadian system). Integrating prior information from psychiatric disorder risk (ie, prioritizing particular SNPs) and neuroscience (ie, collating SNPs involved in a specific neural system/s) may be informative for iGxE research of neural risk mechanisms.
From an atheoretical perspective, various ways to mine GWAS data have been developed. For example, software packages have been developed (eg, GATES (Li et al, 2011); VEGAS (Liu et al, 2010)) that combine single SNP p-values generated from GWAS and assigns each SNP to gene to create a summary statistic representing gene-based associations (Liu et al, 2010). This approach has identified significant gene-based contributions to phenotypes when single SNP GWAS have failed (eg, Agrawal et al, 2014). A similar approach, gene-set enrichment, which can be conducted with GWAS-based single SNP p-values using various software packages (eg, Aligator (Holmans et al, 2009); INRICH (Lee et al, 2012); MAGENTA (Segre et al, 2010), evaluates whether sets of genes in predefined biological processes or functionally-related genes (based upon gene sets in online databases or user specified gene sets) show patterns of enriched associations with a given phenotype. For example, gene-set enrichment analyses have linked voltage-gated cation channel activity-related genes to working memory performance, related brain function, and schizophrenia (Heck et al, 2014).
Lastly, in an attempt to characterize epistatic relationships, machine learning approaches have been applied to data (Andreasen et al, 2012; Arnedo et al, 2015). Although there are promising associations reported, this approach is highly susceptible to false positives and what constitutes replication is unclear as the primarily predictive variables are generated internally from the data set. It will be critical for future regression tree-based exploratory research to ascertain precisely what interactions are driving effects so that these specific interactive pathways can be evaluated within independent data set; this has been done in some (Andreasen et al, 2012), but not other (Arnedo et al, 2015) machine learning approaches. It will be important for future iGxE studies, and imaging genetics research more generally to complement traditional candidate gene approaches with big data approaches to synthesizing complex data sets (eg, gene-set enrichment, machine learning) that are becoming more normative in psychiatric genetics. Moreover, in the context of iGxE, it will be important for iGxE research to use new developments in knowledge to target particular systems, such as transcripts influenced by cortisol signaling (Arloth et al, 2015).
Measuring the Environment and Neural Phenotypes
The ability to conduct research on a given construct is constrained by how precisely it can be measured. With sufficient quality control, genotyping can be measured very reliably. However, measuring stressful experiences and neural phenotypes of interest is fraught with difficulty (for review see Glover, 2011; Monroe, 2008). Measuring life stress in iGxE studies is particularly difficult, because the studies that are powered to detect moderations are typically designed to recruit a large number of participants to complete an imaging protocol. The ability to assess other factors is often limited by resources and the vast majority of these large studies assess the environment with retrospectively reported measures of early life stress and recent life stress checklists. Although there are psychometric data supporting these measures, there is also some evidence that more thorough assessments may better capture stress (Bernstein et al, 2003; Dohrenwend, 2006; Monroe, 2008). Notably, however, such interview based methods such as the Life Events and Difficulties Schedule (LEDS) or Stressful Life Events Schedule (SLES) can often take hours to administer and further hours from multiple trained raters to score (Brown and Harris, 1978; Williamson et al, 2003). This additional burden in already complex studies has largely excluded their use in large-scale imaging genetics studies, despite their potential utility. Notably, new computer-based stress assessments have recently been developed that are less time intensive to administer and score that may prove beneficial for large-scale studies of stress exposure (Slavich and Epel, 2010). A further difficulty confronting large imaging genetics studies incorporating the environment is that, with few exceptions, these studies are generally cross-sectional which prohibits the examination of change in neural phenotypes in the context of a changing environment (Swartz et al, 2014b, 2015). Moreover, within some large cross-sectional studies assaying stress, it is impossible to ascertain the temporality of stress exposure in relation to concurrently measured symptoms; indeed this temporality problem has resulted in many studies evaluating only early life stress, as opposed to recent life stress, when attempting to link environmental experience, brain function, and psychiatric symptoms or related behavior (Moffitt and Caspi, 2014).
A complementary approach to measuring the environment is to acutely manipulate stress to examine its influence on neural function in the context of genetic variation. Although such studies are rare in a neuroscience context, some studies have used stress manipulations while measuring neural phenotypes and assaying genetic variation (Bogdan et al, 2011; Cousijn et al, 2010; Streit et al, 2014; Xu et al, 2013). These within-participant studies are especially powerful because they afford researchers a unique opportunity to experimentally manipulate stress and ensure that each participant is exposed to the same stressor, providing greater reliability and validity of what stress is across participants that is free from reporting biases. However, such studies are intensive to run and exceptionally difficult to have large numbers of participants complete and their ecological validity is questionable. A related powerful and perhaps more difficult methodology to implement is pharmacologically challenging circuits involved in stress response (eg, HPA axis, norepinephrine), ideally in a placebo-controlled crossover design (Bigos et al, 2008; Henckens et al, 2010, 2011). Pharmacologic challenge has identified genetic differences in stress-related gene transcription that are further associated with psychopathology and related brain function (Arloth et al, 2015). However, it has yet to be commonly employed in human genetic research. Moreover, the ecological validity of such challenge studies is reduced due to their specificity as these studies traditionally influence a single receptor that in typical life experience is activated alongside a symphony of other neural cascades.
Lastly, with regard to the environment, theoretical work suggests that some genetic variants that have typically been conceptualized as conferring risk to psychopathology in the context of negative environments may be more accurately conceptualized as conferring sensitivity to the environment, for better or worse (Belsky et al, 2009). This theory has begun to receive empirical support in candidate gene studies showing that some genotypes previously conceptualized as conferring risk to negative environments also confer benefits in the context of positive environments (Hankin et al, 2011). As such, it will be important for iGxE research, to not only measure adverse environmental conditions, which is most common in psychopathology-related research, but to also measure enriching experiences (eg, social support, positive parenting; Hyde et al, 2011b). It is highly probable that such data would also further help untangle the complexity of factors that influence neural phenotypes (eg, buffering effect of supportive social relationships on negative life experiences). There are cautions to be aware of with this research approach, however. Most notably, data suggest that such crossover interactions are more likely to emerge by chance relative to other shapes of interaction (Dick et al, 2015); given that the vast majority of results supporting the plasticity model have been in small samples, it will be important for these effects to be observed in more well-powered studies.
Another outstanding issue in imaging genetics research, and the field of neuroimaging more generally, is the largely unexplored test–retest reliability of neural phenotype measures. Although structural measures such as gray matter volume appear to be highly reliable (Bartzokis et al, 1993), the reliability of functional measures is mixed. With regard to tasks designed to elicit corticolimbic activation, some studies suggest moderate to good reliability whereas other studies suggest relatively poor reliability (Manuck et al, 2007; Plichta et al, 2012, 2014; Sauder et al, 2013). Critically, the vast majority of test–retest reliability research has used exceptionally small samples (ns<30) and further attention to this issue is clearly needed, which may be achieved through collaborative releases of large neuroimaging data sets (eg, Zuo et al, 2014). Moreover, it will be critical for future research to not only assess test–retest reliability but to also understand factors that may reduce reliability (eg, time of day, hydration, fatigue, mood, etc) so that future studies may be optimized (Nakamura et al, 2015). In the context of iGxE research reliable imaging measures are needed to produce replicable associations with genetic variation, environmental experience, and psychopathology symptomatology.
Mechanistic Understanding
One of the primary goals of neuroscience, psychology, and psychiatry is to understand the mechanisms that drive behavioral variability. iGxE complements psychological and psychiatric genetic research by adding a plausible biological mechanism—the brain—through which genetic variation and environmental experience give rise to the vast array human behavior. iGxE is uniquely positioned to explicitly test whether genetic variation and the environment contribute to individual differences in neural phenotypes and whether these neural differences indirectly link GxE to behavior and psychopathology (Hyde et al, 2011a). To do this, iGxE can use structural equation modeling tools such as path analysis, which were developed by geneticist Sewall Wright and are now most commonly used in the social sciences (Figure 2; Duncan, 1966; Hyde et al, 2011a; Wright, 1920). Although testing these models allows one to evaluate theoretical neural mechanisms underlying behavior, there are important considerations when conducting these analyses. First, this analytic approach further emphasizes the need for large samples which is potentiated when considering less common alleles and environmental experiences. Second, the ideal manner in which to conduct mediational analyses is between temporally distinct assessments. However, with few exceptions, the vast majority of iGxE research assesses psychiatric symptoms and behaviors within days of imaging data collection; as such, the strength of interpretation that neural differences or environmental experiences preceded the development of psychiatric symptoms or other behavior differences is an unsupported assumption that needs to be considered in interpretation. Ideally, prospective studies would be conducted (Burghy et al, 2012; Swartz et al, 2015). In addition to examining the brain as a mediator between genetic variation and environmental experience and behavior, it will be further critical to examine whether biomarkers (eg, cortisol, gene expression) mediate links between genetic variation and environmental experience and brain phenotypes (Figure 2; Pagliaccio et al, 2014). Ideally, such measures could be taken in the context of longitudinal assessment and be measured under different conditions (eg, cortisol: diurnal variation, stress-evoked activation).
Transdisciplinary research can be further used to detail mechanisms underlying effects of genetic, environmental, and neural mechanisms on behavior. Natural orthologous genetic variants and the ability to manipulate genetic code in non-human animals (eg, transgenic animals) and cell-based assays are useful for interpreting iGxE research (Caspi et al, 2010). Transgenic mouse models have provided insight into mechanisms through which differences in genetic sequence impact neural function and behavior; these models are particularly powerful in the context of iGxE as the environment can be explicitly controlled and manipulated. For example, a knock-in mouse model that parallels a common human polymorphism, rs324420, in FAAH within the eCB system was recently developed (Dincheva et al, 2015). The rs324240 polymorphism has been linked to functional differences in FAAH and related correlations to behavioral and neural phenotypes (eg, amygdala habituation, stress sensitivity; Gunduz-Cinar et al, 2013a, 2013b; Hariri et al, 2009). This genetic knock-in model shows remarkable parallels in not only biochemistry, but also neurocircuitry and behavior that are consistent with the naturally occurring human polymorphism. Given reciprocal relationships between AEA and the HPA axis, this provides a novel model organism to better understand how this particular variant might influence behavioral susceptibility to stress. Recent developments in genetic engineering using the CRISPR/Cas9 system to disrupt genes across species provides more precise targeted genetic manipulation that may ultimately prove useful for not only clinical treatment but also modeling the effects of polymorphisms (Jinek et al, 2012; Sander and Joung, 2014); however, the recent use of this technology in human embryos has generated significant ethical controversy (Cyranoski and Reardon, 2015; Lanphier et al, 2015).
Lastly, an important potential mechanism underlying iGxE results is epigenetic regulation. For example, genetically-conferred differences in epigenetic regulation may underlie FKBP5-related observations in psychopathology and brain function (Klengel et al, 2013). Moreover, there is evidence that a widely studied variant in COMT, rs4680, confers the presence or absence of a methylation site that may underlie at least some of this polymorphisms effects (Ursini et al, 2011). Indeed, the potential impact of methylation on human brain function has been recently demonstrated (Booij et al, 2015; Dannlowski et al, 2014; Frodl et al, 2015; Klengel et al, 2013; Nikolova and Hariri, 2015; Nikolova et al, 2014; Ziegler et al, 2015). For instance, methylation within the serotonin transporter gene predicts increased threat-related amygdala reactivity, as well as decreased SLC6A4 expression in post mortem brain tissue (Nikolova et al, 2014). These results suggest that considering methylation is critical to understand the impact of the genome on psychiatrically-relevant brain function and suggests that peripheral measures of methylation may, at least in some cases, be of utility in imaging genetics research, provided that such measures find convergence with post mortem tissue. In particular, it will be important to examine epigenetic regulation as a potential mechanism underlying GxE effects (eg, Ursini et al, 2011; Figure 2). In addition to assaying methylation in ongoing studies, novel bioinformatic tools, such as the Wash U Epigenome Browser (http://epigenomegateway.wustl.edu/) may be of utility for understanding the epigenetic landscape within which a genetic polymorphisms reside (Carey et al, in press; Zhou et al, 2011, 2013). This may be particularly useful for understanding the potential impact of polymorphisms that have not been characterized functionally and may provide very preliminary evidence of potential functional consequences of a SNP, even if curated data sets (eg, Brain Cloud: http://braincloud.jhmi.edu/, Colantuoni et al, 2011) suggest the variant is unrelated to expression. Such knowledge could prioritize genetic variants to examine in the context of methylation-related research.
Developmental Timing
Despite evidence that there are vast differences within corticolimbic circuitry according to development, very few studies have examined neural phenotypes across developmental windows (Giedd et al, 2015). Indeed rapid neural growth and pruning in childhood might lead to a developmentally sensitive period wherein the corticolimbic circuitry may be most sensitive to negative and positive experiences (Andersen et al, 2008; Teicher et al, 2003). For example, a host of studies suggest that adolescence is characterized by heightened amygdala responsivity coupled with a low capacity for frontal regulation that may impair emotion regulation and provide a developmental window that is ripe for the development of stress-related psychopathology (Johnson and Casey, 2015). Similarly, there is evidence for differential HPA axis function across development, as well as its association between early life stress and indices of HPA axis function in childhood and adulthood (Lupien et al, 2009; Shirtcliff et al, 2012; Tarullo and Gunnar, 2006). Although rare in neuroimaging studies, prospective studies that allow for dynamic changes across development are clearly needed in which both neural, environmental, and genetic (eg, epigenetic, gene expression) factors are assayed longitudinally; such designs, in large samples, would be ideally positioned to assay the mechanisms through which psychopathology develops (Luby et al, 2014; Pagliaccio et al, 2014; Swartz et al, 2014b).
CONCLUSIONS
The integration of the environment into imaging genetic research has the potential to inform our understanding of the origins of neural mechanisms underlying stress-related psychopathology. Indeed, this research approach has yielded novel insights into the neural mechanisms that may underlie associations between both FKBP5 rs1360780 genotype and BDNF rs6265 genotype with stress-related psychopathology (Gatt et al, 2009; White et al, 2012). Research on additional single variants and polygenic profiles have begun to report significant effects that, if replicated, would help inform the individual difference factors that give rise to variability in stress-related neural phenotypes (Tables 1 and 2). It is critical for iGxE research to address the challenges of: (i) small effects, (ii) measuring the environment and neural phenotypes, (iii) the lack of detailed mechanisms, and (iv) integrating research into a developmental framework. To address the problem of small effects, imaging genetics has begun to rely on larger more well-powered studies and use polygenic approaches such as enrichment to compliment more traditional candidate polymorphism research (Hibar et al, 2015). Imaging genetics has just begun to consider the environment; going forward it will be important for this research to measure environmental experience as accurately as possible and to conduct challenge studies to explicitly target stress-related neural circuitry (Dick et al, 2015). Moreover, it will be important for the test–retest reliability of imaging designs to be evaluated in large studies that attempt to account for confounding factors. To understand mechanisms, it will be important for well-powered iGxE to assess moderated mediation models and for collaboration across iGxE researchers and non-human animal models, while also assessing signatures of epigenetic regulation (Bogdan et al, 2013a; Hyde et al, 2011a). Lastly, given evidence of differences across development in corticolimbic circuitry and HPA axis function (Johnson and Casey, 2015; Lupien et al, 2009), future iGxE studies using longitudinal prospective designs will be particularly useful. Although these challenges are undoubtedly daunting, addressing them is paramount to the potential insight offered by iGxE research. In doing so, iGxE will be uniquely able to identify neural mechanisms through which genetic variation and environmental experience confer vulnerability to psychopathology.
FUNDING AND DISCLOSURE
RB received support from the Klingenstein Third Generation Foundation, McDonnell Center for Systems Neuroscience, and NIA (R01-AG045231). DP was supported by NIGMS (T32 GM081739). DAAB was supported by NIMH (T32-GM008151) and NSF (DGE-1143954). ARH received support through NIDA (DA033369 and DA031579).
References
Agrawal A, Lynskey MT, Bucholz KK, Kapoor M, Almasy L, Dick DM et al (2014). DSM-5 cannabis use disorder: a phenotypic and genomic perspective. Drug Alcohol Depend 134: 362–369.
Alexander N, Klucken T, Koppe G, Osinsky R, Walter B, Vaitl D et al (2012). Interaction of the serotonin transporter-linked polymorphic region and environmental adversity: increased amygdala-hypothalamus connectivity as a potential mechanism linking neural and endocrine hyperreactivity. Biol Psychiatry 72: 49–56.
Alfarez DN, Joels M, Krugers HJ (2003). Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci 17: 1928–1934.
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Publishing, Inc.: Washington, DC, USA.
Andela CD, Van Haalen F, Ragnarsson O, Papakokkinou E, Johannsson G, Santos A et al (2015). Mechanisms in endocrinology: Cushing's syndrome causes irreversible effects on the human brain: a systematic review of structural and functional MRI studies. Eur J Endocrinol 173: R1–R14.
Andersen SL, Tomada A, Vincow ES, Valente E, Polcari A, Teicher MH (2008). Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J Neuropsychiatry Clin Neurosci 20: 292–301.
Andreasen NC, Wilcox MA, Ho BC, Epping E, Ziebell S, Zeien E et al (2012). Statistical epistasis and progressive brain change in schizophrenia: an approach for examining the relationships between multiple genes. Mol Psychiatry 17: 1093–1102.
Anisman H, Zaharia MD, Meaney MJ, Merali Z (1998). Do early-life events permanently alter behavioral and hormonal responses to stressors? Int J Dev Neurosci 16: 149–164.
Arloth J, Bogdan R, Weber P, Frishman G MA, Wagner KV, Schmidt MV et al (2015). Genetically determined differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron 86: 1189–1202.
Arnedo J, Svrakic DM, Del Val C, Romero-Zaliz R, Hernandez-Cuervo H et alMolecular Genetics of Schizophrenia C (2015). Uncovering the hidden risk architecture of the schizophrenias: confirmation in three independent genome-wide association studies. Am J Psychiatry 172: 139–153.
Barch DM, Burgess GC, Harms MP, Petersen SE, Schlaggar BL, Corbetta M et al (2013). Function in the human connectome: task-fMRI and individual differences in behavior. NeuroImage 80: 169–189.
Bartzokis G, Mintz J, Marx P, Osborn D, Gutkind D, Chiang F et al (1993). Reliability of in vivo volume measures of hippocampus and other brain structures using MRI. Magn Reson Imaging 11: 993–1006.
Bastiaansen JA, Servaas MN, Marsman JBC, Ormel J, Nolte IM, Riese H et al (2014). Filling the gap: relationship between the serotonin-transporter-linked polymorphic region and amygdala activation. Psychol Sci 25: 2058–2066.
Belsky J, Jonassaint C, Pluess M, Stanton M, Brummett B, Williams R (2009). Vulnerability genes or plasticity genes? Mol Psychiatry 14: 746–754.
Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T et al (2003). Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl 27: 169–190.
Bigos KL, Pollock BG, Aizenstein HJ, Fisher PM, Bies RR, Hariri AR (2008). Acute 5-HT reuptake blockade potentiates human amygdala reactivity. Neuropsychopharmacology 33: 3221–3225.
Bilder RM, Volavka J, Lachman HM, Grace AA (2004). The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology 29: 1943–1961.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299: 1291–1305.
Binder EB, Nemeroff CB (2010). The CRF system, stress, depression and anxiety-insights from human genetic studies. Mol Psychiatry 15: 574–588.
Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B et al (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36: 1319–1325.
Bogdan R, Agrawal A, Gaffrey MS, Tillman R, Luby JL (2014). Serotonin transporter-linked polymorphic region (5-HTTLPR) genotype and stressful life events interact to predict preschool-onset depression: a replication and developmental extension. J Child Psychol Psychiatry 55: 448–457.
Bogdan R, Hariri AR (2012a). Neural embedding of stress reactivity. Nat Neurosci 15: 1605–1607.
Bogdan R, Hyde LW, Hariri AR (2013a). A neurogenetics approach to understanding individual differences in brain, behavior, and risk for psychopathology. Mol Psychiatry 18: 288–299.
Bogdan R, Nikolova YS, Pizzagalli DA (2013b). Neurogenetics of depression: a focus on reward processing and stress sensitivity. Neurobiol Dis 52: 12–23.
Bogdan R, Santesso DL, Fagerness J, Perlis RH, Pizzagalli DA (2011). Corticotropin-releasing hormone receptor type 1 (CRHR1) genetic variation and stress interact to influence reward learning. J Neurosci 31: 13246–13254.
Bogdan R, Williamson DE, Hariri AR (2012b). Mineralocorticoid receptor Iso/Val (rs5522) genotype moderates the association between previous childhood emotional neglect and amygdala reactivity. Am J Psychiatry 169: 515–522.
Bolton JL, Hayward C, Direk N, Lewis JG, Hammond GL, Hill LA et al (2014). Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin. PLos Genet 10: 7.
Booij L, Szyf M, Carballedo A, Frey EM, Morris D, Dymov S et al (2015). DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: a study in depressed patients and healthy controls. PLoS One 10: e0119061.
Brown GW, Harris TO (1978) Social Origins of Depression: A Study of Psychiatric Disorder in Women. 1st edn. Free Press: New York, USA, xi, 399 pp.
Bruhl AB, Delsignore A, Komossa K, Weidt S (2014). Neuroimaging in social anxiety disorder-a meta-analytic review resulting in a new neurofunctional model. Neurosci Biobehav Rev 47: 260–280.
Bukalo O, Pinard CR, Holmes A (2014). Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol 171: 4690–4718.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR et al (2015). An atlas of genetic correlations across human diseases and traits. bioRxiv (http://dx.doi.org/10.1101/014498).
Burghy CA, Stodola DE, Ruttle PL, Molloy EK, Armstrong JM, Oler JA et al (2012). Developmental pathways to amygdala-prefrontal function and internalizing symptoms in adolescence. Nat Neurosci 15: 1736–1741.
Canli T, Qiu M, Omura K, Congdon E, Haas BW, Amin Z et al (2006). Neural correlates of epigenesis. Proc Natl Acad Sci USA 103: 16033–16038.
Cannon TD, Keller MC (2006). Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol 2: 267–290.
Carballedo A, Morris D, Zill P, Fahey C, Reinhold E, Meisenzahl E et al (2013). Brain-derived neurotrophic factor Val66Met polymorphism and early life adversity affect hippocampal volume. Am J Med Genet B 162B: 183–190.
Carey CE, Agrawal A, Degenhardt L, Heath AC, Montgomery GW, Martin NG et al. (in press). A monoacylglycerol lipase (MGLL) polymorphism rs604300 interacts with childhood adversity to predict cannabis dependence symptoms and amygdala habituation: Evidence from an endocannabinoid system-level analysis. J Abnormal Psychol (e-pub ahead of print).
Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE (2010). Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry 167: 509–527.
Caspi A, Moffitt TE (2006). Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 7: 583–590.
Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT et al (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478: 519–523.
Cousijn H, Rijpkema M, Qin SZ, van Marle HJF, Franke B, Hermans EJ et al (2010). Acute stress modulates genotype effects on amygdala processing in humans. Proc Natl Acad Sci USA 107: 9867–9872.
Cyranoski D, Reardon S (2015). Embryo editing sparks epic debate. Nature 520: 593–594.
Dannlowski U, Kugel H, Redlich R, Halik A, Schneider I, Opel N et al (2014). Serotonin transporter gene methylation is associated with hippocampal gray matter volume. Hum Brain Mapp 35: 5356–5367.
de Kloet ER, Joels M, Holsboer F (2005). Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6: 463–475.
de Kloet ER, Karst H, Joels M (2008). Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrin 29: 268–272.
de Quervain DJ, Kolassa IT, Ertl V, Onyut PL, Neuner F, Elbert T et al (2007). A deletion variant of the alpha2b-adrenoceptor is related to emotional memory in Europeans and Africans. Nat Neurosci 10: 1137–1139.
Dedovic K, Duchesne A, Andrews J, Engert V, Pruessner JC (2009). The brain and the stress axis: the neural correlates of cortisol regulation in response to stress. NeuroImage 47: 864–871.
DeRijk RH (2009). Single nucleotide polymorphisms related to HPA Axis reactivity. Neuroimmunomodulat 16: 340–352.
DeRijk RH, van Leeuwen N, Klok MD, Zitman FG (2008). Corticosteroid receptor-gene variants: modulators of the stress-response and implications for mental health. Eur J Pharmacol 585: 492–501.
DeRijk RH, Wust S, Meijer OC, Zennaro MC, Federenko IS, Hellhammer DH et al (2006). A common polymorphism in the mineralocorticoid receptor modulates stress responsiveness. J Clin Endocr Metab 91: 5083–5089.
Di Iorio DR, Michalski LJ, Corral-Frias NS, Carey CE, Conley EC, Hariri AR et al Hypothalamic-pituitary-adrenal axis genetic variation moderates associations between early life stress and amygdala structure and function. (submitted).
Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S et al (2015). Candidate gene-environment interaction research: reflections and recommendations. Perspective Psychol Sci 10: 37–59.
Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC et al (2015). FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun 6: 6395.
Dohrenwend BP (2000). The role of adversity and stress in psychopathology: some evidence and its implications for theory and research. J Health Social Behav 41: 1–19.
Dohrenwend BP (2006). Inventorying stressful life events as risk factors for psychopathology: toward resolution of the problem of intracategory variability. Psychol Bull 132: 477–495.
Doom JR, Gunnar MR (2013). Stress physiology and developmental psychopathology: past, present, and future. Dev Psychopathol 25: 1359–1373.
Duncan LE, Keller MC (2011). A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry 168: 1041–1049.
Duncan OD (1966). Path analysis: sociological examples. Am J Sociol 72: 1–16.
Duvarci S, Pare D (2014). Amygdala microcircuits controlling learned fear. Neuron 82: 966–980.
Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE et al (2001). Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA 98: 6917–6922.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A et al (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269.
Elzinga BM, Molendijk ML, Voshaar RCO, Bus BAA, Prickaerts J, Spinhoven P et al (2011). The impact of childhood abuse and recent stress on serum brain-derived neurotrophic factor and the moderating role of BDNF Val(66)Met. Psychopharmacology 214: 319–328.
Fani N, Gutman D, Tone EB, Almli L, Mercer KB, Davis J et al (2013). FKBP5 and attention bias for threat associations with hippocampal function and shape. JAMA Psychiatry 70: 392–400.
Fani N, King TZ, Reiser E, Binder EB, Jovanovic T, Bradley B et al (2014). FKBP5 genotype and structural integrity of the posterior cingulum. Neuropsychopharmacology 39: 1206–1213.
Faravelli C, Lo Sauro C, Godini L, Lelli L, Benni L, Pietrini F et al (2012). Childhood stressful events, HPA axis and anxiety disorders. World J Psychiatry 2: 13–25.
Fardet L, Petersen I, Nazareth I (2012). Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry 169: 491–497.
Favaro A, Manara R, Pievani M, Clementi M, Forzan M, Bruson A et al (2014). Neural signatures of the interaction between the 5-HTTLPR genotype and stressful life events in healthy women. Psychiatry Res 223: 157–163.
Federenko IS, Nagamine M, Hellhammer DH, Wadhwa PD, Wust S (2004). The heritability of hypothalamus pituitary adrenal axis responses to psychosocial stress is context dependent. J Clin Endocr Metab 89: 6244–6250.
Flint J, Kendler KS (2014). The genetics of major depression. Neuron 81: 484–503.
Franz CE, York TP, Eaves LJ, Mendoza SP, Hauger RL, Hellhammer DH et al (2010). Genetic and environmental influences on cortisol regulation across days and contexts in middle-aged men. Behav Genet 40: 467–479.
Frodl T, Carballedo A, Frey EM, O'Keane V, Skokauskas N, Morris D et al (2014a). Expression of glucocorticoid inducible genes is associated with reductions in cornu ammonis and dentate gyrus volumes in patients with major depressive disorder. Dev Psychopathol 26: 1209–1217.
Frodl T, Carballedo A, Hughes MM, Saleh K, Fagan A, Skokauskas N et al (2012). Reduced expression of glucocorticoid-inducible genes GILZ and SGK-1: high IL-6 levels are associated with reduced hippocampal volumes in major depressive disorder. Transl Psychiatry 2: e88.
Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C et al (2002). Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry 51: 708–714.
Frodl T, Meisenzahl EM, Zetzsche T, Born C, Jager M, Groll C et al (2003). Larger amygdala volumes in first depressive episode as compared to recurrent major depression and healthy control subjects. Biol Psychiatry 53: 338–344.
Frodl T, O’Keane V (2013). How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol Dis 52: 24–37.
Frodl T, Reinhold E, Koutsouleris N, Donohoe G, Bondy B, Reiser M et al (2010). Childhood stress, serotonin transporter gene and brain structures in major depression. Neuropsychopharmacology 35: 1383–1390.
Frodl T, Skokauskas N, Frey EM, Morris D, Gill M, Carballedo A (2014b). BDNF Val66Met genotype interacts with childhood adversity and influences the formation of hippocampal subfields. Hum Brain Mapp 35: 5776–5783.
Frodl T, Szyf M, Carballedo A, Ly V, Dymov S, Vaisheva F et al (2015). DNA methylation of the serotonin transporter gene (SLC6A4) is associated with brain function involved in processing emotional stimuli. J Psychiatry Neurosci 40: 140180.
Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G et al (2015). Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol 11: 33.
Ganzel BL, Morris PA, Wethington E (2010). Allostasis and the human brain: integrating models of stress from the social and life sciences. Psychol Rev 117: 134–174.
Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR et al (2009). Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry 14: 681–695.
Gee DG, Gabard-Durnam LJ, Flannery J, Goff B, Humphreys KL, Telzer EH et al (2013). Early developmental emergence of human amygdala-prefrontal connectivity after maternal deprivation. Proc Natl Acad Sci USA 110: 15638–15643.
Gerritsen L, Tendolkar I, Franke B, Vasquez AA, Kooijman S, Buitelaar J et al (2012). BDNF Val66Met genotype modulates the effect of childhood adversity on subgenual anterior cingulate cortex volume in healthy subjects. Mol Psychiatry 17: 597–603.
Giedd JN, Raznahan A, Alexander-Bloch A, Schmitt E, Gogtay N, Rapoport JL (2015). Child psychiatry branch of the national institute of mental health longitudinal structural magnetic resonance imaging study of human brain development. Neuropsychopharmacology 40: 43–49.
Glover GH (2011). Overview of functional magnetic resonance imaging. Neurosurg Clin N Am 22: 133–139, vii.
Gorka AX, Hanson JL, Radtke SR, Hariri AR (2014). Reduced hippocampal and medial prefrontal gray matter mediate the association between reported childhood maltreatment and trait anxiety in adulthood and predict sensitivity to future life stress. Biol Mood Anxiety Disord 4: 12.
Gottesman II, Gould TD (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 160: 636–645.
Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB et al (2015). Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci 35: 3879–3892.
Green JG, McLaughlin KA, Berglund PA, Gruber MJ, Sampson NA, Zaslavsky AM et al (2010). Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry 67: 113–123.
Groenewold NA, Opmeer EM, de Jonge P, Aleman A, Costafreda SG (2013). Emotional valence modulates brain functional abnormalities in depression: evidence from a meta-analysis of fMRI studies. Neurosci Biobehav Rev 37: 152–163.
Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A (2013a). Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci 34: 637–644.
Gunduz-Cinar O, MacPherson KP, Cinar R, Gamble-George J, Sugden K, Williams B et al (2013b). Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry 18: 813–823.
Gustafsson PA, Gustafsson PE, Anckarsater H, Lichtenstein P, Ljung T, Nelson N et al (2011). Heritability of cortisol regulation in children. Twin Res Hum Genet 14: 553–561.
Hankin BL, Nederhof E, Oppenheimer CW, Jenness J, Young JF, Abela JRZ et al (2011). Differential susceptibility in youth: evidence that 5-HTTLPR x positive parenting is associated with positive affect 'for better and worse'. Transl Psychiatry 1: e44.
Hanson JL, Nacewicz BM, Sutterer MJ, Cayo AA, Schaefer SM, Rudolph KD et al (2015). Behavioral problems after early life stress: contributions of the hippocampus and amygdala. Biol Psychiatry 77: 314–323.
Hariri AR (2009). The neurobiology of individual differences in complex behavioral traits. Annu Rev Neurosci 32: 225–247.
Hariri AR (2015) Looking Inside the Disordered Brain: An Introduction to the Functional Neuroanatomy of Psychopathology. Sinauer Associates: Sunderland, MA, USA.
Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F et al (2009). Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry 66: 9–16.
Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D et al (2002). Serotonin transporter genetic variation and the response of the human amygdala. Science 297: 400–403.
Hariri AR, Weinberger DR (2003). Imaging genomics. Br Med Bull 65: 259–270.
Hasler G, Drevets WC, Manji HK, Charney DS (2004). Discovering endophenotypes for major depression. Neuropsychopharmacology 29: 1765–1781.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM (2006). Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets 5: 453–479.
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM (2009). Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann Ny Acad Sci 1179: 120–143.
Hayes JP, Vanelzakker MB, Shin LM (2012). Emotion and cognition interactions in PTSD: a review of neurocognitive and neuroimaging studies. Front Integr Neurosci 6: 89.
Heck A, Fastenrath M, Ackermann S, Auschra B, Bickel H, Coynel D et al (2014). Converging genetic and functional brain imaging evidence links neuronal excitability to working memory, psychiatric disease, and brain activity. Neuron 81: 1203–1213.
Henckens MJ, van Wingen GA, Joels M, Fernandez G (2010). Time-dependent effects of corticosteroids on human amygdala processing. J Neurosci 30: 12725–12732.
Henckens MJ, van Wingen GA, Joels M, Fernandez G (2011). Time-dependent corticosteroid modulation of prefrontal working memory processing. Proc Natl Acad Sci USA 108: 5801–5806.
Henckens MJ, van Wingen GA, Joels M, Fernandez G (2012). Corticosteroid induced decoupling of the amygdala in men. Cereb Cortex 22: 2336–2345.
Hernaus D, van Winkel R, Gronenschild E, Habets P, Kenis G, Marcelis M et al (2014). Brain-derived neurotrophic factor/FK506-binding protein 5 genotype by childhood trauma interactions do not impact on hippocampal volume and cognitive performance. PLoS One 9: 3.
Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N et al (2015). Common genetic variants influence human subcortical brain structures. Nature 520: 224–229.
Hilbert K, Lueken U, Beesdo-Baum K (2014). Neural structures, functioning and connectivity in generalized anxiety disorder and interaction with neuroendocrine systems: a systematic review. J Affect Disord 158: 114–126.
Hill MN, Hellemans KG, Verma P, Gorzalka BB, Weinberg J (2012). Neurobiology of chronic mild stress: parallels to major depression. Neurosci Biobehav Rev 36: 2085–2117.
Holmans P, Green EK, Pahwa JS, Ferreira MAR, Purcell SM, Sklar P et al (2009). Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 85: 13–24.
Holmes AJ, Hollinshead MO, O’Keefe TM, Petrov VI, Fariello GR, Wald LL et al (2015). Brain Genomics Superstruct Project initial data release with structural, functional, and behavioral measures. Scientific Data 2, article number: 150031.
Holmes AJ, Lee PH, Hollinshead MO, Bakst L, Roffman JL, Smoller JW et al (2012). Individual differences in amygdala-medial prefrontal anatomy link negative affect, impaired social functioning, and polygenic depression risk. J Neurosci 32: 18087–18100.
Holz NE, Buchmann AF, Boecker R, Blomeyer D, Baumeister S, Wolf I et al (2014). Role of FKBP5 in emotion processing: results on amygdala activity, connectivity and volume. Brain Struct Funct 220: 1355–1368.
Horowitz MA, Zunszain PA (2015). Neuroimmune and neuroendocrine abnormalities in depression: two sides of the same coin. Ann NY Acad Sci (e-pub ahead of print).
Hyde LW, Bogdan R, Hariri AR (2011a). Understanding risk for psychopathology through imaging gene-environment interactions. Trends Cogn Sci 15: 417–427.
Hyde LW, Gorka A, Manuck SB, Hariri AR (2011b). Perceived social support moderates the link between threat-related amygdala reactivity and trait anxiety. Neuropsychologia 49: 651–656.
Iacono WG (2014). The molecular genetic basis for psychophysiological endophenotypes. Psychophsyiology 51, Special issue.
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K et al (2010). Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 167: 748–751.
Ising M, Holsboer F (2007). CRH1 receptor antagonists for the treatment of depression and anxiety. Exp Clin Psychopharm 15: 519–528.
Janak PH, Tye KM (2015). From circuits to behaviour in the amygdala. Nature 517: 284–292.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821.
Johnson DC, Casey BJ (2015). Easy to remember, difficult to forget: the development of fear regulation. Dev Cogn Neurosci 11: 42–55.
Karg K, Burmeister M, Shedden K, Sen S (2011). The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 68: 444–454.
Kaufman J (2015). The serotonin transporter gene and amygdala activation: research in context. Biol Psychiatry (e-pub ahead of print).
Kehne JH, Cain CK (2010). Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharmacol Therapeut 128: 460–487.
Keller MC (2014). Gene x environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry 75: 18–24.
Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S et al (2011). Structural neuroimaging studies in major depressive disorder meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 68: 675–690.
Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM et al (2010). Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br J Psychiatry 197: 378–385.
Kim MJ, Loucks RA, Palmer AL, Brown AC, Solomon KM, Marchante AN et al (2011). The structural and functional connectivity of the amygdala: from normal emotion to pathological anxiety. Behav Brain Res 223: 403–410.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Klok MD, Alt SR, AJMI Lafitte, Turner JD, EAJF Lakke, Huitinga I et al (2011a). Decreased expression of mineralocorticoid receptor mRNA and its splice variants in postmortem brain regions of patients with major depressive disorder. J Psychiat Res 45: 871–878.
Klok MD, Giltay EJ, Van der Does AJW, Geleijnse JM, Antypa N, BWJH Penninx et al (2011b). A common and functional mineralocorticoid receptor haplotype enhances optimism and protects against depression in females. Transl Psychiatry 1: e62.
Kolber BJ, Muglia LJ (2009). Defining brain region-specific glucocorticoid action during stress by conditional gene disruption in mice. Brain Res 1293: 85–90.
Lanphier E, Urnov F, Haecker SE, Werner M, Smolenski J (2015). Don't edit the human germ line. Nature 519: 410–411.
Lee PH, O'Dushlaine C, Thomas B, Purcell SM (2012). INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28: 1797–1799.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S et al (1996). Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274: 1527–1531.
Lewinsohn PM, Rohde P, Seeley JR (1998). Major depressive disorder in older adolescents: prevalence, risk factors, and clinical implications. Clin Psychol Rev 18: 765–794.
Li MX, Gui HS, Kwan JSH, Sham PC (2011). GATES: a rapid and powerful gene-based association test using extended simes procedure. Am J Hum Genet 88: 283–293.
Li S, Weerda R, Milde C, Wolf OT, Thiel CM (2015). ADRA2B genotype differentially modulates stress-induced neural activity in the amygdala and hippocampus during emotional memory retrieval. Psychopharmacology 232: 755–764.
Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM et al (2010). A versatile gene-based test for genome-wide association studies. AmJ Hum Genet 87: 139–145.
Luby J, Belden A, Botteron K, Marrus N, Harms MP, Babb C et al (2013). The effects of poverty on childhood brain development: the mediating effect of caregiving and stressful life events. JAMA Pediatr 167: 1135–1142.
Luby JL, Gaffrey MS, Tillman R, April LM, Belden AC (2014). Trajectories of preschool disorders to full DSM depression at school age and early adolescence: continuity of preschool depression. Am J Psychiatry 171: 768–776.
Lucassen PJ, Pruessner J, Sousa N, Almeida OF, Van Dam AM, Rajkowska G et al (2014). Neuropathology of stress. Acta Neuropathol 127: 109–135.
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10: 434–445.
Maheu FS, Mazzone L, Merke DP, Keil MF, Stratakis CA, Pine DS et al (2007). A fMRI study of amygdala and hippocampus function in adolescents with cushing syndrome. Biol Psychiatry 61: 62s–62s.
Maheu FS, Mazzone L, Merke DP, Keil M, Stratakis CA, Pine DS et al (2008). Altered amygdala and hippocampus function in adolescents with hypercortisolemia: a functional magnetic resonance imaging study of Cushing syndrome. Dev Psychopathol 20: 1177–1189.
Manuck SB, Brown SM, Forbes EE, Hariri AR (2007). Temporal stability of individual differences in amygdala reactivity. Am J Psychiatry 164: 1613–1614.
Marques AH, Silverman MN, Sternberg EM (2009). Glucocorticoid dysregulations and their clinical correlates from receptors to therapeutics. Ann NY Acad Sci 1179: 1–18.
McCrory E, De Brito SA, Viding E (2011). The impact of childhood maltreatment: a review of neurobiological and genetic factors. Front Psychiatry 2: 48.
McEwen BS (2000). Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology 22: 108–124.
McEwen BS (2003). Mood disorders and allostatic load. Biol Psychiatry 54: 200–207.
McEwen BS, Gianaros PJ (2010). Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann NY Acad Sci 1186: 190–222.
McEwen BS, Gianaros PJ (2011). Stress- and allostasis-induced brain plasticity. Annu Rev Med 62: 431–445.
McEwen BS, Morrison JH (2013). The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79: 16–29.
McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M et al (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 12: 342–348.
Mehta MA, Golembo NI, Nosarti C, Colvert E, Mota A, Williams SC et al (2009). Amygdala, hippocampal and corpus callosum size following severe early institutional deprivation: the English and Romanian Adoptees study pilot. J Child Psychol Psychiatry 50: 943–951.
Menke A, Arloth J, Putz B, Weber P, Klengel T, Mehta D et al (2012). Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 37: 1455–1464.
Meyer-Lindenberg A, Weinberger DR (2006). Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci 7: 818–827.
Miller GE, Chen E, Zhou ES (2007). If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol Bull 133: 25–45.
Mitra R, Ferguson D, Sapolsky RM (2009). Mineralocorticoid receptor overexpression in basolateral amygdala reduces corticosterone secretion and anxiety. Biol Psychiatry 66: 686–690.
Moffitt TE, Caspi A (2014). Bias in a protocol for a meta-analysis of 5-HTTLPR, stress, and depression. BMC Psychiatry 14: 179.
Molendijk ML, Bus BAA, Spinhoven P, Kaimatzoglou A, Voshaar RCO, BW Pennix et al (2012). A systematic review and meta-analysis on the association between BDNF val(66)met and hippocampal volume-a genuine effect or a winners curse? Am J Med Genet B 159B: 731–740.
Monroe SM (2008). Modern approaches to conceptualizing and measuring human life stress. Annu Rev Clin Psychol 4: 33–52.
Monroe SM, Reid MW (2009). Life stress and major depression. Curr Dir Psychol Sci 18: 68–72.
Munafo MR, Bowes L, Clark TG, Flint J (2005). Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Mol Psychiatry 10: 765–770.
Murphy SE, Norbury R, Godlewska BR, Cowen PJ, Mannie ZM, Harmer CJ et al (2013). The effect of the serotonin transporter polymorphism (5-HTTLPR) on amygdala function: a meta-analysis. Mol Psychiatry 18: 512–520.
Nakamura K, Brown RA, Narayanan S, Collins DL, Arnold DL, Alzheimer's Disease Neuroimaging I (2015). Diurnal fluctuations in brain volume: statistical analyses of MRI from large populations. NeuroImage 118: 126–132.
Nikolova YS, Bogdan R, Brigidi BD, Hariri AR (2012). Ventral striatum reactivity to reward and recent life stress interact to predict positive affect. Biol Psychiatry 72: 157–163.
Nikolova YS, Ferrell RE, Manuck SB, Hariri AR (2011). Multilocus genetic profile for dopamine signaling predicts ventral striatum reactivity. Neuropsychopharmacology 36: 1940–1947.
Nikolova YS, Hariri AR (2015). Can we observe epigenetic effects on human brain function? Trends Cogn Sci 19: 366–373.
Nikolova YS, Koenen KC, Galea S, Wang CM, Seney ML, Sibille E et al (2014). Beyond genotype: serotonin transporter epigenetic modification predicts human brain function. Nat Neurosci 17: 1153–1155.
Otte C, Hinkelmann K, Moritz S, Yassouridis A, Jahn H, Wiedemann K et al (2010). Modulation of the mineralocorticoid receptor as add-on treatment in depression: a randomized, double-blind, placebo-controlled proof-of-concept study. J Psychiat Res 44: 339–346.
Pagliaccio D, Luby JL, Bogdan R, Agrawal A, Gaffrey MS, Belden AC et al (2014). Stress-system genes and life stress predict cortisol levels and amygdala and hippocampal volumes in children. Neuropsychopharmacology 39: 1245–1253.
Pagliaccio D, Luby JL, Bogdan R, Agrawal A, Gaffrey MS, Belden AC et al (2015). HPA axis genetic variation, pubertal status, and sex interact to predict amygdala and hippocampus responses to negative emotional faces in school-age children. NeuroImage 109C: 1–11.
Pariante CM, Lightman SL (2008). The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31: 464–468.
Patten SB, Neutel CI (2000). Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Safety 22: 111–122.
Pearson R, McGeary JE, Beevers CG (2014). Association between serotonin cumulative genetic score and the behavioral approach system (BAS): moderation by early life environment. Pers Individ Dif 70: 140–144.
Peciña M, Martínez-Jauand M, Love T, Heffernan J, Montoya P, Hodgkinson C et al (2014). Valence-specific effects of BDNF Val66Met polymorphism on dopaminergic stress and reward processing in humans. J Neurosci 34: 5874–5881.
Pelizza L, Ferrari A (2009). Anhedonia in schizophrenia and major depression: state or trait? Ann Gen Psychiatry 8: 22.
Plichta MM, Grimm O, Morgen K, Mier D, Sauer C, Haddad L et al (2014). Amygdala habituation: a reliable fMRI phenotype. NeuroImage 103: 383–390.
Plichta MM, Schwarz AJ, Grimm O, Morgen K, Mier D, Haddad L et al (2012). Test-retest reliability of evoked BOLD signals from a cognitive-emotive fMRI test battery. NeuroImage 60: 1746–1758.
Price JL, Drevets WC (2012). Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci 16: 61–71.
Purcell SM, Wray WR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460: 748–752.
Qin S, Cousijn H, Rijpkema M, Luo J, Franke B, Hermans EJ et al (2012). The effect of moderate acute psychological stress on working memory-related neural activity is modulated by a genetic variation in catecholaminergic function in humans. Front Integr Neurosci 6: 16.
Rabl U, Meyer BM, Diers K, Bartova L, Berger A, Mandorfer D et al (2014). Additive gene-environment effects on hippocampal structure in healthy humans. J Neurosci 34: 9917–9926.
Rasch B, Spalek K, Buholzer S, Luechinger R, Boesiger P, Papassotiropoulos A et al (2009). A genetic variation of the noradrenergic system is related to differential amygdala activation during encoding of emotional memories. Proc Natl Acad Sci USA 106: 19191–19196.
Rose EJ, Donohoe G (2013). Brain vs behavior: an effect size comparison of neuroimaging and cognitive studies of genetic risk for schizophrenia. Schizophrenia Bull 39: 518–526.
Sander JD, Joung JK (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355.
Sauder CL, Hajcak G, Angstadt M, Phan KL (2013). Test-retest reliability of amygdala response to emotional faces. Psychophysiology 50: 1147–1156.
Scharf SH, Liebl C, Binder EB, Schmidt MV, Muller MB (2011). Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS One 6: e16883.
Schizophrenia Working Group of the Psychiatric Genomics C (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427.
Schumann G, Loth E, Banaschewski T, Barbot A, Barker G, Buchel C et al (2010). The IMAGEN study: reinforcement-related behaviour in normal brain function and psychopathology. Mol Psychiatry 15: 1128–1139.
Segre AV, Groop L, Mootha VK, Daly MJ, Altshuler D, Consortium D et al (2010). Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6: pii.
Selye H (1936). A syndrome produced by diverse nocuous agents. Nature 138: 32–32.
Sharpley CF, Palanisamy SK, Glyde NS, Dillingham PW, Agnew LL (2014). An update on the interaction between the serotonin transporter promoter variant (5-HTTLPR), stress and depression, plus an exploration of non-confirming findings. Behav Brain Res 273: 89–105.
Sheridan MA, Fox NA, Zeanah CH, McLaughlin KA, Nelson CA 3rd (2012). Variation in neural development as a result of exposure to institutionalization early in childhood. Proc Natl Acad Sci USA 109: 12927–12932.
Shirtcliff EA, Allison AL, Armstrong JM, Slattery MJ, Kalin NH, Essex MJ (2012). Longitudinal stability and developmental properties of salivary cortisol levels and circadian rhythms from childhood to adolescence. Dev Psychobiol 54: 493–502.
Slavich GM, Epel ES (2010). The Stress and Adversity Inventory (STRAIN): an automated system for assessing cumulative stress exposure. Laboratory for Stress Assessment and Research. UCLA: Los Angeles, CA, USA.
Slavich GM, Tartter MA, Brennan PA, Hammen C (2014). Endogenous opioid system influences depressive reactions to socially painful targeted rejection life events. Psychoneuroendocrinology 49: 141–149.
Small KM, Brown KM, Forbes SL, Liggett SB (2001). Polymorphic deletion of three intracellular acidic residues of the alpha 2B-adrenergic receptor decreases G protein-coupled receptor kinase-mediated phosphorylation and desensitization. J Biol Chem 276: 4917–4922.
Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI et al (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381: 1371–1379.
Sousa N, Lukoyanov NV, Madeira MD, Almeida OFX, Paula-Barbosa MM (2000). Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 97: 253–266.
Streit F, Haddad L, Paul T, Frank J, Schafer A, Nikitopoulos J et al (2014). A functional variant in the neuropeptide S receptor 1 gene moderates the influence of urban upbringing on stress processing in the amygdala. Stress 17: 352–361.
Swartz JR, Knodt AR, Radtke SR, Hariri AR (2015). A neural biomarker of psychological vulnerability to future life stress. Neuron 85: 505–511.
Swartz JR, Monk CS (2014a). The role of corticolimbic circuitry in the development of anxiety disorders in children and adolescents. Curr Top Behav Neurosci 16: 133–148.
Swartz JR, Williamson DE, Hariri AR (2014b). Developmental change in amygdala reactivity during adolescence: effects of family history of depression and stressful life events. Am J Psychiatry 172: 276–283, appiajp2 01414020195.
Tan HY, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF et al (2007). Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci USA 104: 12536–12541.
Tarullo AR, Gunnar MR (2006). Child maltreatment and the developing HPA axis. Horm Behav 50: 632–639.
Teicher MH, Andersen SL, Polcari A, Anderson CM, Navalta CP, Kim DM (2003). The neurobiological consequences of early stress and childhood maltreatment. Neurosci Biobehav Rev 27: 33–44.
Teicher MH, Samson JA (2013). Childhood maltreatment and psychopathology: a case for ecophenotypic variants as clinically and neurobiologically distinct subtypes. Am J Psychiatry 170: 1114–1133.
Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME et al (2014). The ENIGMA consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imag Behav 8: 153–182.
Thomson F, Craighead M (2008). Innovative approaches for the treatment of depression: targeting the HPA axis. Neurochem Res 33: 691–707.
Tottenham N, Hare TA, Millner A, Gilhooly T, Zevin JD, Casey BJ (2011). Elevated amygdala response to faces following early deprivation. Dev Sci 14: 190–204.
Tottenham N, Hare TA, Quinn BT, McCarry TW, Nurse M, Gilhooly T et al (2010). Prolonged institutional rearing is associated with atypically large amygdala volume and difficulties in emotion regulation. Dev Sci 13: 46–61.
Tottenham N, Sheridan MA (2009). A review of adversity, the amygdala and the hippocampus: a consideration of developmental timing. Front Hum Neurosci 3: 68.
Turecki G, Meaney MJ (in press). Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol Psychiatry.
Tyrka AR, Burgers DE, Philip NS, Price LH, Carpenter LL (2013). The neurobiological correlates of childhood adversity and implications for treatment. Acta Psychiat Scand 128: 434–447.
Ulrich-Lai YM, Herman JP (2009). Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10: 397–409.
Urry HL, van Reekum CM, Johnstone T, Kalin NH, Thurow ME, Schaefer HS et al (2006). Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci 26: 4415–4425.
Ursini G, Bollati V, Fazio L, Porcelli A, Iacovelli L, Catalani A et al (2011). Stress-related methylation of the catechol-O-methyltransferase val(158) allele predicts human prefrontal cognition and activity. J Neurosci 31: 6692–6698.
Van Hulle CA, Shirtcliff EA, Lemery-Chalfant K, Goldsmith HH (2012). Genetic and environmental influences on individual differences in cortisol level and circadian rhythm in middle childhood. Horm Behav 62: 36–42.
van Leeuwen N, Bellingrath S, de Kloet ER, Zitman FG, DeRijk RH, Kudielka BM et al (2011). Human mineralocorticoid receptor (MR) gene haplotypes modulate MR expression and transactivation: implication for the stress response. Psychoeuroendocrinology 36: 699–709.
van Leeuwen N, Kumsta R, Entringer S, de Kloet ER, Zitman FG, DeRijk RH et al (2010). Functional mineralocorticoid receptor (MR) gene variation influences the cortisol awakening response after dexamethasone. Psychoneuroendocrinology 35: 339–349.
Verhagen M, van der Meij A, van Deurzen PAM, Janzing JGE, Arias-Vasquez A, Buitelaar JK et al (2010). Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry 15: 260–271.
Vogel S, Klumpers F, Krugers HJ, Fang Z, Oplaat KT, Oitzl MS et al (2015). Blocking the mineralocorticoid receptor in humans prevents the stress-induced enhancement of centromedial amygdala connectivity with the dorsal striatum. Neuropsychopharmacology 40: 947–956.
Wada K, Yamada N, Suzuki H, Lee Y, Kuroda S (2000). Recurrent cases of corticosteroid-induced mood disorder: clinical characteristics and treatment. J Clin Psychiatry 61: 261–267.
Wardle MC, de Wit H, Penton-Voak I, Lewis G, Munafo MR (2013). Lack of association between COMT and working memory in a population-based cohort of healthy young adults. Neuropsychopharmacology 38: 1253–1263.
Weaver ICG, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Jr S et al (2004). Epigenetic programming by maternal behavior. Nat Neurosci 7: 847–854.
Weaver ICG, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ et al (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 25: 11045–11054.
Whalen PJ, Phelps EA (2009) The Human Amygdala. Guilford Press: New York, USA, xiv, 429 p, 416 p of plates.
White MG, Bogdan R, Fisher PM, Munoz KE, Williamson DE, Hariri AR (2012). FKBP5 and emotional neglect interact to predict individual differences in amygdala reactivity. Genes Brain Behav 11: 869–878.
Williams LM, Gatt JM, Schofield PR, Olivieri G, Peduto A, Gordon E (2009). 'Negativity bias' in risk for depression and anxiety: brain-body fear circuitry correlates, 5-HTT-LPR and early life stress. NeuroImage 47: 804–814.
Williamson DE, Birmaher B, Ryan ND, Shiffrin TP, Lusky JA, Protopapa J et al (2003). The stressful life events schedule for children and adolescents: development and validation. Psychiatry Res 119: 225–241.
Wright S (1920). Correlation and causation part I. Method of path coefficients. J Agric Res 20: 0557–0585.
Wust S, Federenko I, Hellhammer DH, Kirschbaum C (2000). Genetic factors, perceived chronic stress, and the free cortisol response to awakening. Psychoneuroendocrinology 25: 707–720.
Xu K, Seo D, Hodgkinson C, Hu Y, Goldman D, Sinha R (2013). A variant on the kappa opioid receptor gene (OPRK1) is associated with stress response and related drug craving, limbic brain activation and cocaine relapse risk. Transl Psychiatry 3.
Zannas AS, Binder EB (2014). Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav 13: 25–37.
Zannas AS, McQuoid DR, Payne ME, Steffens DC, MacFall JR, Ashley-Koch A et al (2013). Negative life stress and longitudinal hippocampal volume changes in older adults with and without depression. J Psychiat Res 47: 829–834.
Zhou X, Lowdon RF, Li D, Lawson HA, Madden PAF, Costello JF et al (2013). Exploring long-range genome interactions using the WashU epigenome browser. Nat Methods 10: 375–376.
Zhou X, Maricque B, Xie MC, Li DF, Sundaram V, Martin EA et al (2011). The human epigenome browser at Washington university. Nat Methods 8: 989–990.
Ziegler C, Dannlowski U, Brauer D, Stevens S, Laeger I, Wittmann H et al (2015). Oxytocin receptor gene methylation: converging multilevel evidence for a role in social anxiety. Neuropsychopharmacology 40: 1528–1538.
Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, Uhr M et al (2011). Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry 168: 1107–1116.
Zuo XN, Anderson JS, Bellec P, Birn RM, Biswal BB, Blautzik J et al (2014). An open science resource for establishing reliability and reproducibility in functional connectomics. Scientific Data 1: 140049.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bogdan, R., Pagliaccio, D., Baranger, D. et al. Genetic Moderation of Stress Effects on Corticolimbic Circuitry. Neuropsychopharmacol 41, 275–296 (2016). https://doi.org/10.1038/npp.2015.216
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.216
This article is cited by
-
Brain Reactions to Opening and Closing the Eyes: Salivary Cortisol and Functional Connectivity
Brain Topography (2022)
-
Title: “Labels Matter: Is it stress or is it Trauma?”
Translational Psychiatry (2021)
-
The role of childhood adversities, FKBP5, BDNF, NRN1, and generalized self-efficacy in suicide attempts in alcohol-dependent patients
Pharmacological Reports (2020)
-
At-risk individuals display altered brain activity following stress
Neuropsychopharmacology (2018)
-
Intensified vmPFC surveillance over PTSS under perturbed microRNA-608/AChE interaction
Translational Psychiatry (2016)
