
Abstract
Research into the causes of psychopathology has largely focused on two broad etiologic factors: genetic vulnerability and environmental stressors. An important role for familial/heritable factors in the etiology of a broad range of psychiatric disorders was established well before the modern era of genomic research. This review focuses on the genetic basis of three disorder categories—posttraumatic stress disorder (PTSD), major depressive disorder (MDD), and the anxiety disorders—for which environmental stressors and stress responses are understood to be central to pathogenesis. Each of these disorders aggregates in families and is moderately heritable. More recently, molecular genetic approaches, including genome-wide studies of genetic variation, have been applied to identify specific risk variants. In this review, I summarize evidence for genetic contributions to PTSD, MDD, and the anxiety disorders including genetic epidemiology, the role of common genetic variation, the role of rare and structural variation, and the role of gene–environment interaction. Available data suggest that stress-related disorders are highly complex and polygenic and, despite substantial progress in other areas of psychiatric genetics, few risk loci have been identified for these disorders. Progress in this area will likely require analysis of much larger sample sizes than have been reported to date. The phenotypic complexity and genetic overlap among these disorders present further challenges. The review concludes with a discussion of prospects for clinical translation of genetic findings and future directions for research.
Similar content being viewed by others

Main
Genes and stress are the two most widely acknowledged contributors to psychopathology. The ‘diathesis-stress’ hypothesis has been the leading etiologic model for psychiatric disorders for decades (although alternatives have been proposed, see Boyce, this issue). The essence of this model is that genes and adversity, independently and in combination, increase the liability to disorder, which in turn results once a threshold of sufficient liability is crossed. In this review, I will focus on the ‘diathesis’ (that is, genetic) component as it relates to disorders that are most commonly thought of as stress-related disorders: posttraumatic stress disorder (PTSD), major depressive disorder (MDD), and anxiety disorders. In reality, stress (especially early adversity and later stressful life events) has been identified as a risk factor for a broader range of psychiatric disorders, including bipolar disorder (Gilman et al, 2015) and schizophrenia (Matheson et al, 2013). However, the role of stressful environments and the physiology of stress response systems have been most closely linked to depressive, anxiety, and traumatic stress disorders.
The role of genes in psychiatric illness was suggested well before the modern genomic era through family and twin studies. All psychiatric disorders that have been studied by these methods have been reported to be familial and heritable to varying degrees. More recently, molecular genetic studies have begun to identify specific DNA variations associated with neuropsychiatric disorders and related phenotypes. In the sections that follow, I briefly introduce and summarize the methods and study designs used in psychiatric genetics. I will then review in more detail the status of genetic studies of PTSD, MDD, and anxiety disorders, including evidence that the heritable component of these disorders overlaps and is shared to varying degrees with other disorders. Finally, I will discuss prospects for clinical translation of these findings and future directions for research.
THE TOOLKIT OF PSYCHIATRIC GENETICS
Defining the genetic contribution to psychiatric disorders (as with other complex disorders) typically involves a series of questions and study designs as depicted in Figure 1 and reviewed in Smoller et al (2015), from which this section is adapted. The first question is usually whether the disorder aggregates in families. To answer this, family studies compare the prevalence of disorder among first-degree relatives of affected individuals (probands) with that among either first-degree relatives of unaffected controls or to the population prevalence. When the ratio of these prevalences, known as the familial recurrence ratio, is significantly greater than 1.0, we conclude that the disorder is familial. Because family members share both genetic and environmental influences, family studies by themselves cannot establish a genetic contribution. Twin studies can be used to distinguish genetic and environmental components by comparing disorder concordance rates between genetically identical (monozygotic, MZ) and non-identical (dizygotic, DZ) twin pairs. Assuming that environmental risk factors do not differ systematically for MZ vs DZ twins, a significantly increased concordance in MZ vs DZ pairs implies a role for genetic variation. Variance component modeling methods are often used to dissect genetic, shared environmental, and individual-specific variance components. The ratio of the genetic variance component to the full phenotypic variance provides the heritability of the phenotype. Typically, additive (narrow-sense) heritability is reported (denoted h2) (Visscher et al, 2008). Heritability may range from 0 (no contribution of genetic variation to phenotypic variation) to 100% (phenotypic variation entirely attributable to genetic variation).

Summary of psychiatric genetic methods. (Adapted from Smoller et al, 2015.)
Once a trait or disorder is shown to be heritable, molecular genetic studies can be undertaken to localize and identify the genetic influences at the level of DNA variation. Linkage studies aim to map the chromosomal location of disease genes. Co-inheritance of a DNA marker with the disease of interest in families is evidence that a disease-related gene is ‘linked to’ (physically close to) that DNA marker. However, linkage studies are best-suited to Mendelian disorders where disease is caused by one or more rare mutations of large effect. In contrast, psychiatric disorders are complex disorders thought to result from the contribution of many, often common and low-penetrance, susceptibility loci in combination with environmental risk factors. For complex disorders, association studies are more powerful for identifying risk loci and have become the dominant strategy for genetic studies of psychiatric disorders. Association studies typically utilize a case–control design to determine whether specific genetic variants (alleles) are more common among affected (cases) than among unaffected individuals (controls). For continuous traits, association analysis examines whether specific alleles are associated with quantitative variation in the trait of interest.
Association studies have been used to evaluate different classes of DNA variation relevant to psychopathology. The genetic architecture of a phenotype refers to entire complement of underlying genetic risks factors including their number, allele frequencies, and effect sizes of contributing variants. The allelic spectrum underlying complex phenotypes includes at least three major classes of DNA variation: common single-nucleotide variants (common SNVs or ‘SNPs’, allele frequencies ⩾1%), rarer single-nucleotide variation (rare SNVs, allele frequencies <1%), and structural variations, including copy number variants (CNVs), insertion/deletions, and balanced translocations. In addition to inherited variation, rare pathogenic variants can occur de novo, arising in a parental gamete, a fertilized embryo, or the developing fetus. In general, penetrance is inversely related to allele frequency because selection pressure suppresses the frequency of highly penetrant deleterious loci. Thus, rare pathogenic variants are typically more highly penetrant (ie, have a larger effect size) than common variants. More recently, a potential role for somatic mutation (in which a mutation arises in a subpopulation of developing or mature cells including neurons) has been suggested for neuropsychiatric phenotypes (Insel, 2014).
In general, two strategies for association analysis of SNVs are widely used: candidate gene studies and genome-wide association studies (GWAS). The first relies on genes that have been implicated in a phenotype based on prior evidence. Biological candidate genes are usually selected based on prior evidence from animal models (eg, knockout mice) or human studies (eg, genes that are targets of psychotropic medications). More recently, loci identified from GWAS (see below) have become popular candidates. Biological candidate gene studies have well-documented limitations. Perhaps most importantly, they are derived from existing hypotheses about the biological basis of psychiatric disorders. Because our knowledge of disorder biology is incomplete, the prior probability that any given candidate gene is truly relevant is low. Not surprisingly, then, candidate gene studies have a poor track record of replication and many candidate gene associations are likely to be spurious (Sullivan, 2007). In addition, by their very nature, association studies of biological candidates can at best validate existing hypotheses rather than discover novel risk loci.
In contrast, GWAS enable a so-called ‘unbiased’ search for risk loci by examining variants across the genome instead of limiting the search to hypothesized candidates. GWAS of common SNPs became possible with the development of DNA microarrays that interrogate up to 1 million or more SNPs across the genome. Alleles of many SNPs in a given genomic region are correlated and inherited together (due to linkage disequilibrium, or LD). Therefore, a given SNP may provide information about other correlated SNPs, so that only a subset of the millions of SNPs in the genome need to be genotyped directly to cover genome-wide variation. The conventional threshold for ‘genome-wide significance’ is a P-value less than 5 × 10−8, a Bonferroni correction for the effective number (one million) of independent statistical tests that are conducted in a typical GWAS. More recently, advances in DNA sequencing technologies have enabled exome-wide (and even genome-wide) analysis of rare variants.
Very large sample sizes (on the order of 25 000 or more cases) are needed to adequately power genome-wide analyses of either common or rare variants. In the case of common variants, the requirement for large samples is due to the small effect sizes expected-–typically an allelic odds ratio of less than 1.20. In the case of rare variants, effect sizes may be larger, but their rarity again entails large sample sizes to allow detection of a sufficient number of risk variant carriers (Moutsianas et al, 2015; Zuk et al, 2014). Gene–environment interaction (G × E) studies examine whether the effect of a genetic variant is modified by an environmental exposure. To date, most G × E association studies have focused on a limited set of biological candidate genes and have been underpowered to detect robust effects (Duncan et al, 2014b). The availability of established genetic risk alleles (eg, from recent genome-wide studies) has created a new set of candidate variants that can be examined in G × E studies.
GWAS data can also be used to study genetic effects beyond the simple association of a single variant or gene with a disorder or trait. Pathway (or gene set enrichment) analyses can implicate sets of genes—for example, genes in a given biological pathway. Pathway analyses can be more powerful than single marker tests because they aggregate association signals across groups of genes. By implicating functionally related genes, they can also provide insights into the biological mechanisms by translating association signals into sets of cellular functions or molecular pathways that may underlie disease pathogenesis. So-called polygene risk scores (PRS) provide another index of aggregate genetic effects. Psychiatric disorders are now known to be highly polygenic—that is the genetic component includes hundreds or thousands of individual rare and common variants. The large majority of common risk alleles will not show genome-wide significant evidence of association given available sample sizes. A PRS can be derived from a GWAS by selecting all SNPs that exceed a nominal P-value threshold (eg, P<0.05). These SNPs are then used to generate a score that is simply a sum of the allele count weighted by the effect size (eg, log odds ratio) for each SNP. In other words, the PRS provides a single measure of genetic loading for common risk alleles for a given phenotype. The PRS can then be used as a predictor or a covariate in future studies of a disease in independent samples for which GWAS data are available. GWAS data for a given phenotype can also be used to estimate the variance attributable to common variation (so-called SNP-Chip heritability, h2SNP). A variety of approaches have been developed to estimate such heritability (Bulik-Sullivan et al, 2015; Ge et al, 2015; Yang et al, 2011; Zaitlen et al, 2014). Aggregate cross-disorder effects of common variants can be estimated using PRS and bivariate analyses of h2SNP (Wray et al, 2014).
Establishing that a genetic variant, gene, or gene set is associated with a disorder of interest is only the first step in answering the question of how specific genes contribute to the disorder. A wide range of molecular, cellular, and clinical research studies may be needed to characterize the pathogenetic mechanisms involved. These include studies of gene expression, animal and cellular models in which genes may be experimentally altered to study functional effects, and clinical neuroscience studies (eg, neuroimaging and neurophysiology) examining the effect of genetic variation on brain structure and function.
The next section briefly highlights findings from psychiatric genetic research, most notably the discovery of scores of genetic associations that exceed stringent genome-wide thresholds for statistical significance.
PROGRESS IN PSYCHIATRIC GENETICS
In the 1980s and 1990s, linkage studies implicated a large number of chromosomal regions that might harbor risk loci for a range of psychotic, mood, and anxiety disorders. However, linkage findings were difficult to replicate, and by the late 1990s, it became clear that association studies would be more powerful than linkage studies for discovering common, low penetrance risk alleles. During the next decade, hundreds of candidate gene association studies were reported, but replication of findings from these studies proved elusive.
A turning point for psychiatric genetics occurred with the advent of large GWAS. Whereas few if any loci could be confidently said to be associated with a psychiatric disorder before 2008, GWAS has, at this writing, yielded nearly 200. In 2007, the international Psychiatric Genomics Consortium (PGC) was formed as investigators came together to share GWAS data sets and pursue centralized analyses (Sullivan, 2010). To date, large GWAS by the PGC and other groups have identified numerous genome-wide significant loci, including more than 100 for schizophrenia alone (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
Another success story has been the identification of large (>100 kilobases) CNVs that confer risk to a range of neuropsychiatric disorders, including ASD, SCZ, and, to a lesser extent, BD (Green et al, 2015; Malhotra and Sebat, 2012). Recently, exome sequencing studies and family-based studies of rare mutations have implicated rare SNVs in ASD (Geschwind and State, 2015) and SCZ (Purcell et al, 2014). As expected, the common variants have modest effect sizes (allelic odds ratios ~1.05–1.2) while those for rare CNVs and mutations can be more substantial (odds ratios of ~2.0–30.0). Studies of common variants, rare mutations, and structural variations have also documented genetic overlap among a range of psychiatric disorders (Smoller, 2013). Beyond the growing catalog of established risk variants, pathway analyses have begun to implicate functional sets of genes in multiple disorders, including genes involved in immune function, glial cell function, calcium channel signaling, glutamatergic neurotransmission, histone methylation, structural elements of the postsynaptic density, and targets of the fragile X mental retardation protein (FMRP) (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Duncan et al, 2014a; Lee et al, 2012; Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium et al, 2015; Nurnberger et al, 2014; Pedroso et al, 2012; Purcell et al, 2014; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Uddin et al, 2014).
However, progress in genetic studies of the stress-related disorders reviewed here has been slower. The following sections will review genetic findings for the three categories of stress-related disorders: traumatic-stress disorders (ie, PTSD), MDD, and anxiety disorders (generalized, panic, and phobic disorders). For each, I will first review their genetic epidemiology (family and twin studies), studies of common SNVs, rare SNVs, and structural variation, and finally G × E studies.
GENETICS OF POSTTRAUMATIC STRESS DISORDER
PTSD occurs in a subset of individuals exposed to traumatic events and comprises four clusters of symptoms: intrusive re-experiencing of the trauma, avoidance of stimuli associated with the trauma, negative cognitions and mood associated with the trauma, and excessive arousal or reactivity. In DSM-5 (American Psychiatric Association, 2013), PTSD was moved from the anxiety disorder group to a new category of ‘trauma- and stressor-related disorders’. While a majority (50–85%) of Americans experience a traumatic event during their lifetimes, the lifetime prevalence of PTSD is approximately 7% (Kessler and Wang, 2008), indicating that a minority of individuals exposed to trauma develop the disorder. Genetic studies may provide important clues to these individual differences in vulnerability.
Family and Twin Studies
Because the expression of PTSD requires exposure to a significant trauma, estimating the familial aggregation of PTSD is complicated by the need to account for varying degrees of trauma exposure among relatives. Thus, in a standard case–control design, it is impossible to determine whether relatives who have not experienced significant trauma might have otherwise been affected. Risk of PTSD has been shown to be elevated among offspring of parents with PTSD in some but not in all studies (Leen-Feldner et al, 2013). The largest study (Roth et al, 2014) comprised a cohort of 6924 mothers and their children and found a significant increase in offspring PTSD in a dose–response relationship with maternal posttraumatic stress symptoms (ORs 1.2–1.6). Interestingly, parent–offspring transmission was largely mediated through an increase in trauma exposure among offspring of mothers with PTSD. PTSD-related twin studies have addressed the complex genetic relationship between trauma exposure and disorder risk by distinguishing their heritable components. Genetic factors can contribute to PTSD liability in at least four ways: by influencing the liability to (criterion A) trauma exposure; by influencing PTSD risk independent of trauma exposure; by influencing other risk factors for PTSD (eg, personality traits, or comorbidities); or by a pleiotropic effect on two or more of these.
A series of twin studies treating trauma exposure as the phenotype of interest has documented the heritability of this pre-condition for PTSD. In a study of the Vietnam Era Twin Registry (True et al, 1993) all individual DSM-IIIR PTSD symptoms were heritable (h2=0.32–0.45). In addition, combat exposure was itself similarly heritable (h2=0.35). PTSD and combat exposure were also genetically correlated. In a civilian twin sample, Stein et al (2002) found evidence for the heritability of PTSD symptoms (h2=0.28–0.36) and assaultive trauma (h2=0.20) but not non-assaultive trauma. The genetic correlation between PTSD symptoms and assaultive trauma was substantial (rg=0.76). Interestingly, this genetic correlation declined as the number of assaultive traumas reached high levels, consistent with the hypothesis that large doses of trauma can cause PTSD regardless of genetic vulnerability (Jang et al, 2007). In a large population-based twin sample in which the heritability of PTSD was 0.46, even larger estimates were observed for the heritability of high-risk trauma (h2=0.60), and the genetic correlation between high-risk trauma and PTSD was again substantial (rg=0.89) (Sartor et al, 2012).
Thus, twin studies have documented moderately heritabilities for PTSD (in the range of 30–50%) and high-risk trauma exposure (in the range of 30–60%) and that genetic influences on them are highly overlapping. The mechanism underlying genetic effects on trauma exposure remains unclear, though they may be mediated by personality traits (such as risk-taking) that increase the probability of exposure to dangerous environments.
Common Variation
Candidate gene studies
Efforts to identify specific genetic variants contributing to the heritability of PTSD have overwhelmingly focused on biological candidate genes (Cornelis et al, 2010). In particular, genes involved in monoaminergic neurotransmission and elements of the HPA axis have been commonly investigated. The most widely studied polymorphism has been a variable number tandem repeat (VNTR) in the promoter region of the serotonin transporter gene (SLC6A4), which encodes the target of serotonin reuptake inhibitors. This functional polymorphism, referred to as the 5HTTLPR, is common in European ancestry populations, with a frequency of approximately 45% for the so-called short (S) allele that is associated with reduced transcription of SLC6A4. Association studies of the 5HTTLPR and PTSD have been inconclusive, and a recent meta-analysis of 12 studies found no evidence of association overall, though homozygosity for the S allele was associated with PTSD in samples classified as having high-trauma exposure (Gressier et al, 2013).
Nominally significant associations have been reported between PTSD symptoms and other neurotransmitter- and neuropeptide-related genes including HRT2A, SLC6A3, DRD3, NPY, CNR1, and RGS2 (Cornelis et al, 2010; Sah and Geracioti, 2013; Wolf et al, 2014a). Recent data from the Grady Trauma Project, a study of predominantly African-American inner-city individuals with high trauma exposure (N=2698), found evidence for association between PTSD and variants in the proinflammatory C-reactive protein gene (CRP) (Michopoulos et al, 2015). Risk SNPs were also associated with serum CRP levels which, in turn, were associated with PTSD symptoms and fear-potentiated startle. In the same cohort, a variant in the opioid receptor-like 1 gene (OPRL1) was associated with PTSD symptoms and fear-potentiated startle as well as amygdala-insula connectivity by fMRI (Andero et al, 2013). This gene encodes the amygdala nociceptin (NOP)/orphanin FQ receptor (NOP-R) and rodent studies have demonstrated its importance to fear conditioning. Amygdala expression of the mouse ortholog oprl1 was reduced after fear conditioning in a PTSD-like mouse model. Further, injection of an NOP-R agonist into the central amygdala impaired fear consolidation (Andero et al, 2013). Of note, administration of the opioid agonist morphine shortly after trauma exposure has been found to reduce the incidence of PTSD (Holbrook et al, 2010), further supporting a role for opioidergic systems in PTSD.
More compelling results have been reported for two genes related to neuroendocrine function. Pituitary adenylate cyclase-activating polypeptide (PACAP) is a hypothalamic neuropeptide with modulatory effects on a broad range of neurohormones including stimulation of corticotropin releasing hormone (CRH) secretion (Vaudry et al, 2009). In mice, deletion of the PACAP receptor PAC-R1 is associated with decreases in anxiety behavior and fear conditioning. Ressler et al (2011) reported that variation in ADCYAP1R1, the gene encoding PAC-R1, is associated with PTSD in females. In particular, an SNP (rs2267735) in a putative estrogen response element (ERE) was associated with PTSD risk as both a main effect and a gene–environment interaction with trauma. While several subsequent association analyses did not support a main effect of rs2267735 with PTSD (Chang et al, 2012), gene–environment interactions for this SNP have been observed with childhood adversity and high levels of lifetime trauma in females (Almli et al, 2013; Uddin et al, 2013). Further analyses revealed associations between the PTSD risk allele at this SNP and expression of PAC-R1 in human brain, increased amygdala reactivity to threat stimuli, reduced amygdala/hippocampal connectivity, and decreased hippocampal activation during contextual fear conditioning by fMRI (Pohlack et al, 2015; Ressler et al, 2011; Stevens et al, 2014).
Perhaps the most most comprehensively-studied candidate to date has been the FK506 binding protein 5 gene, FKBP5. Binder, Ressler and colleagues (Binder et al, 2004, 2008; Fani et al, 2013; Klengel et al, 2013; Mehta et al, 2011) have demonstrated a role for FKBP5 variation in stress-related phenotypes and disorders. However, the evidence linking this gene to PTSD suggests that its effect is mainly in the form of G × E, and so it is discussed in the G × E section below. In general, candidate gene and GWAS studies have not supported a main effect of FKBP5 variation (Binder et al, 2008; Logue et al, 2013; Nievergelt et al, 2015; Solovieff et al, 2014).
For the most part, candidate gene studies of PTSD, like those for the other stress-related disorders reviewed here, have been underpowered, making both positive and negative findings difficult to interpret. One of the largest candidate gene studies, comprising 845 PTSD cases and 1693 trauma-exposed controls from the Nurses Health Study II, examined 3742 SNPs spanning more than 300 genes (Solovieff et al, 2014). With one exception, no genes showed significant evidence of association. The exception was SLC18A2 (aka VMAT2) whose product transports monoamine neurotransmitters into synaptic vesicles. An intronic SNP (rs323276) was associated with PTSD after correction for multiple testing of candidate SNPs and phenotypes. In an independent African-American sample drawn from the Detroit Neighborhood Health Study, a 9-SNP haplotype containing the rs323276 risk allele was again associated with PTSD, though rs323276 itself was not associated. In another study of a similar set of 295 candidate genes, no main effect associations were identified in a sample of 810 National Guard soldiers (Liberzon et al, 2014).
Genome-wide association studies
The first GWAS of PTSD, in military veterans and their intimate partners, reported a genome-wide significant association between PTSD (295 cases and 196 controls) and an SNP (rs8042149) in the retinoid-related orphan receptor gene (RORA) (Logue et al, 2013). While this SNP was not significantly associated in two replication samples in the same publication, further analysis of RORA SNPs in a subset of the discovery and replication samples (N=540) reported an association between rs17303244 and a fear component of PTSD (Miller et al, 2013). Examination of rs8042149 among individuals assessed for PTSD symptoms after hurricane exposure detected an association with PTSD symptom severity (Amstadter et al, 2013). However, associations between RORA SNPs (including rs8042149) were not detected in two larger independent replication samples from the Nurses’ Health Study II and a cohort of Iraq and Afghanistan-era US veterans (Guffanti et al, 2014).
Genome-wide significant associations have also been reported with PTSD in the Tolloid-Like 1 gene (TLL1), the cordon-bleu WH2 repeat protein gene (COBL) (Xie et al, 2013), and in the lincRNA gene AC068718.1 (Guffanti et al, 2013). In a sample of 3215 trauma-exposed individuals, no significant association between the COBL SNP rs406001 with PTSD symptoms was found, but a significant interaction between this SNP with childhood trauma was reported (Almli et al, 2014).
Nievergelt et al (2015) recently conducted a GWAS of PTSD in a multi-ethnic cohort (N=3494) of male combat-exposed US Marines and Sailors from the Marine Resiliency Study. A total of 940 met criteria for partial or full PTSD. Meta-analysis of GWAS results across ethnicities identified a genome-wide significant SNP (rs6482463) in intron 3 of the phosphoribosyl transferase domain containing 1 gene (PRTFDC1). However, there was no clear evidence of association in a smaller replication sample of veterans and their intimate partners.
A single study surveyed SNPs across the mitochondrial genome (Flaquer et al, 2015) in light of the key role of mitochondria in cellular stress responses to a range of external challenges (Manoli et al, 2007). Mitochondrial dysfunction has been linked to abnormalities in synaptic plasticity and cellular resilience and has been implicated in mood and other psychiatric disorders (Manji et al, 2012). Flaquer et al reported association between PTSD and two mitochondrial SNPs (mtSNPs): mt8414C→T, a missense variant located in MT-ATP8, and mt12501G→A a synonymous variant located in MT-ND5. Both genes are involved in the regulation of reactive oxygen species. Finally, a GWAS of dissociative symptoms associated with PTSD, and another recent GWAS of PTSD among combat veterans reported no genome-wide significant hits (Ashley-Koch et al, 2015; Wolf et al, 2014b).
Rare and Structural Variation
To date, no systematic search for rare SNVs or structural variation related to PTSD has been reported.
Gene–Environment Interaction
With the exception of substance use disorders, PTSD is the only psychiatric disorder for which an environmental exposure is required by definition. In addition other environmental exposures, including childhood maltreatment, poverty, and low social support, have been shown to increase the risk of PTSD among trauma-exposed individuals (DiGangi et al, 2013b; Ozer et al, 2003). In a 2013 systematic review, Digangi et al (2013a) examined the 28 candidate gene association studies of PTSD that compared cases and trauma-exposed controls. They concluded that, in addition to low statistical power, G × E studies of PTSD have suffered from a range of limitations including a restricted focus on a small number of polymorphisms and inadequate consideration of the timing, chronicity, or differential effect sizes of multiple exposures. Notable G × E associations in PTSD with at least some replication in independent samples have included interactions between childhood adversity and the PACAP receptor (described above) (Almli et al, 2013; Ressler et al, 2011; Uddin et al, 2013) as well as a promoter region polymorphism in the β2 adrenergic gene (ADRB2) (Liberzon et al, 2014).
The most extensive G × E analyses have focused on FKBP5 and have elucidated a mechanism by which genetic variation in FKBP5 may interact with early adversity to increase the risk of stress-related disorders. The gene product of FKBP5 acts as a co-chaperone in the Hsp-90-steroid receptor complex and inhibits glucocorticoid receptor (GR) activity by several mechanisms including reducing GR binding affinity and nuclear translocation (Zannas and Binder, 2014). FKBP5 transcription is itself rapidly induced by GR activation through steroid-response elements in the FKBP5 promoter region. These reciprocal effects create an ultra-short negative feedback loop that modulates GR sensitivity (Zannas and Binder, 2014). Several studies have reported that variation in FKBP5 moderates the effect of adversity and trauma on risk of PTSD (Binder et al, 2004, 2008; Fani et al, 2013; Klengel et al, 2013; Mehta et al, 2011; Xie et al, 2010). In particular, the FKBP5 SNP rs1360780, located in an enhancer region 488 bp away from a glucocorticoid response element (GRE) in intron 2, appears to be functionally related to GR-mediated expression of FKBP5 (Klengel et al, 2013). The minor T allele enhances FKBP5 transcription in response to GR activation by inducing chromatin-mediated conformational changes that enhance contact between the intron 2 GRE and the transcription start site (TSS). Exposure to early-life adversity and trauma leads to GR activation and, in the presence of the T (risk) allele, induces excessive hippocampal FKBP5 expression resulting in sustained cortisol levels. This effect is accompanied by demethylation of a GRE in intron 7 that also interacts with the TSS, further amplifying FKBP5 expression in the presence of GR activation (Zannas and Binder, 2014). Thus, FKBP5 genotype interacts with childhood trauma to induce epigenetically-mediated hippocampal glucocorticoid resistance. This effect was not observed with adult trauma. Klengel et al (2013) demonstrated that this G × E is associated with increased risk for stress-related disorders including PTSD and depression. In addition, FKBP5 risk variants have been associated with neuroimaging markers implicated in stress-related disorders, including amygdala and hippocampal volume and reactivity (Fani et al, 2013; Holz et al, 2015; Pagliaccio et al, 2014; White et al, 2012). Several important features of this story are worth highlighting. First, they provide some of the most direct evidence of both statistical and biological gene–environment interaction. Second, they highlight the importance of developmental sensitive periods in G × E studies: the FKBP5 risk allele effects on gene expression and PTSD risk appear to interact specifically with childhood but not later-life adversity.
Genetic and G × E studies of PTSD face an important challenge which is often under-appreciated. Trauma exposure is a precondition for the expression of PTSD. However, we need to distinguish two types of adversity exposure that have different etiologic roles. We might call the first type ‘index traumas’—that is, traumatic events that fulfill Criterion A of the DSM diagnostic criteria as causes of PTSD (see Figure 2). The second type, which I’ll call ‘risk exposures’, would include traumas and other adversities (eg, early or chronic stressors) that are risk factors for PTSD (Koenen et al, 2007; Ozer et al, 2003) but were not sufficient to cause PTSD. The outcome of main-effect genetic studies and G × E studies of PTSD will depend on how these two types of trauma are ascertained in case–control studies. In addition, as noted earlier, genetic influences on trauma exposure and PTSD overlap. This gene-by-environment correlation (rGE) suggests that some of the genetic variants influencing PTSD risk may act through their effect on exposure to causal traumas. That is, a genetic variant may increase the risk for PTSD by increasing the risk of exposure to index traumatic events. Genetic association studies of PTSD often compare cases with controls matched on trauma exposure. By design, then, such analyses would have limited ability to detect the fraction of PTSD risk alleles whose effect is mediated by increasing exposure to an ‘index trauma’. At the same time, studies that include controls unscreened for index trauma exposure would have reduced power to detect loci that act independent of an effect on trauma exposure compared with studies in which controls are matched for trauma exposure. In examining G × E for risk exposures, matching cases and controls with respect to index traumas becomes less relevant. G × E studies of PTSD where the environmental exposure is an index trauma could compare cases with varying severity of index trauma, for example. On the other hand, G × E studies of risk exposures could examine controls matched (or not) for index trauma. However, the interpretation of both main effect and G × E studies are further complicated by the fact that index traumas and risk exposures are correlated (Koenen et al, 2007). Unfortunately, it is often difficult to determine whether a trauma exposure is a sufficient cause or merely risk-enhancing and few studies have endeavored to distinguish these two classes.

Complexity of causal relationships between genetic risk and environmental trauma/adversity. Only a subset of possible relationships are depicted. Genes (G) may (path a) influence PTSD directly or (b) by increasing the risk of index traumas (Ti) which in turn cause PTSD (c). In addition, index traumas may increase PTSD risk independent of genetic effects (d). In addition, trauma/adversity may act as risk exposures (Re) that amplify risk (e and f) but are not a primary cause of PTSD. These risk exposures may themselves be under genetic influence (d). Finally, Ti and Re may be correlated and each may modify the effect of risk genes (representing gene–environment interactions).
Summary
Twin studies have documented that both trauma exposure and PTSD itself are moderately heritable and genetically correlated. To date, however, the molecular genetic basis of PTSD remains largely uncharted (Table 1). Candidate gene studies have focused primarily on a subset of genes involved in serotonergic, dopaminergic, and neuroendocrine function with decidedly mixed results. GWAS have identified a handful of common variants associated with PTSD, but none have been widely replicated. More convincing evidence has been reported for a G × E between early adversity and variation in FKBP5, and functional studies have provided the basics of a mechanistic account of how this genetic variation alters stress responses by epigenetic alterations of gene expression and downstream effects on glucocorticoid function. In general, common variant studies of PTSD have been underpowered to detect the relatively modest effects that have been validated in large-scale GWAS studies of other complex phenotypes. In addition, the role of rare and structural variation remains unexplored.
In the coming years, progress in identifying the genetic basis of PTSD is expected to take a leap forward with analyses of larger samples. In particular, the PGC’s PTSD Workgroup is bringing together GWAS data sets from around the world and participating sites have genotyping completed or underway for approximately 20 000 cases and 50 000 controls (Logue et al, 2015).
GENETICS OF DEPRESSION
MDD is a common condition with lifetime prevalence estimates from US population-based studies of 11% among adolescents (Avenevoli et al, 2015) and more than 16% among adults (Kessler and Wang, 2008). In addition, depressive disorders including MDD and dysthymia are the second leading cause of disability worldwide (Ferrari et al, 2013). A variety of stressful life events have been shown to increase the risk for depression (Hammen, 2005; Kessler, 1997) including poverty (Brooks-Gunn and Duncan, 1997; McLeod and Shanahan, 1996), negative family relationships and parental divorce (Gilman et al, 2003; Repetti et al, 2002), and childhood maltreatment (Chapman et al, 2004; Widom et al, 2007) Unlike PTSD, MDD and depressive symptoms have been investigated extensively in genetic studies, with genetic epidemiologic studies dating back to the 1920s (Tsuang and Faraone, 1990). Recent genomic studies have implicated several risk loci, although few have been replicated, as detailed below.
Family and Twin Studies
A meta-analysis of high quality family studies confirmed that MDD aggregates in families, with a summary odds ratio of 2.84 (95% CI: 2.31–3.49) among first-degree relatives of affected probands compared with those of controls (Sullivan et al, 2000). More recent family studies have produced similar recurrence risk ratio estimates (Merikangas et al, 2014). Twin studies have shown that the familial aggregation of MDD is largely due to genetic factors, with a heritability of approximately 40% (Kendler et al, 2006; Sullivan et al, 2000), though higher estimates (up to 70%) have been reported in clinically-ascertained or longitudinal twin samples (Kendler et al, 1993b; McGuffin et al, 1996).
Family and twin studies have also examined subtypes of MDD that may confer greater familial risk (Smoller and Perlis, 2004). In particular, increased familial recurrence risk and heritability have been associated with earlier-onset and recurrent depression (Bland et al, 1986; Kendler et al, 1999a, 2005; Weissman et al, 1984) as well as greater depression severity or impairment (Klein et al, 2002; Lyons et al, 1998), though these findings have not been universally observed.
Common Genetic Variation
Candidate gene studies
Before the availability of GWAS, common variation studies of MDD were largely limited to biological candidate genes in neurotransmitter, neuropeptide, and neuroendocrine systems related to antidepressant targets or implicated in animal models. The results of these studies have been mixed and most have been underpowered to detect the modest effects expected for common variants. Meta-analyses of biological candidate gene studies have provided some support for association between MDD and specific variants (Gatt et al, 2015). In particular, nominal associations have been reported in meta-analyses for variants in several serotonergic genes (HTR2A, SLC6A4 HTR1A,TPH2) (Gao et al, 2012; Kishi et al, 2013; Lopez-Leon et al, 2007; Zhao et al, 2014), as well as APOE, DRD4, GNB3, SLC6A3, and MTHFR (Clarke et al, 2010; Lopez-Leon et al, 2007; Wu et al, 2013). Of note, however, none of these genes has been implicated in large GWAS (Wray et al, 2012). As Flint and Kendler (2014) have demonstrated, candidate gene meta-analyses of MDD have been underpowered and the data thus far are consistent with a lack of significant findings in any meta-analysis. Using a different approach, Lee et al (2012) examined 188 MDD candidate genes and 178 biological gene sets related to these genes in a pathway analysis encompassing three GWAS data sets. Genes involved in glutamatergic synaptic neurotransmission were significantly associated with MDD after correction for multiple testing.
GWAS
The first GWAS of MDD comprised 1738 cases and 1802 controls (Sullivan et al, 2009). Although no genome-wide significant loci were detected, 11 of the top signals localized to a 167-kb region encompassing PCLO, which encodes piccolo, a presynaptic protein involved in monoamingeric neurotransmitter release. A larger series of replication samples did not support this association but an exploratory analysis of the subset of samples most similar to the discovery sample yielded nearly significant association for a non-synonymous PCLO SNP (rs2522833). Although subsequent candidate gene studies have implicated this or other PCLO SNPs in depression (Hek et al, 2010; Minelli et al, 2012), bipolar disorder (Choi et al, 2011), and HPA axis function (Kuehner et al, 2011; Schuhmacher et al, 2011), the gene has not shown association in independent GWAS of MDD. A subsequent GWAS (Kohli et al, 2011) reported a significant recessive association for an SNP (rs1545843) in SLC6A15, a gene involved in transporting neutral amino acids. Risk alleles were also correlated with reduced hippocampal SLC6A15 expression and reduced hippocampal volume and neuronal integrity measured by magnetic resonance spectroscopy. Mice susceptible to chronic stress were also found to have reduced hipoocampal SLC6A15 expression. Subsequent research suggested that the putative risk allele is associated with enhanced cortisol secretion on the dexamethasone/CRH test and impairments in memory and attention in patients with MDD (Schuhmacher et al, 2013). Of note, however, this locus has not been supported in subsequent larger depression GWAS (Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium et al, 2013; Wray et al, 2012) or in GWAS meta-analyses of hippocampal volume by the international ENIGMA consortium (Hibar et al, 2015).
More than 15 GWAS of MDD have now been reported, but success in establishing risk variants has been limited (Dunn et al, 2015). The largest case–control GWAS to date was reported by the PGC, comprising a discovery sample of 9240 cases and 9519 controls (Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium et al, 2013). No SNPs reached genome-wide significance in this sample or in a joint analysis with independent replication samples. The study found that loci accounting for as little as 0.5% of the phenotypic variance in MDD could be excluded with 90% power. Secondary analyses of depressive subtypes (by sex, recurrent depression, recurrent early-onset, and latent class analysis of ‘typical’ MDD) also produced no significant findings, though smaller sample sizes limited the power of these analyses.
A GWAS meta-analysis of 17 independent samples (N=34 549) from the CHARGE Consortium (Hek et al, 2013) examined depressive symptoms measured by the Center for Epidemiologic Studies Depression Scale (CES-D). No genome-wide significant associations were identified in the discovery sample. However, one locus on chromosome 5q21 did show significant evidence of association when top SNPs from this analysis were meta-analyzed with an independent replication sample (total n=51 258). Because the CES-D assesses depressive symptoms in the past week, it may be that phenotypic misclassification contributed to the largely null results of this study. Other loci that appear to influence depression risk have emerged from cross-disorder genetic studies (discussed in more detail later), including SYNE1 (Green et al, 2013) (a gene associated with BPD) and CACNA1C (also associated with BPD and SCZ) (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Green et al, 2010).
The most compelling GWAS results for MDD were recently reported by the CONVERGE Consortium using low-coverage whole genome sequencing (CONVERGE Consortium, 2015). In a sample of 5303 Han Chinese women with recurrent MDD and 5337 controls, they identified two genome-wide significant loci: one on chromosome 10 near SIRT1 and a second in an intron of LHPP. These loci were replicated in an independent Chinese sample, and in a combined analysis of the discovery and replication samples, the top SIRT1 SNP (rs12415800) and the top LHPP SNP (rs35936514) achieved P-values of 2.37 × 10−10 and 6.43 × 10−12, respectively. The investigators attributed their success to reducing heterogeneity by focusing on a Chinese sample with severe MDD. Indeed, the SIRT1 association was even stronger when cases were restricted to those with the more severe melancholic subtype despite a smaller sample size. The associated SNPs are much less common in European ancestry samples and were not associated in the larger PGC-MDD mega-analysis (European ancestry), suggesting that these loci may have population-specific effects.
In addition to single locus analyses, gene set enrichment analyses of GWAS data for MDD have begun to implicate numerous biological pathways (Kao et al, 2012; Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015; Song et al, 2013). The largest of these, by the PGC, included GWAS data on more than 60 000 individuals across multiple psychiatric disorders (Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015). With respect to MDD, enriched pathways included those involved in protein phosphatase type 2A regulator activity, cell junction organization, and histone modification.
Rare and Structural Variation
Depressive symptoms are among the phenotypic manifestations of several medical genetic syndromes caused by rare mutations in specific genes. These include autosomal dominant disorders such as Huntington disease (gene: HTT) and acute intermittent porphyria (HBMS), recessive inborn errors of metabolism such as homocystinuria (CBS) and other recessive disorders such as Wolfram syndrome (WFS1), X-linked disorders such as Fabry disease (GLA), and mitochondrial disorders such as MELAS (MT-TL1 and MT-ND5) (Anglin et al, 2012; Ebert et al, 2010). An excess of expanded triplet repeat alleles in HTT was also reported in a study examining more than 11 000 chromosomes from MDD cases and controls, with a reported carrier frequency of 3 per 1000 among MDD patients (Perlis et al, 2010).
Pre-dating the recent era of common variant and rare variant association studies, a large number of linkage analyses of MDD were reported. Although linkage studies are less powerful than association analysis for studying common risk variants, they can detect rare variant loci that segregate in families. Linkage signals have been reported for 15 of the 22 autosomes, with some regions observed in independent studies (Flint and Kendler, 2014), but none of these have led to the identification of specific mutations. Few rare variant association studies have been reported for non-syndromic forms of MDD (Kato, 2015) and no well-powered whole exome or whole genome studies are available. In sum, then non-syndromic forms of MDD have not been convincingly associated with rare SNVs, though efforts to identify such variants have been limited.
Several genome-wide studies have examined rare CNVs in relation to MDD. An initial report (Glessner et al, 2010) identified a 646-kb duplication in 5 of 1693 cases but none of 4506 controls. The duplication, on chromosome 5q35.1, encompassed exons of SLIT3, CCDC99, and DOCK2; however, subsequent studies have not observed enrichment of this duplication. In one study, though, exome sequencing of 40 families ascertained for probands with ASD identified putatively damaging SLIT3 mutations in two families in which carriers presented with MDD (Cukier et al, 2014). The gene is of interest because of its role in axon guidance. Another study found a significantly greater burden of copy number deletions of >100 kb in cases with recurrent MDD (N=3106) compared with controls (N=459 screened and 5619 unscreened controls) (Rucker et al, 2013), but this was not supported in a re-analysis of the same cases with more stringent CNV calling algorithms (Rucker et al, 2015). Additional studies have also failed to find significant evidence for enrichment of CNVs in recurrent MDD (O'Dushlaine et al, 2014) or among patients who attempted suicide (Perlis et al, 2012). Finally, whole genome sequencing in a sample of children and adolescents referred to a neuropsychiatric evaluation clinic and observed two gene-disrupting CNVs in children with MDD and anxiety (Brand et al, 2014). Larger-scale studies will be needed to clarify whether CNVs represent a risk factor for MDD.
Gene-Environment Interaction
There is no question that both genes and environment influence risk for depression, and recent evidence suggests that exposure to stressful life events is, itself, heritable (Power et al, 2013). However, G × E studies ask the more specific question of whether the joint effect of genetic variation and environmental risk factors is significantly different from the product of their individual effects. The existence of important G × E effects could help account for the largely negative findings of main effect candidate gene and GWAS studies. Perhaps the most influential and controversial association finding related to depression was a report that stressful life events interact with the 5HTTLPR to increase the risk of depression (Caspi et al, 2003). That study examined 847 members of the longitudinal Dunedin Multidisciplinary Health and Development Study at age 26 and found a dose-response interaction between the 5HTTLPR short allele and number of stressful life events over the past 5 years on past-year major depressive episodes, depressive symptoms, and suicide attempt. The same interaction was seen when considering childhood maltreatment as the environmental exposure. These findings seemed to fit with convergent evidence from animal models (Champoux et al, 2002; Holmes et al, 2003). They also spawned a large number of similar studies and attempts at replication, though findings have been mixed. In fact, meta-analyses of the 5HTTLPR G × E have come to conflicting conclusions. Weighing against the validity of an interaction, the two meta-analyses that used more stringent study inclusion criteria did not support the interaction (Munafo et al, 2009; Risch et al, 2009) while those with more liberal criteria (and using one-tailed P-values) did (Karg et al, 2011; Sharpley et al, 2014). In addition, the most direct effort to replicate the original findings (using a similar measure in a longitudinal birth cohort from New Zealand) found no evidence for the interaction even after conducting 104 separate regression models (Fergusson et al, 2011). Some have suggested that the inconsistent picture of G × E studies of the 5HTTLPR is attributable to heterogeneity of measurement and outcomes across studies, and that the effect is predominantly seen in females, young people, and studies using structured diagnostic interviews to assess exposures and depression (Uher and McGuffin, 2008). Nevertheless, the fact that better-powered replication studies have been less likely to detect the interaction and that evidence of publication bias exists have cast doubt on the robustness of the finding (Duncan et al, 2014b).
Many other candidate gene G × E studies of depression have been reported, largely focusing on monoaminergic and HPA axis genes (Dunn et al, 2011; Mandelli and Serretti, 2013). Of these, the strongest evidence has emerged for genes involved in stress hormone function—in particular, CRHR1 and FKBP5. CRHR1 is a G protein-coupled receptor that has a key role in stress responses through its role in the HPA axis well as by mediating CRH effects on extrahypothalamic sites. An extensive preclinical literature has supported a role for CRHR1 in depressive and anxiety behaviors (Heim et al, 2009). CRHR1 SNPs have also been associated with anxious temperament and emotion circuit activity in a rhesus macaque model (Rogers et al, 2013) as well as MDD and cortisol reactivity in human studies (Ishitobi et al, 2012; Mahon et al, 2013; Sheikh et al, 2013). In a predominantly African-American sample, Bradley et al (2008) reported an interaction between variants in intron 1 of CRHR1 and a history of child abuse for risk of depressive symptoms, with supportive evidence in a smaller clinical sample of predominantly European-American women. Several subsequent studies have supported a CRHR1 genotype × childhood maltreatment effect for depression (Heim et al, 2009; Laucht et al, 2013; Ressler et al, 2010) and cortisol reactivity (Heim et al, 2009; Sumner et al, 2014; Tyrka et al, 2009). As reviewed above, childhood adversity or trauma has also been shown to interact with FKBP5 variation to increase the risk for stress-related disorders including depression (Appel et al, 2011; Zimmermann et al, 2011).
As noted earlier, candidate gene studies have limitations that may be overcome by well-powered genome-wide studies. To date, however, genome-wide G × E studies have only begun to appear. Using a PRS derived from the PGC-MDD meta-analysis, Peyrot et al (2014) observed a PRS-by-childhood trauma interaction for risk of depression in an European sample. Specifically, the penetrance of polygenic loading for MDD was greater among those with a history of childhood trauma. A US population-based study of older adults (N=8761) found no interaction between the MDD PRS and recent stressful life events on depressive symptoms, though the life events measure was limited and the outcome measure (CES-D) captures only current symptoms (Musliner et al, 2014).
Summary
MDD aggregates in families and is moderately heritable (~40%). The voluminous body of biological candidate gene studies has implicated several loci but none of these genes has been supported by unbiased genome-wide studies. In fact, the largest GWAS mega-analysis of MDD failed to identify any genome-wide significant common variants. More recently, two genome-wide significant loci were identified in a sample of Han Chinese women with recurrent MDD.
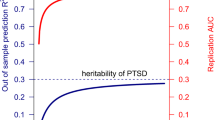
The difficulty in identifying loci associated with depression from large-scale GWAS suggests that the genetic architecture of depression is highly complex. In particular, the common variant component of MDD is likely to be extremely polygenic with thousands of contributing variants of individually small effect (Chang et al, 2014; Major Depressive Disorder Working Group of the Psychiatric GC et al, 2013). The hSNP2 of MDD has been estimated at 0.21–0.32 (Cross-Disorder Group of the Psychiatric Genomics Consortium et al, 2013; Lubke et al, 2012), confirming that common variants are causally related to risk. However, simulations accounting for disorder prevalence and heritability indicate that GWAS of MDD would need to have sample sizes up to five-fold larger than those for SCZ to have comparable power to identify risk loci (Wray et al, 2012; Yang et al, 2010). Experience with GWAS of complex traits suggests that there is typically a sample size threshold or inflection point beyond which the number of genome-wide significant loci increases linearly (Levinson et al, 2014). For SCZ, the inflection point was observed at approximately 15 000 cases. Extrapolating from these results, the inflection point for MDD is not likely to be reached before GWAS include 75 000 to 100 000 cases. These predictions will soon be put to the test as analyses on this scale are currently being planned by the PGC. However, as recently discussed (Levinson et al, 2014), sample size requirements could even be greater in the presence of misdiagnosis or if MDD is more genetically heterogenous than SCZ. Thus, strategies to enhance the genetic homogeneity of MDD samples may be needed to advance our understanding of the genetic basis of depression. Numerous variables have been proposed to index more genetically homogenous subtypes of MDD, including age of onset, recurrence, comorbid anxiety, chronicity, severity, suicidality, postpartum onset, and others. In addition, a recent large twin study suggested that MDD risk may reflect three orthogonal genetic factors, influencing, respectively, a general liability to internalizing disorders, core symptoms of MDD, and neurovegetative symptoms (Kendler et al, 2013). The recent successful GWAS by the CONVERGE Consortium supports the utility of examining more homogenous subtypes of depression.
Larger-scale genome-wide studies will also facilitate studies of gene–environment interplay (Dunn et al, 2015). For example, risk loci established by GWAS would provide a set of robust candidate variants for G × E studies. In addition, genome-wide G × E analyses may be useful for discovering novel interactions. Finally, the contribution of rare and structural variation remains to be seen.
GENETICS OF ANXIETY DISORDERS
According to epidemiologic surveys, anxiety disorders represent the most common class of psychiatric disorders, though these estimates include PTSD and obsessive-compulsive disorder (OCD) which are no longer grouped with anxiety disorders in DSM-5 (Kessler and Wang, 2008). The burden of these disorders is substantial, owing in part to their early onset and persistence (Costello et al, 2005). Early stress and trauma are important risk factors for anxiety disorders (Gibb et al, 2007; Stein et al, 1996) and dysregulation of HPA axis function has been demonstrated in generalized, panic, and phobic anxiety disorders (Abelson et al, 2007; Dieleman et al, 2015).
Family and Twin Studies
The risk of anxiety disorders is approximately 4–6 times higher in first-degree relatives of affected probands compared with relatives of unaffected probands (Hettema et al, 2001). In twin studies, heritability estimates have been in the range of 30–50% (Hettema et al, 2001; Scaini et al, 2012, 2014; Smoller et al, 2009; Tambs et al, 2009a) though higher estimates (~40–65%) have been reported from analyses that include repeated assessments and correct for diagnostic unreliability (Kendler et al, 1999b). In addition, family and twin studies have suggested that genetic contributions cross the boundaries among anxiety disorders and between normal and pathologic anxiety. First-degree relatives of probands with one anxiety disorder are at risk for a range of other anxiety disorders, and twin studies have documented genetic overlap among most if not all anxiety disorders (Hettema et al, 2005; Roberson-Nay et al, 2012; Smoller et al, 2008a; Tambs et al, 2009b). Factor analyses of twin data show genetic overlap of phobic fears that do not respect DSM boundaries (Loken et al, 2014). In fact, the shared genetic component among panic disorder (PD), phobias, generalized anxiety disorder (GAD), OCD, and PTSD is substantially larger than their disorder-specific genetic components (Tambs et al, 2009b).
Common Genetic Variation
As with PTSD and MDD, the majority of genetic association studies of the anxiety disorders have been candidate gene studies based on a limited number of biological hypotheses. These have commonly focused on genes related to monoaminergic neurotransmitter systems, neuropeptides, and HPA axis function. Of the anxiety disorders, PD has received the most attention.
Panic disorder
In candidate gene studies of PD, several loci have shown nominal association in independent samples. These include the 5HTTLPR polymorphism of SLC6A4, the val158met polymorphism (rs4680) of COMT, and a promoter length polymorphism of MAOA; however, conflicting results have been reported and the direction of allelic effect has been inconsistent. Nominally significant results have also been reported between panic disorder or panic attacks with variants in other monoaminergic genes (eg, HTR1A and HTR2A), GABA receptor genes (GABRB3 and GABRA5) stress hormone genes (CRHR1), neuropeptidergic/neurotrophic genes (BDNF, NPSR1, ACE), and many others, but a recent comprehensive meta-analysis of the 23 most widely-studied candidate variants found no robust associations (Howe et al, 2015).
The relevance of several of these genes has been supported by imaging-genetic studies in which putative anxiety or panic risk alleles have been associated with reactivity or structural differences in the amygdala or related components of fear circuitry including functional variants in SLC6A4 (5HTTLPR), MAOA (μVNTR), BDNF (val66met), COMT (val158met), HTR1A (−1019C/G), NPRS1 (Asn107Ile), and RGS2 (rs4606) (Domschke and Dannlowski, 2010); again, however, association results are inconsistent. The most widely-studied of these has been the 5HTTLPR for which the short allele has been associated with amygdala reactivity in response to emotional faces in numerous studies. However, the most recent meta-analysis of 35 studies failed to support this association (Bastiaansen et al, 2014).
Another approach has been to focus on biologic systems that may be specific to PD. For example, heightened sensitivity to carbon dioxide (CO2) is an established biological correlate of PD. Inhaled CO2 triggers panic attacks in a majority of individuals with PD, but only a minority of unaffected controls (Battaglia and Ogliari, 2005). In rodents, CO2 inhalation reduces amygdala pH, inducing acidosis and fear behaviors (Coryell et al, 2007; Wemmie et al, 2003; Ziemann et al, 2009). More specifically, the rodent amygdala acts as a chemosensor for the detection of hypercarbia, a function mediated by the acid sensing ion channel-1a subunit (ASIC1a) (Ziemann et al, 2009). Disrupting the asic1a gene in mice decreases acidosis-induced fear behavior, which can be restored through transgenic expression of asic1a in the amygdala (Ziemann et al, 2009). Similar findings have been reported for asic1a at the level of the bed nucleus of the stria terminalis (Taugher et al, 2014). We previously reported evidence of linkage between PD and a region of chromosome 12q encompassing the human ortholog of asic1a (ACCN2) (Smoller et al, 2001). More recently, we observed association between variants in ACCN2 and PD that appeared to be stronger in cases with the respiratory subtype of PD (Smoller et al, 2014). In healthy volunteers, PD-associated alleles were also associated with amygdala volume and amygdala reactivity to emotional faces (Smoller et al, 2014). In a subsequent report, an intronic ACCN2 SNP was associated with respiratory rate and anxiety sensitivity (but not panic symptoms) in response to inhalation of CO2-enriched air among healthy volunteers (Savage et al, 2015).
In the first GWAS of PD (N=200 cases/200 controls) (Otowa et al, 2009), genome-wide significant variants were reported in TMEM16B and PKP1, but these findings were not supported in a replication attempt by the same investigators in a larger independent sample (Otowa et al, 2010). However, more robust support has emerged for transmembrane protein 132D (TMEM132D). An initial GWAS found association of a TMEM132D SNP (rs7309727) as well as a haplotype combining this and another SNP (rs11060369). Risk genotypes were associated with higher TMEM132D mRNA expression in human post-mortem frontal cortex, results further supported by a mouse model in which high anxiety-related behavior was associated with a Tmem132d SNP and correlated with expression of Tmem132d mRNA in the anterior cingulate cortex (Erhardt et al, 2011). In a subsequent meta-analysis of eight independent case-control samples, genome-wide significant associations of rs7309727 and the haplotype of rs7309727-rs11060369 were reported when the analysis was restricted to European ancestry cases with primary PD (Erhardt et al, 2012). The function of this gene is not fully understood, but it has been suggested that it has a role in threat processing (Haaker et al, 2014).
Other anxiety disorder-related phenotypes
Fewer candidate gene studies and no GWAS have been reported for social anxiety disorder (SAD), phobic disorders, or GAD. Nominally significant associations with social anxiety or SAD have been reported for several candidate loci including COMT (Hettema et al, 2015), catenin-δ2 (CTNND2) (Nivard et al, 2014), and CNTNAP2 (Stein et al, 2011) and several loci involved in circadian rhythm function (Sipila et al, 2010). Given the strong genetic correlations among anxiety disorders, several studies have examined broad phenotypes including multiple anxiety disorders with nominal associations reported for glutamic acid decarboxylase 1 (GAD1) (Donner et al, 2012; Hettema et al, 2006a), DRD2 (Sipila et al, 2010), COMT, BDNF (Enoch et al, 2008), and PPARGC1A (Hettema et al, 2011).
In addition to the genetic and phenotypic overlap among anxiety disorder phenotypes, the boundary between pathologic and subsyndromal anxiety is indistinct, consistent with the hypothesis that anxiety disorders represent more extreme variations of a continuously distributed set of traits. For the most part, studies of the genetics of anxiety related traits have focused on temperamental and personality traits associated with a liability to develop various anxiety disorders in children and adults, including behavioral inhibition (BI), neuroticism, introversion, and harm avoidance (Smoller et al, 2015). For example, BI, a stable temperamental tendency to be cautious and behaviorally restrained in novel social and non-social situations, is a familial and developmental risk factor for SAD and other anxiety disorders (McGrath et al, 2012). We found that genetic variants in RGS2, a gene implicated in mouse models of anxious temperament (Yalcin et al, 2004), are associated with BI in childhood, introversion in adults and amygdala and insular cortex activation in response to emotional faces, brain phenotypes implicated in liability to social and other anxiety disorders (Smoller et al, 2008b). The relevance of RGS2 to pathologic anxiety is supported by several studies that have reported association to a variety of anxiety disorders, including GAD, PD, and PTSD (Amstadter et al, 2009; Hohoff et al, 2015; Koenen et al, 2009; Leygraf et al, 2006; Mouri et al, 2009; Otowa et al, 2011) as well as SSRI response in patients with SAD (Stein et al, 2014), although studies to date have been small and negative results have been reported (Hettema et al, 2015).
GWAS of anxiety-related traits have not yet established any genome-wide significant loci. Indeed, meta-analyses of up to 17 000 subjects have found no genome-wide significant associations for neuroticism or harm avoidance (de Moor et al, 2012; Service et al, 2012). A GWAS of phobic anxiety measured by the 8-item Crown-Crisp index in more than 11 000 health professionals also failed to identify significant associations (Walter et al, 2013). Estimates of hSNP2 from aggregate genome-wide SNP data have been substantially lower than those reported in twin studies: 6% for neuroticism, 1–19% for anxiety-related behaviors as measured by the Anxiety-Related Behaviors Questionnaire, and 17% for phobic anxiety (Trzaskowski et al, 2013; Vinkhuyzen et al, 2012; Walter et al, 2013). These results suggest that common variants influencing these traits exist, but have very modest effects, consistent with the hypothesis that anxiety-related personality traits are highly polygenic and that environmental influences have a substantial role.
Rare and Structural Variation
Anxiety is a prominent feature of Williams syndrome, a neurodevelopmental disorder caused by a copy number deletion of approximately 3 megabases on chromosome 7q. Individuals with Williams syndromes exhibit high rates of GAD and specific phobias (though not SAD), suggesting that one or more genes influencing non-social anxiety may reside in the deleted region (Leyfer et al, 2009). Similarly high rates of anxiety disorders (along with psychotic and mood disorders) are seen in individuals with 22q11.2 deletion syndrome (also known as DiGeorge or velocardiofacial syndrome) (Fung et al, 2010; Schneider et al, 2014; Zarchi et al, 2014). Few studies have examined the role of CNVs in more common, non-syndromic anxiety disorders, however. A study of PD in a Japanese sample (535 cases and 1520 controls) found no association of rare CNVs, but a statistical excess of common duplications in the 16p11.2 region (Kawamura et al, 2011).
Several neurodevelopmental genetic syndromes due to rare mutations have been reported to include prominent anxiety symptoms including Rett syndrome (due to mutations in MECP2) (Cianfaglione et al, 2015) and Fragile X syndrome (due to triplet repeat expansion of FMR1) in which social avoidance is particularly common (Wadell et al, 2013). Carriers of the FMR1 premutation also have elevated rates of social and other anxiety disorder phenotypes (Boyle and Kaufmann, 2010).
As noted earlier, linkage analyses can detect rare mutations of large effect in affected pedigrees. Numerous linkage studies of anxiety disorders have been reported, though results have been inconsistent. A meta-analysis of anxiety disorder linkage studies comprising 162 families found modest, but not genome-wide significant, evidence of linkage at 24 different chromosomal regions (Webb et al, 2012). To date, efforts to identify rare SNVs associated with anxiety disorders by sequencing methods have been limited. Following the association of TMEM132D in GWAS analyses of PD, the gene was resequenced in 300 anxiety disorder cases (85% with a diagnosis of PD) and 300 healthy controls (Quast et al, 2012). An overrepresentation of putatively functional coding variants in controls was reported, suggesting that rare alleles in TMEM132D may have a protective effect. This finding awaits further replication and functional analyses.
Gene-Environment Interaction
Very few G × E studies have been reported for panic, phobic, or generalized anxiety disorders. These have been largely limited to the same candidate genes studied in main effect association analyses, with some nominally significant associations reported but conflicting results and inadequate power overall (Amstadter et al, 2010; Blaya et al, 2010; Choe et al, 2013; Laucht et al, 2009; Reinelt et al, 2014).
Summary
Anxiety disorders are familial and moderately heritable. To date, however, efforts to identify the specific DNA variations contributing to the heritability of these disorders have yet to establish any unequivocal susceptibility loci. Genetic studies of anxiety face a number of obstacles. First, the line between normal and pathologic anxiety is often unclear, and the phenotypic and genetic boundaries between clinical anxiety syndromes are indistinct. Second, the discovery of anxiety-related genetic variants has been hampered by the relatively small sample sizes of existing studies and focus on a limited set of biological candidate genes. Like other psychiatric disorders, anxiety disorders are likely to be highly polygenic, involving thousands of genetic variants of modest effect. Identifying such variants may require genome-wide studies on a large scale. At the same time, genetic research on anxiety can make use of several resources that are less well developed for other psychiatric disorders. In particular, well-established experimental animal models capture key aspects of human anxiety and fear behavior, and neuroimaging studies have made substantial progress in mapping the structural and functional components of anxiety/fear circuitry (Smoller, 2011). These resources create opportunities for focusing genetic studies on relevant biological pathways as well as platforms for evaluating the functional significance of risk loci that may be identified in the future.
CROSS-PHENOTYPE GENETICS OF STRESS-RELATED DISORDERS
Family and twin studies have documented familial coaggregation and co-heritability of multiple psychiatric disorders (Smoller, 2013). Of relevance here, substantial familial and genetic overlap has been found between stress-related disorders and between these disorders and other psychiatric phenotypes (Figure 3). As noted earlier, family and twin studies have shown cross-disorder familial risk and high degrees of genetic correlation among the anxiety disorders, supporting the hypothesis that liability to these disorders involves a general anxiety diathesis (Hettema et al, 2005; Loken et al, 2014; Smoller et al, 2008a; Tambs et al, 2009b). In addition, offspring of parents with anxiety have a nearly three-fold increased odds of having depressive disorders compared with non-psychiatric control offspring (Micco et al, 2009). Conversely, first-degree relatives of individuals with MDD are at increased risk of BPD, ADHD, and anxiety disorders (Faraone and Biederman, 1997; Smoller, 2013; Smoller and Finn, 2003). Twin studies have shown that familial co-aggregation is largely due to heritable factors. For example, in twin samples, genetic variation underlying the risk of MDD and GAD overlaps completely (Kendler et al, 2007; Roy et al, 1995). Significant genetic correlations have also been reported between MDD and, respectively, PTSD (0.77–1.0), BPD (0.65), alcohol dependence (0.40–0.60), ADHD (0.67–0.77), and nicotine dependence (0.21), and neuroticism (0.43) (Cole et al, 2009; Edwards and Kendler, 2012; Kendler et al, 1993a; Kendler and Myers, 2010; Koenen et al, 2008, 2003; McGuffin et al, 2003; Sartor et al, 2012). Anxiety symptoms and ADHD symptoms also show substantial genetic correlations (0.45–0.58) (Michelini et al, 2015). In addition, shared genetic factors have been found between MDD and non-psychiatric disorders including obesity and migraine headache (Afari et al, 2010; Ligthart et al, 2010; Schur et al, 2009).

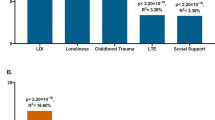
Cross-disorder genetic relationships for stress-related disorders. (a) Genetic correlations derived from twin studies. (Chantarujikapong et al, 2001; Hettema et al, 2006b; Sartor et al, 2012). (b) Common variant overlap derived from GWAS (numbers represent genetic correlations, PRS: polygene risk score) (Cross-Disorder Group of the Psychiatric Genomics Consortium et al, 2013; Nievergelt et al, 2015; Solovieff et al, 2014). (c) Example loci with effects reported across multiple disorders (Cross Disorder Group of the Psychiatric GWAS Consortium, 2013; Green et al, 2013). ADHD, attention deficit/hyperactivity disorder; AG, agoraphobia; BPD, bipolar disorder; GAD, generalized anxiety disorder; MDD, major depressive disorder; PD, panic disorder; PTSD, posttraumatic stress disorder; SAD, social anxiety disorder; SCZ, schizophrenia.
Molecular genetic studies have recently extended the study of cross-disorder effects to the level of DNA variation. The phenomenon of pleiotropy—the association of one genetic locus to more than one phenotype—is widespread in many areas of medicine including autoimmune, metabolic, and cardiovascular diseases (Solovieff et al, 2013). In the realm of neuropsychiatric disorders, pleiotropic effects have been identified for common and rare SNVs and rare CNVs and other structural variants (Smoller, 2013). For example, a GWAS meta-analysis including more than 60 000 cases and controls identified several loci with genome-wide significant effects across five disorders: MDD, SCZ, BPD, ASD, and ADHD (Cross Disorder Group of the Psychiatric GWAS Consortium, 2013). In particular, genes involved in calcium channel signaling were associated with all five disorders. A subsequent analysis of SNP heritability found significant genetic correlations between MDD and SCZ (0.43), BPD (0.47), and ADHD (0.37), respectively, supporting prior evidence from family and twin studies (Cross-Disorder Group of the Psychiatric Genomics Consortium et al, 2013). PRS analyses have also documented common variant overlap between MDD and anxiety symptoms as well as neuroticism (Demirkan et al, 2011; Luciano et al, 2012; Middeldorp et al, 2011) and between PTSD and BPD (Nievergelt et al, 2015; Solovieff et al, 2014). In addition, pathway analyses by the PGC have revealed enrichment of multiple biological pathways across MDD, BPD, and SCZ (Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015). In particular, pathways related histone methylation, synapse-related genes, and genes involved in immune and neurotrophic function exhibited shared enrichment across these disorders.
Cross-disorder effects of rare SNVs and structural variants have been well documented for neurodevelopmental disorders (especially ASD, SCZ, and intellectual disability) (Fromer et al, 2014; Smoller, 2013), but less so for stress-related disorders. Recurrent MDD is one of several psychiatric disorders (including psychotic disorders and ASD) linked to the balanced translocation between chromosomes 1 and 11 (1;11)(q42.1;q14.3) that disrupts DISC1 (Blackwood et al, 2001). As noted earlier, CNVs have shown little evidence of association with stress-related disorders, although the 22q11.2 deletion may be variably expressed in a broad range of psychopathologies including depression and anxiety disorders (Squarcione et al, 2013).
While it is clear that genetic influences on stress-related and other disorders overlap to varying degrees, the biological basis of this overlap is largely unknown. Filling this gap may require investigation at multiple levels including characterization of pleiotropic allelic effects in model systems (cellular and animal models) and human clinical neuroscience (eg, neuroimaging and neurophysiology). Nevertheless, several notable examples of such research are beginning to emerge. The pleiotropic effect of FKBP5 variants on stress-related disorders—resulting from transcriptional and epigenetic effects on HPA axis dysregulation—has already been discussed. Another relevant example involves experimental animal model studies in which human genetic variation can be introduced, creating ‘humanized mice’. The functional vall66met polymorphism of BDNF alters intracellular trafficking activity-dependent BDNF secretion and has been associated with a range of neuropsychiatric phenotypes including mood and anxiety disorders and PTSD, as well as brain structural variation (eg, hippocampal volume), although genetic association studies have had mixed results (see Notaras et al, 2015 for review). Casey et al (2013) have demonstrated that mice engineered to express the human Met allele exhibit a range of phenotypes relevant to stress-related disorders including increased anxiety-related behaviors and resistance to SSRI treatment. They further found that the Met allele is associated with impaired fear extinction in mice and humans and neuroimaging measures of amygdala-prefrontal cortical imbalance in humans.
Summary
Convergent evidence from genetic epidemiologic, molecular genetics, and neuroscience studies has demonstrated overlapping genetic influences on stress-related and other psychiatric disorders. These studies are beginning to highlight shared biological underprinnings that transcend diagnostic boundaries. They are also consistent with the rationale behind the Research Domain Criteria (RDOC) initiative whose goals are to develop ‘new ways of classifying psychopathology based on dimensions of observable behavior and neurobiological measures’ by studying ‘basic dimensions of functioning…across multiple levels of analysis, from genes to neural circuits to behaviors, cutting across disorders as traditionally defined’ (Insel and Cuthbert, 2009). Genetic studies are essential to such a bottom-up approach and may facilitate the goal of an etiology-based nosology.
PROSPECTS FOR CLINICAL TRANSLATION
Although few specific genetic risk variants for stress-related disorders have been established, progress in this area may provide important opportunities for translation to clinical and public health applications. In particular, genetic discoveries may facilitate advances in therapeutics, risk prediction, and diagnostic classification.
Pharmacologic treatments for stress-related disorders are still based on serendipitous discoveries and biological insights that are half a century old. The discovery that a specific gene or set of genes confers risk for illness raises the possibility that drugs targeting that gene or pathway may have therapeutic effects. A principal advantage of unbiased genome-wide studies and pathway analyses is their capacity to identify novel targets for therapeutic development. For several complex disorders, including cardiovascular and autoimmune diseases, GWAS findings include known targets of effective drugs, providing a proof-of-concept that genetic discoveries have therapeutic relevance (Okada et al, 2014; Plenge et al, 2013). Notably, among the strongest associations emerging from SCZ GWAS are variants in DRD2, the target of all approved antipsychotic drugs. Nevertheless, some risk loci present more ‘drug-able’ targets than others. The most promising are risk variants that represent gain-of-function mutations and protective variants that cause loss-of-function effects (Flannick et al, 2014; Kathiresan, 2015). Inhibition of these gene products might be expected to be therapeutic. A prominent example are mutations in PCSK9, an enzyme involved in the degradation of LDL receptors. Loss-of-function variants in PCSK9 are associated with reduced LDL cholesterol concentrations and risk of coronary disease, while rare gain-of-function mutations are associated with increased LDL and premature heart disease (Dadu and Ballantyne, 2014). In 2015, the first PCSK9 inhibitors obtained FDA approval for the treatment of hypercholesterolemia. The translation of genetic findings to therapeutic strategies for stress-related disorders must await robust genetic associations—though as reviewed above, several loci (eg, FKBP5, ADCYAP1R1) and pathways (neurotrophic, opioidergic, and glucorticoid receptor function) have been implicated in stress responses and fear behavior. Several of these findings converge on biological systems that may be responsive to therapeutic modulation (Andero and Ressler, 2012). However, even with validated genetic associations in hand, the road to therapeutic translation may be long and costly, beginning with functional studies (eg, in cellular or experimental animal models) needed for target validation (Smoller, 2014).
Apart from providing new molecular targets, genetic findings may also translate to improved therapeutic options by identifying new phenotypic targets: etiologically-based subgroups (eg, based on early age of onset, comorbidities, and other factors) that may be useful for matching patients to effective treatments (Smoller, 2014). Relatedly, pharmacogenetic approaches aim to identify genetic profiles that can be used to select the most effective and least toxic therapies for a given patient—a central goal of ‘precision medicine’. While validated genetic predictors of treatment response have been identified in other areas of medicine, none has yet been established for stress-related disorders (Smoller, 2014).
A second hope for translation of genetic findings is the prospect for early identification of risk and the development of primary or secondary prevention strategies. However, given the polygenicity and modest penetrance of most genetic risk variants, individual common variants will have insufficient predictive value to be clinically useful for risk prediction or diagnosis (Keyes et al, 2015). Multilocus predictors, including PRS that include thousands of common variants, can capture greater proportions of risk, but their clinical utility is likely to remain limited for common disorders (Chatterjee et al, 2013). However, PRS may be more useful in the epidemiologic context, functioning as a summary measure of genetic vulnerability in studies that may reveal environmental main or interaction effects. Such approaches could identify modifiable exposures that should be prioritized for prevention or intervention strategies, particularly among genetically at-risk individuals. Risk stratification for stress-related disorders may also be more feasible if rare, highly penetrant risk variants are discovered or with the development of integrative methods that can incorporate common and rare variants, other biomarkers and environmental risk factors.
Finally, as noted earlier, genetic findings may inform efforts, along the lines of the RDOC initiative, to move beyond a psychiatric nosology based on symptom-based, categorically-defined syndromes. We have seen that family, twin, and genomic studies have demonstrated substantial genetic correlations among stress-related and other disorders as well as with subsyndromal dimensions of symptoms and personality and neuroimaging phenotypes. Nevertheless, the project of creating a bottom-up, etiologically-based nosology may be a long, iterative process and is unlikely to supplant our syndromic approach for clinical purposes without a much more developed evidence base than is foreseeable in the near term. In addition, this effort will need to address several challenges along the way. For example, while many psychiatric phenotypes are more accurately modeled as quantitative dimensions, continuous with normal variation, adapting this to the needs of clinical decision making may be difficult. Moreover, the complexity and multiplicity of causes and phenotypes will continue to complicate nosologic boundaries.
CONCLUSIONS
Stress-related disorders, like other psychiatric disorders, are familial and heritable. However, efforts to identify the specific genetic variants underlying this heritability have thus far met with limited success. Throughout this review, I have highlighted several recurrent themes that have contributed to this state-of-the-science. These include an over-reliance on candidate gene studies, and the lack of sufficient power to detect the kind of modest main and G × E effects expected for complex phenotypes. In addition, the role of rare and structural variation remains largely unexplored, making it difficult to characterize the full allelic spectrum underlying disorder risk.
These conclusions immediately suggest directions for future research. First, candidate gene studies will be most informative when they have adequate power to detect small effect sizes and are supported by compelling, convergent biological evidence (such as that accumulated for FKBP5). Second, there is a need for the kind of large-scale unbiased genome-wide searches that have already proven successful for other psychiatric disorders including ASD, SCZ, and BPD. As noted earlier, genetic associations for depression, PTSD, and anxiety disorders may require even larger samples to achieve power comparable to that required for SCZ—on the order of tens of thousands of cases. Fortunately, analyses approaching this scale are now becoming feasible for MDD and PTSD through the PGC. Third, as GWAS establishes variants with significant main effects, environmental modifiers of these effects can be examined in G × E studies. In parallel, genome-wide G × E studies may reveal novel components of the stress–diathesis relationship.
Funding and Disclosure
The authors declare no conflict of interest.
References
Abelson JL, Khan S, Liberzon I, Young EA (2007). HPA axis activity in patients with panic disorder: review and synthesis of four studies. Depress Anxiety 24: 66–76.
Afari N, Noonan C, Goldberg J, Roy-Byrne P, Schur E, Golnari G et al (2010). Depression and obesity: do shared genes explain the relationship? Depress Anxiety 27: 799–806.
Almli LM, Mercer KB, Kerley K, Feng H, Bradley B, Conneely KN et al (2013). ADCYAP1R1 genotype associates with post-traumatic stress symptoms in highly traumatized African-American females. Am J Med Genet B Neuropsychiatr Genet 162B: 262–272.
Almli LM, Srivastava A, Fani N, Kerley K, Mercer KB, Feng H et al (2014). Follow-up and extension of a prior genome-wide association study of posttraumatic stress disorder: gene x environment associations and structural magnetic resonance imaging in a highly traumatized African-American civilian population. Biol Psychiatry 76: e3–e4.
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th edn. American Psychiatric Association: Arlington, VA, xliv p, 947 pp.
Amstadter AB, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Kilpatrick DG et al (2009). Variant in RGS2 moderates posttraumatic stress symptoms following potentially traumatic event exposure. J Anxiety Disord 23: 369–373.
Amstadter AB, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Kilpatrick DG et al (2010). NPY moderates the relation between hurricane exposure and generalized anxiety disorder in an epidemiologic sample of hurricane-exposed adults. Depress Anxiety 27: 270–275.
Amstadter AB, Sumner JA, Acierno R, Ruggiero KJ, Koenen KC, Kilpatrick DG et al (2013). Support for association of RORA variant and post traumatic stress symptoms in a population-based study of hurricane exposed adults. Mol Psychiatry 18: 1148–1149.
Andero R, Brothers SP, Jovanovic T, Chen YT, Salah-Uddin H, Cameron M et al (2013). Amygdala-dependent fear is regulated by Oprl1 in mice and humans with PTSD. Sci Transl Med 5: 188ra173.
Andero R, Ressler KJ (2012). Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav 11: 503–512.
Anglin RE, Mazurek MF, Tarnopolsky MA, Rosebush PI (2012). The mitochondrial genome and psychiatric illness. Am J Med Genet B Neuropsychiatr Genet 159B: 749–759.
Appel K, Schwahn C, Mahler J, Schulz A, Spitzer C, Fenske K et al (2011). Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology 36: 1982–1991.
Ashley-Koch AE, Garrett ME, Gibson J, Liu Y, Dennis MF, Kimbrel NA et al (2015). Genome-wide association study of posttraumatic stress disorder in a cohort of Iraq-Afghanistan era veterans. J Affect Disord 184: 225–234.
Avenevoli S, Swendsen J, He JP, Burstein M, Merikangas KR (2015). Major depression in the national comorbidity survey-adolescent supplement: prevalence, correlates, and treatment. J Am Acad Child Adolesc Psychiatry 54: 37–44, e32.
Bastiaansen JA, Servaas MN, Marsman JB, Ormel J, Nolte IM, Riese H et al (2014). Filling the gap: relationship between the serotonin-transporter-linked polymorphic region and amygdala activation. Psychol Sci 25: 2058–2066.
Battaglia M, Ogliari A (2005). Anxiety and panic: from human studies to animal research and back. Neurosci Biobehav Rev 29: 169–179.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299: 1291–1305.
Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B et al (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36: 1319–1325.
Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ (2001). Schizophrenia and affective disorders—cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 69: 428–433.
Bland RC, Newman SC, Orn H (1986). Recurrent and nonrecurrent depression: a family study. Arch Gen Psychiatry 43: 1085–1089.
Blaya C, Salum GA, Moorjani P, Seganfredo AC, Heldt E, Leistner-Segal S et al (2010). Panic disorder and serotonergic genes (SLC6A4, HTR1A and HTR2A): association and interaction with childhood trauma and parenting. Neurosci Lett 485: 11–15.
Boyle L, Kaufmann WE (2010). The behavioral phenotype of FMR1 mutations. Am J Med Genet C Semin Med Genet 154C: 469–476.
Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W et al (2008). Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry 65: 190–200.
Brand H, Pillalamarri V, Collins RL, Eggert S, O'Dushlaine C, Braaten EB et al (2014). Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders. Am J Hum Genet 95: 454–461.
Brooks-Gunn J, Duncan GJ (1997). Effects of poverty on children. Future Child 7: 55–71.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J et alSchizophrenia Working Group of the Psychiatric Genomics Consortium (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295.
Casey BJ, Pattwell SS, Glatt CE, Lee FS (2013). Treating the developing brain: implications from human imaging and mouse genetics. Ann Rev Med 64: 427–439.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301: 386–389.
Champoux M, Bennett A, Shannon C, Higley JD, Lesch KP, Suomi SJ (2002). Serotonin transporter gene polymorphism, differential early rearing, and behavior in rhesus monkey neonates. Mol Psychiatry 7: 1058–1063.
Chang SC, Glymour MM, Walter S, Liang L, Koenen KC, Tchetgen EJ et al (2014). Genome-wide polygenic scoring for a 14-year long-term average depression phenotype. Brain Behav 4: 298–311.
Chang SC, Xie P, Anton RF, De Vivo I, Farrer LA, Kranzler HR et al (2012). No association between ADCYAP1R1 and post-traumatic stress disorder in two independent samples. Mol Psychiatry 17: 239–241.
Chantarujikapong SI, Scherrer JF, Xian H, Eisen SA, Lyons MJ, Goldberg J et al (2001). A twin study of generalized anxiety disorder symptoms, panic disorder symptoms and post-traumatic stress disorder in men. Psychiatry Res 103: 133–145.
Chapman DP, Whitfield CL, Felitti VJ, Dube SR, Edwards VJ, Anda RF (2004). Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord 82: 217–225.
Chatterjee N, Wheeler B, Sampson J, Hartge P, Chanock SJ, Park JH (2013). Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat Genet 45: 400–405, 405e401-403.
Choe AY, Kim B, Lee KS, Lee JE, Lee JY, Choi TK et al (2013). Serotonergic genes (5-HTT and HTR1A) and separation life events: gene-by-environment interaction for panic disorder. Neuropsychobiology 67: 192–200.
Choi KH, Higgs BW, Wendland JR, Song J, McMahon FJ, Webster MJ (2011). Gene expression and genetic variation data implicate PCLO in bipolar disorder. Biol Psychiatry 69: 353–359.
Cianfaglione R, Clarke A, Kerr M, Hastings RP, Oliver C, Moss J et al (2015). A national survey of Rett syndrome: behavioural characteristics. J Neurodev Disord 7: 11.
Clarke H, Flint J, Attwood AS, Munafo MR (2010). Association of the 5- HTTLPR genotype and unipolar depression: a meta-analysis. Psychol Med 40: 1767–1778.
Cole J, Ball HA, Martin NC, Scourfield J, McGuffin P (2009). Genetic overlap between measures of hyperactivity/inattention and mood in children and adolescents. J Am Acad Child Adolesc Psychiatry 48: 1094–1101.
CONVERGE Consortium (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523: 588–591.
Cornelis MC, Nugent NR, Amstadter AB, Koenen KC (2010). Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep 12: 313–326.
Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha XM et al (2007). Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry 62: 1140–1148.
Costello EJ, Egger HL, Angold A (2005). The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am 14: 631–648, vii.
Cross Disorder Group of the Psychiatric GWAS Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381: 1371–1379.
Cross-Disorder Group of the Psychiatric Genomics Consortium Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al (2013). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45: 984–994.
Cukier HN, Dueker ND, Slifer SH, Lee JM, Whitehead PL, Lalanne E et al (2014). Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism 5: 1.
Dadu RT, Ballantyne CM (2014). Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol 11: 563–575.
de Moor MH, Costa PT, Terracciano A, Krueger RF, de Geus EJ, Toshiko T et al (2012). Meta-analysis of genome-wide association studies for personality. Mol Psychiatry 17: 337–349.
Demirkan A, Penninx BW, Hek K, Wray NR, Amin N, Aulchenko YS et al (2011). Genetic risk profiles for depression and anxiety in adult and elderly cohorts. Mol Psychiatry 16: 773–783.
Dieleman GC, Huizink AC, Tulen JH, Utens EM, Creemers HE, van der Ende J et al (2015). Alterations in HPA-axis and autonomic nervous system functioning in childhood anxiety disorders point to a chronic stress hypothesis. Psychoneuroendocrinology 51: 135–150.
Digangi J, Guffanti G, McLaughlin KA, Koenen KC (2013a). Considering trauma exposure in the context of genetics studies of posttraumatic stress disorder: a systematic review. Biol Mood Anxiety Disord 3: 2.
DiGangi JA, Gomez D, Mendoza L, Jason LA, Keys CB, Koenen KC (2013b). Pretrauma risk factors for posttraumatic stress disorder: a systematic review of the literature. Clin Psychol Rev 33: 728–744.
Domschke K, Dannlowski U (2010). Imaging genetics of anxiety disorders. NeuroImage 53: 822–831.
Donner J, Sipila T, Ripatti S, Kananen L, Chen X, Kendler KS et al (2012). Support for involvement of glutamate decarboxylase 1 and neuropeptide Y in anxiety susceptibility. Am J Med Genet B Neuropsychiatr Genet 159B: 316–327.
Duncan LE, Holmans PA, Lee PH, O'Dushlaine CT, Kirby AW, Smoller JW et al (2014a). Pathway analyses implicate glial cells in schizophrenia. PLoS One 9: e89441.
Duncan LE, Pollastri AR, Smoller JW (2014b). Mind the gap: why many geneticists and psychological scientists have discrepant views about gene-environment interaction (GxE) research. Am Psychol 69: 249–268.
Dunn EC, Brown RC, Dai Y, Rosand J, Nugent NR, Amstadter AB et al (2015). Genetic determinants of depression: recent findings and future directions. Harv Rev Psychiatry 23: 1–18.
Dunn EC, Uddin M, Subramanian SV, Smoller JW, Galea S, Koenen KC (2011). Research review: gene-environment interaction research in youth depression—a systematic review with recommendations for future research. J Child Psychol Psychiatry 52: 1223–1238.
Ebert DH, Finn CT, Stoler JM, Smoller JW (2010) Genetics and psychiatry. In: Stern TA, Fricchione GL, Cassem NH, Jellinek MS, Rosenbaum JF (eds). Massacusetts General Hospital Handbook of General Hospital Psychiatry, 6th edn. Elsevier, Inc.: Philadelphia, PA, pp 409–423.
Edwards AC, Kendler KS (2012). A twin study of depression and nicotine dependence: shared liability or causal relationship? J Affect Disord 142: 90–97.
Enoch MA, White KV, Waheed J, Goldman D (2008). Neurophysiological and genetic distinctions between pure and comorbid anxiety disorders. Depress Anxiety 25: 383–392.
Erhardt A, Akula N, Schumacher J, Czamara D, Karbalai N, Muller-Myhsok B et al (2012). Replication and meta-analysis of TMEM132D gene variants in panic disorder. Transl Psychiatry 2: e156.
Erhardt A, Czibere L, Roeske D, Lucae S, Unschuld PG, Ripke S et al (2011). TMEM132D, a new candidate for anxiety phenotypes: evidence from human and mouse studies. Mol Psychiatry 16: 647–663.
Fani N, Gutman D, Tone EB, Almli L, Mercer KB, Davis J et al (2013). FKBP5 and attention bias for threat: associations with hippocampal function and shape. JAMA Psychiatry 70: 392–400.
Faraone SV, Biederman J (1997). Do attention deficit hyperactivity disorder and major depression share familial risk factors? J Nerv Ment Dis 185: 533–541.
Fergusson DM, Horwood LJ, Miller AL, Kennedy MA (2011). Life stress, 5-HTTLPR and mental disorder: findings from a 30-year longitudinal study. Br J Psychiatry 198: 129–135.
Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ et al (2013). Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med 10: e1001547.
Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP et al (2014). Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46: 357–363.
Flaquer A, Baumbach C, Ladwig KH, Kriebel J, Waldenberger M, Grallert H et al (2015). Mitochondrial genetic variants identified to be associated with posttraumatic stress disorder. Transl Psychiatry 5: e524.
Flint J, Kendler KS (2014). The genetics of major depression. Neuron 81: 484–503.
Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P et al (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506: 179–184.
Fung WL, McEvilly R, Fong J, Silversides C, Chow E, Bassett A (2010). Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. Am J Psychiatry 167: 998.
Gao J, Pan Z, Jiao Z, Li F, Zhao G, Wei Q et al (2012). TPH2 gene polymorphisms and major depression—a meta-analysis. PLoS One 7: e36721.
Gatt JM, Burton KL, Williams LM, Schofield PR (2015). Specific and common genes implicated across major mental disorders: a review of meta-analysis studies. J Psychiatr Res 60: 1–13.
Ge T, Nichols TE, Lee PH, Holmes AJ, Roffman JL, Buckner RL et al (2015). Massively expedited genome-wide heritability analysis (MEGHA). Proc Natl Acad Sci USA 112: 2479–2484.
Geschwind DH, State MW (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol pii: S1474-4422(15)00044-7.
Gibb BE, Chelminski I, Zimmerman M (2007). Childhood emotional, physical, and sexual abuse, and diagnoses of depressive and anxiety disorders in adult psychiatric outpatients. Depress Anxiety 24: 256–263.
Gilman SE, Kawachi I, Fitzmaurice GM, Buka SL (2003). Family disruption in childhood and risk of adult depression. Am J Psychiatry 160: 939–946.
Gilman SE, Ni MY, Dunn EC, Breslau J, McLaughlin KA, Smoller JW et al (2015). Contributions of the social environment to first-onset and recurrent mania. Mol Psychiatry 20: 329–336.
Glessner JT, Wang K, Sleiman PM, Zhang H, Kim CE, Flory JH et al (2010). Duplication of the SLIT3 locus on 5q35.1 predisposes to major depressive disorder. PLoS One 5: e15463.
Green EK, Grozeva D, Forty L, Gordon-Smith K, Russell E, Farmer A et al (2013). Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol Psychiatry 18: 614–617.
Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S et al (2010). The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 15: 1016–1022.
Green EK, Rees E, Walters JT, Smith KG, Forty L, Grozeva D et al (2015). Copy number variation in bipolar disorder. Mol Psychiatry (doi:10.1038/mp.2014.174; e-pub ahead of print).
Gressier F, Calati R, Balestri M, Marsano A, Alberti S, Antypa N et al (2013). The 5-HTTLPR polymorphism and posttraumatic stress disorder: a meta-analysis. J Trauma Stress 26: 645–653.
Guffanti G, Ashley-Koch AE, Roberts AL, Garrett ME, Solovieff N, Ratanatharathorn A et al (2014). No association between RORA polymorphisms and PTSD in two independent samples. Mol Psychiatry 19: 1056–1057.
Guffanti G, Galea S, Yan L, Roberts AL, Solovieff N, Aiello AE et al (2013). Genome-wide association study implicates a novel RNA gene, the lincRNA AC068718.1, as a risk factor for post-traumatic stress disorder in women. Psychoneuroendocrinology 38: 3029–3038.
Haaker J, Lonsdorf TB, Raczka KA, Mechias ML, Gartmann N, Kalisch R (2014). Higher anxiety and larger amygdala volumes in carriers of a TMEM132D risk variant for panic disorder. Transl Psychiatry 4: e357.
Hammen C (2005). Stress and depression. Annu Rev Clin Psychol 1: 293–319.
Heim C, Bradley B, Mletzko TC, Deveau TC, Musselman DL, Nemeroff CB et al (2009). Effect of childhood trauma on adult depression and neuroendocrine function: sex-specific moderation by CRH receptor 1 gene. Front Behav Neurosci 3: 41.
Hek K, Demirkan A, Lahti J, Terracciano A, Teumer A, Cornelis MC et al (2013). A genome-wide association study of depressive symptoms. Biol Psychiatry 73: 667–678.
Hek K, Mulder CL, Luijendijk HJ, van Duijn CM, Hofman A, Uitterlinden AG et al (2010). The PCLO gene and depressive disorders: replication in a population-based study. Hum Mol Genet 19: 731–734.
Hettema JM, An SS, Neale MC, Bukszar J, van den Oord EJ, Kendler KS et al (2006a). Association between glutamic acid decarboxylase genes and anxiety disorders, major depression, and neuroticism. Mol Psychiatry 11: 752–762.
Hettema JM, Chen X, Sun C, Brown TA (2015). Direct, indirect and pleiotropic effects of candidate genes on internalizing disorder psychopathology. Psychol Med 1–10.
Hettema JM, Neale MC, Kendler KS (2001). A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 158: 1568–1578.
Hettema JM, Neale MC, Myers JM, Prescott CA, Kendler KS (2006b). A population-based twin study of the relationship between neuroticism and internalizing disorders. Am J Psychiatry 163: 857–864.
Hettema JM, Prescott CA, Myers JM, Neale MC, Kendler KS (2005). The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch Gen Psychiatry 62: 182–189.
Hettema JM, Webb BT, Guo AY, Zhao Z, Maher BS, Chen X et al (2011). Prioritization and association analysis of murine-derived candidate genes in anxiety-spectrum disorders. Biol Psychiatry 70: 888–896.
Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N et al (2015). Common genetic variants influence human subcortical brain structures. Nature 520: 224–229.
Hohoff C, Weber H, Richter J, Domschke K, Zwanzger PM, Ohrmann P et al (2015). RGS2 genetic variation: association analysis with panic disorder and dimensional as well as intermediate phenotypes of anxiety. Am J Med Genet B Neuropsychiatr Genet 168: 211–222.
Holbrook TL, Galarneau MR, Dye JL, Quinn K, Dougherty AL (2010). Morphine use after combat injury in Iraq and post-traumatic stress disorder. N Engl J Med 362: 110–117.
Holmes A, Lit Q, Murphy DL, Gold E, Crawley JN (2003). Abnormal anxiety-related behavior in serotonin transporter null mutant mice: the influence of genetic background. Genes Brain Behav 2: 365–380.
Holz NE, Buchmann AF, Boecker R, Blomeyer D, Baumeister S, Wolf I et al (2015). Role of FKBP5 in emotion processing: results on amygdala activity, connectivity and volume. Brain Struct Funct 220: 1355–1368.
Howe AS, Buttenschon HN, Bani-Fatemi A, Maron E, Otowa T, Erhardt A et al (2015). Candidate genes in panic disorder: meta-analyses of 23 common variants in major anxiogenic pathways. Mol Psychiatry (doi:10.1038/mp.2015.138; e-pub ahead of print).
Insel TR (2014). Brain somatic mutations: the dark matter of psychiatric genetics? Mol Psychiatry 19: 156–158.
Insel TR, Cuthbert BN (2009). Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry 66: 988–989.
Ishitobi Y, Nakayama S, Yamaguchi K, Kanehisa M, Higuma H, Maruyama Y et al (2012). Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a Japanese population. Am J Med Genet B Neuropsychiatr Genet 159B: 429–436.
Jang KL, Taylor S, Stein MB, Yamagata S (2007). Trauma exposure and stress response: exploration of mechanisms of cause and effect. Twin Res Hum Genet 10: 564–572.
Kao CF, Jia P, Zhao Z, Kuo PH (2012). Enriched pathways for major depressive disorder identified from a genome-wide association study. Int J Neuropsychopharmacol 15: 1401–1411.
Karg K, Burmeister M, Shedden K, Sen S (2011). The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 68: 444–454.
Kathiresan S (2015). Developing medicines that mimic the natural successes of the human genome: lessons from NPC1L1, HMGCR, PCSK9, APOC3, and CETP. J Am Coll Cardiol 65: 1562–1566.
Kato T (2015). Whole genome/exome sequencing in mood and psychotic disorders. Psychiatry Clin Neurosci 69: 65–76.
Kawamura Y, Otowa T, Koike A, Sugaya N, Yoshida E, Yasuda S et al (2011). A genome-wide CNV association study on panic disorder in a Japanese population. J Hum Genet 56: 852–856.
Kendler KS, Aggen SH, Neale MC (2013). Evidence for multiple genetic factors underlying DSM-IV criteria for major depression. JAMA Psychiatry 70: 599–607.
Kendler KS, Gardner CO, Gatz M, Pedersen NL (2007). The sources of co-morbidity between major depression and generalized anxiety disorder in a Swedish national twin sample. Psychol Med 37: 453–462.
Kendler KS, Gardner CO, Prescott CA (1999a). Clinical characteristics of major depression that predict risk of depression in relatives. Arch Gen Psychiatry 56: 322–327; erratum 2000;2057:2094-2095.
Kendler KS, Gatz M, Gardner CO, Pedersen NL (2005). Age at onset and familial risk for major depression in a Swedish national twin sample. Psychol Med 35: 1573–1579.
Kendler KS, Gatz M, Gardner CO, Pedersen NL (2006). A Swedish national twin study of lifetime major depression. Am J Psychiatry 163: 109–114.
Kendler KS, Heath AC, Neale MC, Kessler RC, Eaves LJ (1993a). Alcoholism and major depression in women. A twin study of the causes of comorbidity. Arch Gen Psychiatry 50: 690–698.
Kendler KS, Karkowski LM, Prescott CA (1999b). Fears and phobias: reliability and heritability. Psychol Med 29: 539–553.
Kendler KS, Myers J (2010). The genetic and environmental relationship between major depression and the five-factor model of personality. Psychol Med 40: 801–806.
Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ (1993b). A longitudinal twin study of personality and major depression in women. Arch Gen Psychiatry 50: 853–862.
Kessler RC (1997). The effects of stressful life events on depression. Annu Rev Psychol 48: 191–214.
Kessler RC, Wang PS (2008). The descriptive epidemiology of commonly occurring mental disorders in the United States. Annu Rev Public Health 29: 115–129.
Keyes KM, Smith GD, Koenen KC, Galea S (2015). The mathematical limits of genetic prediction for complex chronic disease. J Epidemiol Commun Health 69: 574–579.
Kishi T, Yoshimura R, Fukuo Y, Okochi T, Matsunaga S, Umene-Nakano W et al (2013). The serotonin 1A receptor gene confer susceptibility to mood disorders: results from an extended meta-analysis of patients with major depression and bipolar disorder. Eur Arch Psychiatry Clin Neurosci 263: 105–118.
Klein DN, Lewinsohn PM, Rohde P, Seeley JR, Durbin CE (2002). Clinical features of major depressive disorder in adolescents and their relatives: impact on familial aggregation, implications for phenotype definition, and specificity of transmission. J Abnorm Psychol 111: 98–106.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Koenen KC, Amstadter AB, Ruggiero KJ, Acierno R, Galea S, Kilpatrick DG et al (2009). RGS2 and generalized anxiety disorder in an epidemiologic sample of hurricane-exposed adults. Depress Anxiety 26: 309–315.
Koenen KC, Fu QJ, Ertel K, Lyons MJ, Eisen SA, True WR et al (2008). Common genetic liability to major depression and posttraumatic stress disorder in men. J Affect Disord 105: 109–115.
Koenen KC, Lyons MJ, Goldberg J, Simpson J, Williams WM, Toomey R et al (2003). A high risk twin study of combat-related PTSD comorbidity. Twin Res 6: 218–226.
Koenen KC, Moffitt TE, Poulton R, Martin J, Caspi A (2007). Early childhood factors associated with the development of post-traumatic stress disorder: results from a longitudinal birth cohort. Psychol Med 37: 181–192.
Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K et al (2011). The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron 70: 252–265.
Kuehner C, Huffziger S, Witt SH, Rietschel M (2011). PCLO rs2522833 impacts HPA system activity in healthy young adults. Transl Psychiatry 1: e10.
Laucht M, Treutlein J, Blomeyer D, Buchmann AF, Schmid B, Becker K et al (2009). Interaction between the 5-HTTLPR serotonin transporter polymorphism and environmental adversity for mood and anxiety psychopathology: evidence from a high-risk community sample of young adults. Int J Neuropsychopharmacol 12: 737–747.
Laucht M, Treutlein J, Blomeyer D, Buchmann AF, Schmidt MH, Esser G et al (2013). Interactive effects of corticotropin-releasing hormone receptor 1 gene and childhood adversity on depressive symptoms in young adults: findings from a longitudinal study. Eur Neuropsychopharmacol 23: 358–367.
Lee PH, Perlis RH, Jung JY, Byrne EM, Rueckert E, Siburian R et al (2012). Multi-locus genome-wide association analysis supports the role of glutamatergic synaptic transmission in the etiology of major depressive disorder. Transl Psychiatry 2: e184.
Leen-Feldner EW, Feldner MT, Knapp A, Bunaciu L, Blumenthal H, Amstadter AB (2013). Offspring psychological and biological correlates of parental posttraumatic stress: review of the literature and research agenda. Clin Psychol Rev 33: 1106–1133.
Levinson DF, Mostafavi S, Milaneschi Y, Rivera M, Ripke S, Wray NR et al (2014). Genetic studies of major depressive disorder: why are there no genome-wide association study findings and what can we do about it? Biol Psychiatry 76: 510–512.
Leyfer O, Woodruff-Borden J, Mervis CB (2009). Anxiety disorders in children with williams syndrome, their mothers, and their siblings: implications for the etiology of anxiety disorders. J Neurodev Disord 1: 4–14.
Leygraf A, Hohoff C, Freitag C, Willis-Owen SA, Krakowitzky P, Fritze J et al (2006). Rgs 2 gene polymorphisms as modulators of anxiety in humans? J Neural Transm 113: 1921–1925.
Liberzon I, King AP, Ressler KJ, Almli LM, Zhang P, Ma ST et al (2014). Interaction of the ADRB2 gene polymorphism with childhood trauma in predicting adult symptoms of posttraumatic stress disorder. JAMA Psychiatry 71: 1174–1182.
Ligthart L, Nyholt DR, Penninx BW, Boomsma DI (2010). The shared genetics of migraine and anxious depression. Headache 50: 1549–1560.
Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, Liberzon I et al (2015). The Psychiatric Genomics Consortium Posttraumatic Stress Disorder Workgroup: posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology 40: 2287–2297.
Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, Reardon AF et al (2013). A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol Psychiatry 18: 937–942.
Loken EK, Hettema JM, Aggen SH, Kendler KS (2014). The structure of genetic and environmental risk factors for fears and phobias. Psychol Med 44: 2375–2384.
López-León S, Janssens AC, González-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA et al (2007). Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry 13: 772–785.
Lubke GH, Hottenga JJ, Walters R, Laurin C, de Geus EJ, Willemsen G et al (2012). Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biol Psychiatry 72: 707–709.
Luciano M, Huffman JE, Arias-Vasquez A, Vinkhuyzen AA, Middeldorp CM, Giegling I et al (2012). Genome-wide association uncovers shared genetic effects among personality traits and mood states. Am J Med Genet B Neuropsychiatr Genet 159B: 684–695.
Lyons M, Eisen S, Goldberg J, True W, Lin N, Meyer J et al (1998). A registry-based twin study of depression in men. Arch Gen Psychiatry 55: 468–472.
Mahon PB, Zandi PP, Potash JB, Nestadt G, Wand GS (2013). Genetic association of FKBP5 and CRHR1 with cortisol response to acute psychosocial stress in healthy adults. Psychopharmacology (Berl) 227: 231–241.
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium Ripke S Wray NR Lewis CM Hamilton SP Weissman MM et al (2013). A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 18: 497–511.
Malhotra D, Sebat J (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148: 1223–1241.
Mandelli L, Serretti A (2013). Gene environment interaction studies in depression and suicidal behavior: an update. Neurosci Biobehav Rev 37: 2375–2397.
Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M et al (2012). Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13: 293–307.
Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP (2007). Mitochondria as key components of the stress response. Trends Endocrinol Metab 18: 190–198.
Matheson SL, Shepherd AM, Pinchbeck RM, Laurens KR, Carr VJ (2013). Childhood adversity in schizophrenia: a systematic meta-analysis. Psychol Med 43: 225–238.
McGrath LM, Weill S, Robinson EB, Macrae R, Smoller JW (2012). Bringing a developmental perspective to anxiety genetics. Dev Psychopathol 24: 1179–1193.
McGuffin P, Katz R, Watkins S, Rutherford J (1996). A hospital-based twin register of the heritability of DSM-IV unipolar depression. Arch Gen Psychiatry 53: 129–136.
McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A (2003). The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 60: 497–502.
McLeod JD, Shanahan MJ (1996). Trajectories of poverty and children's mental health. J Health Social Behav 37: 207–220.
Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J et al (2011). Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch Gen Psychiatry 68: 901–910.
Merikangas KR, Cui L, Heaton L, Nakamura E, Roca C, Ding J et al (2014). Independence of familial transmission of mania and depression: results of the NIMH family study of affective spectrum disorders. Mol Psychiatry 19: 214–219.
Micco JA, Henin A, Mick E, Kim S, Hopkins CA, Biederman J et al (2009). Anxiety and depressive disorders in offspring at high risk for anxiety: a meta-analysis. J Anxiety Disord 23: 1158–1164.
Michelini G, Eley TC, Gregory AM, McAdams TA (2015). Aetiological overlap between anxiety and attention deficit hyperactivity symptom dimensions in adolescence. J Child Psychol Psychiatry 56: 423–431.
Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO et al (2015). Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry 172: 353–362.
Middeldorp CM, de Moor MH, McGrath LM, Gordon SD, Blackwood DH, Costa PT et al (2011). The genetic association between personality and major depression or bipolar disorder. A polygenic score analysis using genome-wide association data. Transl Psychiatry 1: e50.
Miller MW, Wolf EJ, Logue MW, Baldwin CT (2013). The retinoid-related orphan receptor alpha (RORA) gene and fear-related psychopathology. J Affect Disord 151: 702–708.
Minelli A, Scassellati C, Cloninger CR, Tessari E, Bortolomasi M, Bonvicini C et al (2012). PCLO gene: its role in vulnerability to major depressive disorder. J Affect Disord 139: 250–255.
Mouri K, Hishimoto A, Fukutake M, Nishiguchi N, Shirikawa O, Maeda K (2009). Association study of RGS2 gene polymorphisms with panic disorder in Japanese. Kobe J Med Sci 55: e116–e121.
Moutsianas L, Agarwala V, Fuchsberger C, Flannick J, Rivas MA, Gaulton KJ et al (2015). The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet 11: e1005165.
Munafo MR, Durrant C, Lewis G, Flint J (2009). Gene X environment interactions at the serotonin transporter locus. Biol Psychiatry 65: 211–219.
Musliner KL, Seifuddin F, Judy JA, Pirooznia M, Goes FS, Zandi PP (2014). Polygenic risk, stressful life events and depressive symptoms in older adults: a polygenic score analysis. Psychol Med 45: 1709–1720.
Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium (2015). Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 18: 199–209.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW et al (2015). Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: a genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology 51: 459–471.
Nivard MG, Mbarek H, Hottenga JJ, Smit JH, Jansen R, Penninx BW et al (2014). Further confirmation of the association between anxiety and CTNND2: replication in humans. Genes Brain Behav 13: 195–201.
Notaras M, Hill R, van den Buuse M (2015). The BDNF gene Val66Met polymorphism as a modifier of psychiatric disorder susceptibility: progress and controversy. Mol Psychiatry. 20: 916–930.
Nurnberger JI Jr, Koller DL, Jung J, Edenberg HJ, Foroud T, Guella I et al (2014). Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry 71: 657–664.
O'Dushlaine C, Ripke S, Ruderfer DM, Hamilton SP, Fava M, Iosifescu DV et al (2014). Rare copy number variation in treatment-resistant major depressive disorder. Biol Psychiatry 76: 536–541.
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K et al (2014). Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506: 376–381.
Otowa T, Shimada T, Kawamura Y, Sugaya N, Yoshida E, Inoue K et al (2011). Association of RGS2 variants with panic disorder in a Japanese population. Am J Med Genet B Neuropsychiatr Genet 156B: 430–434.
Otowa T, Tanii H, Sugaya N, Yoshida E, Inoue K, Yasuda S et al (2010). Replication of a genome-wide association study of panic disorder in a Japanese population. J Hum Genet 55: 91–96.
Otowa T, Yoshida E, Sugaya N, Yasuda S, Nishimura Y, Inoue K et al (2009). Genome-wide association study of panic disorder in the Japanese population. J Hum Genet 54: 122–126.
Ozer EJ, Best SR, Lipsey TL, Weiss DS (2003). Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 129: 52–73.
Pagliaccio D, Luby JL, Bogdan R, Agrawal A, Gaffrey MS, Belden AC et al (2014). Stress-system genes and life stress predict cortisol levels and amygdala and hippocampal volumes in children. Neuropsychopharmacology 39: 1245–1253.
Pedroso I, Lourdusamy A, Rietschel M, Nothen MM, Cichon S, McGuffin P et al (2012). Common genetic variants and gene-expression changes associated with bipolar disorder are over-represented in brain signaling pathway genes. Biol Psychiatry 72: 311–317.
Perlis RH, Ruderfer D, Hamilton SP, Ernst C (2012). Copy number variation in subjects with major depressive disorder who attempted suicide. PLoS One 7: e46315.
Perlis RH, Smoller JW, Mysore J, Sun M, Gillis T, Purcell S et al (2010). Prevalence of incompletely penetrant Huntington's disease alleles among individuals with major depressive disorder. Am J Psychiatry 167: 574–579.
Peyrot WJ, Milaneschi Y, Abdellaoui A, Sullivan PF, Hottenga JJ, Boomsma DI et al (2014). Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry 205: 113–119.
Plenge RM, Scolnick EM, Altshuler D (2013). Validating therapeutic targets through human genetics. Nat Rev Drug Discov 12: 581–594.
Pohlack ST, Nees F, Ruttorf M, Cacciaglia R, Winkelmann T, Schad LR et al (2015). Neural mechanism of a sex-specific risk variant for posttraumatic stress disorder in the type I receptor of the pituitary adenylate cyclase activating polypeptide. Biol Psychiatry pii: S0006-3223(15)00002-5.
Power RA, Wingenbach T, Cohen-Woods S, Uher R, Ng MY, Butler AW et al (2013). Estimating the heritability of reporting stressful life events captured by common genetic variants. Psychol Med 43: 1965–1971.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506: 185–190.
Quast C, Altmann A, Weber P, Arloth J, Bader D, Heck A et al (2012). Rare variants in TMEM132D in a case-control sample for panic disorder. Am J Med Genet B Neuropsychiatr Genet 159B: 896–907.
Reinelt E, Aldinger M, Stopsack M, Schwahn C, John U, Baumeister SE et al (2014). High social support buffers the effects of 5-HTTLPR genotypes within social anxiety disorder. Eur Arch Psychiatry Clin Neuroscience 264: 433–439.
Repetti RL, Taylor SE, Seeman TE (2002). Risky families: family social environments and the mental and physical health of offspring. Psychol Bull 128: 330–366.
Ressler KJ, Bradley B, Mercer KB, Deveau TC, Smith AK, Gillespie CF et al (2010). Polymorphisms in CRHR1 and the serotonin transporter loci: gene x gene x environment interactions on depressive symptoms. Am J Med Genet B Neuropsychiatr Genet 153B: 812–824.
Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K et al (2011). Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470: 492–497.
Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J et al (2009). Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 301: 2462–2471.
Roberson-Nay R, Eaves LJ, Hettema JM, Kendler KS, Silberg JL (2012). Childhood separation anxiety disorder and adult onset panic attacks share a common genetic diathesis. Depress Anxiety 29: 320–327.
Rogers J, Raveendran M, Fawcett GL, Fox AS, Shelton SE, Oler JA et al (2013). CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol Psychiatry 18: 700–707.
Roth M, Neuner F, Elbert T (2014). Transgenerational consequences of PTSD: risk factors for the mental health of children whose mothers have been exposed to the Rwandan genocide. Int J Mental Health Syst 8: 12.
Roy MA, Neale MC, Pedersen NL, Mathe AA, Kendler KS (1995). A twin study of generalized anxiety disorder and major depression. Psychol Med 25: 1037–1049.
Rucker JJ, Breen G, Pinto D, Pedroso I, Lewis CM, Cohen-Woods S et al (2013). Genome-wide association analysis of copy number variation in recurrent depressive disorder. Mol Psychiatry 18: 183–189.
Rucker JJ, Tansey KE, Rivera M, Pinto D, Cohen-Woods S, Uher R et al (2015). Phenotypic association analyses with copy number variation in recurrent depressive disorder. Biol Psychiatry pii: S0006-3223(15)00150-X.
Sah R, Geracioti TD (2013). Neuropeptide Y and posttraumatic stress disorder. Mol Psychiatry 18: 646–655.
Sartor CE, Grant JD, Lynskey MT, McCutcheon VV, Waldron M, Statham DJ et al (2012). Common heritable contributions to low-risk trauma, high-risk trauma, posttraumatic stress disorder, and major depression. Archives of general psychiatry 69: 293–299.
Savage JE, McMichael O, Gorlin EI, Beadel JR, Teachman B, Vladimirov VI et al (2015). Validation of candidate anxiety disorder genes using a carbon dioxide challenge task. Biol Psychol 109: 61–66.
Scaini S, Belotti R, Ogliari A (2014). Genetic and environmental contributions to social anxiety across different ages: a meta-analytic approach to twin data. J Anxiety Disord 28: 650–656.
Scaini S, Ogliari A, Eley TC, Zavos HM, Battaglia M (2012). Genetic and environmental contributions to separation anxiety: a meta-analytic approach to twin data. Depress Anxiety 29: 754–761.
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427.
Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M et al (2014). Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 171: 627–639.
Schuhmacher A, Lennertz L, Wagner M, Hofels S, Pfeiffer U, Guttenthaler V et al (2013). A variant of the neuronal amino acid transporter SLC6A15 is associated with ACTH and cortisol responses and cognitive performance in unipolar depression. Int J Neuropsychopharmacol 16: 83–90.
Schuhmacher A, Mossner R, Hofels S, Pfeiffer U, Guttenthaler V, Wagner M et al (2011). PCLO rs2522833 modulates HPA system response to antidepressant treatment in major depressive disorder. Int J Neuropsychopharmacol 14: 237–245.
Schur EA, Noonan C, Buchwald D, Goldberg J, Afari N (2009). A twin study of depression and migraine: evidence for a shared genetic vulnerability. Headache 49: 1493–1502.
Service SK, Verweij KJ, Lahti J, Congdon E, Ekelund J, Hintsanen M et al (2012). A genome-wide meta-analysis of association studies of Cloninger's Temperament Scales. Transl Psychiatry 2: e116.
Sharpley CF, Palanisamy SK, Glyde NS, Dillingham PW, Agnew LL (2014). An update on the interaction between the serotonin transporter promoter variant (5-HTTLPR), stress and depression, plus an exploration of non-confirming findings. Behav Brain Res 273: 89–105.
Sheikh HI, Kryski KR, Smith HJ, Hayden EP, Singh SM (2013). Corticotropin-releasing hormone system polymorphisms are associated with children's cortisol reactivity. Neuroscience 229: 1–11.
Sipila T, Kananen L, Greco D, Donner J, Silander K, Terwilliger JD et al (2010). An association analysis of circadian genes in anxiety disorders. Biol Psychiatry 67: 1163–1170.
Smoller J, Cerrato F, Weatherall S (2015) The genetics of anxiety disorders. In: Pine D, Rothbaum B, Ressler K, Muskin A (eds). Anxiety Disorders: Translational Perspectives on Diagnosis and Treatment. Oxford University Press: Oxford, UK, pp 47–61.
Smoller JW (2011). Who's afraid of anxiety genetics? Biol Psychiatry 69: 506–507.
Smoller JW (2013). Disorders and borders: psychiatric genetics and nosology. Am J Med Genet B Neuropsychiatr Genet 162B: 559–578.
Smoller JW (2014). Psychiatric genetics and the future of personalized treatment. Depress Anxiety 31: 893–898.
Smoller JW, Acierno JS Jr, Rosenbaum JF, Biederman J, Pollack MH, Meminger S et al (2001). Targeted genome screen of panic disorder and anxiety disorder proneness using homology to murine QTL regions. Am J Med Genet 105: 195–206.
Smoller JW, Block SR, Young MM (2009). Genetics of anxiety disorders: the complex road from DSM to DNA. Depress Anxiety 26: 965–975.
Smoller JW, Finn CT (2003). Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet 123: 48–58.
Smoller JW, Gallagher PJ, Duncan LE, McGrath LM, Haddad SA, Holmes AJ et al (2014). The human ortholog of acid-sensing ion channel gene ASIC1a is associated with panic disorder and amygdala structure and function. Biol Psychiatry. 76: 902–910.
Smoller JW, Gardner-Schuster E, Covino J (2008a). The genetic basis of panic and phobic anxiety disorders. Am J Med Genet C Semin Med Genet 148C: 118–126.
Smoller JW, Paulus MP, Fagerness JA, Purcell S, Yamaki LH, Hirshfeld-Becker D et al (2008b). Influence of RGS2 on anxiety-related temperament, personality, and brain function. Arch Gen Psychiatry 65: 298–308.
Smoller JW, Perlis RH (2004) Family and genetic studies of depression. In: Alpert JE, Fava M (eds). Handbook of Chronic Depression. Marcel Dekker, Inc.: New York, pp 97–138.
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW (2013). Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 14: 483–495.
Solovieff N, Roberts AL, Ratanatharathorn A, Haloosim M, De Vivo I, King AP et al (2014). Genetic association analysis of 300 genes identifies a risk haplotype in SLC18A2 for post-traumatic stress disorder in two independent samples. Neuropsychopharmacology 39: 1872–1879.
Song GG, Kim JH, Lee YH (2013). Genome-wide pathway analysis in major depressive disorder. J Mol Neurosci 51: 428–436.
Squarcione C, Torti MC, Di Fabio F, Biondi M (2013). 22q11 deletion syndrome: a review of the neuropsychiatric features and their neurobiological basis. Neuropsychiatr Dis Treat 9: 1873–1884.
Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ (2002). Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry 159: 1675–1681.
Stein MB, Keshaviah A, Haddad SA, Van Ameringen M, Simon NM, Pollack MH et al (2014). Influence of RGS2 on sertraline treatment for social anxiety disorder. Neuropsychopharmacology 39: 1340–1346.
Stein MB, Walker JR, Anderson G, Hazen AL, Ross CA, Eldridge G et al (1996). Childhood physical and sexual abuse in patients with anxiety disorders and in a community sample. Am J Psychiatry 153: 275–277.
Stein MB, Yang BZ, Chavira DA, Hitchcock CA, Sung SC, Shipon-Blum E et al (2011). A common genetic variant in the neurexin superfamily member CNTNAP2 is associated with increased risk for selective mutism and social anxiety-related traits. Biol Psychiatry 69: 825–831.
Stevens JS, Almli LM, Fani N, Gutman DA, Bradley B, Norrholm SD et al (2014). PACAP receptor gene polymorphism impacts fear responses in the amygdala and hippocampus. Proc Natl Acad Sci USA 111: 3158–3163.
Sullivan PF (2007). Spurious genetic associations. Biol Psychiatry 61: 1121–1126.
Sullivan PF (2010). The psychiatric GWAS consortium: big science comes to psychiatry. Neuron 68: 182–186.
Sullivan PF, de Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T et al (2009). Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry 14: 359–375.
Sullivan PF, Neale MC, Kendler KS (2000). Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 157: 1552–1562.
Sumner JA, McLaughlin KA, Walsh K, Sheridan MA, Koenen KC (2014). CRHR1 genotype and history of maltreatment predict cortisol reactivity to stress in adolescents. Psychoneuroendocrinology 43: 71–80.
Tambs K, Czajkowsky N, Roysamb E, Neale MC, Reichborn-Kjennerud T, Aggen SH et al (2009a). Structure of genetic and environmental risk factors for dimensional representations of DSM-IV anxiety disorders. Br J Psychiatry 195: 301–307.
Tambs K, Ronning T, Prescott CA, Kendler KS, Reichborn-Kjennerud T, Torgersen S et al (2009b). The Norwegian Institute of Public Health twin study of mental health: examining recruitment and attrition bias. Twin Res Human Genet 12: 158–168.
Taugher RJ, Lu Y, Wang Y, Kreple CJ, Ghobbeh A, Fan R et al (2014). The bed nucleus of the stria terminalis is critical for anxiety-related behavior evoked by CO2 and acidosis. J Neurosci 34: 10247–10255.
True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ et al (1993). A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms [see comments]. Arch Gen Psychiatry 50: 257–264.
Trzaskowski M, Eley TC, Davis OS, Doherty SJ, Hanscombe KB, Meaburn EL et al (2013). First genome-wide association study on anxiety-related behaviours in childhood. PLoS One 8: e58676.
Tsuang MT, Faraone SV (1990) The Genetics of Mood Disorders. The Johns Hopkins University Press: Baltimore.
Tyrka AR, Price LH, Gelernter J, Schepker C, Anderson GM, Carpenter LL (2009). Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol Psychiatry 66: 681–685.
Uddin M, Chang SC, Zhang C, Ressler K, Mercer KB, Galea S et al (2013). Adcyap1r1 genotype, posttraumatic stress disorder, and depression among women exposed to childhood maltreatment. Depress anxiety 30: 251–258.
Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z et al (2014). Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet 46: 742–747.
Uher R, McGuffin P (2008). The moderation by the serotonin transporter gene of environmental adversity in the aetiology of mental illness: review and methodological analysis. Mol Psychiatry 13: 131–146.
Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O et al (2009). Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev 61: 283–357.
Vinkhuyzen AA, Pedersen NL, Yang J, Lee SH, Magnusson PK, Iacono WG et al (2012). Common SNPs explain some of the variation in the personality dimensions of neuroticism and extraversion. Transl Psychiatry 2: e102.
Visscher PM, Hill WG, Wray NR (2008). Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet 9: 255–266.
Wadell PM, Hagerman RJ, Hessl DR (2013). Fragile X syndrome: psychiatric manifestations, assessment and emerging therapies. Curr Psychiatry Rev 9: 53–58.
Walter S, Glymour MM, Koenen K, Liang L, Tchetgen Tchetgen EJ, Cornelis M et al (2013). Performance of polygenic scores for predicting phobic anxiety. PLoS One 8: e80326.
Webb BT, Guo AY, Maher BS, Zhao Z, van den Oord EJ, Kendler KS et al (2012). Meta-analyses of genome-wide linkage scans of anxiety-related phenotypes. Eur J Hum Genet 20: 1078–1084.
Weissman MM, Wickramaratne P, Merikangas KR, Leckman JF, Prusoff BA, Caruso KA et al (1984). Onset of major depression in early adulthood: increased familial loading and specificity. Arch Gen Psychiatry 41: 1136–1143.
Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH Jr, Welsh MJ (2003). Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci 23: 5496–5502.
White MG, Bogdan R, Fisher PM, Munoz KE, Williamson DE, Hariri AR (2012). FKBP5 and emotional neglect interact to predict individual differences in amygdala reactivity. Genes Brain Behav 11: 869–878.
Widom CS, DuMont K, Czaja SJ (2007). A prospective investigation of major depressive disorder and comorbidity in abused and neglected children grown up. Arch Gen Psychiatry 64: 49–56.
Wolf EJ, Mitchell KS, Logue MW, Baldwin CT, Reardon AF, Aiello A et al (2014a). The dopamine D3 receptor gene and posttraumatic stress disorder. J Trauma Stress 27: 379–387.
Wolf EJ, Rasmusson AM, Mitchell KS, Logue MW, Baldwin CT, Miller MW (2014b). A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depress Anxiety 31: 352–360.
Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM (2014). Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 55: 1068–1087.
Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR et al (2012). Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 17: 36–48.
Wu YL, Ding XX, Sun YH, Yang HY, Chen J, Zhao X et al (2013). Association between MTHFR C677T polymorphism and depression: an updated meta-analysis of 26 studies. Prog Neuropsychopharmacol Biol Psychiatry 46: 78–85.
Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA et al (2010). Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology 35: 1684–1692.
Xie P, Kranzler HR, Yang C, Zhao H, Farrer LA, Gelernter J (2013). Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol Psychiatry 74: 656–663.
Yalcin B, Willis-Owen SA, Fullerton J, Meesaq A, Deacon RM, Rawlins JN et al (2004). Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat Genet 36: 1197–1202.
Yang J, Lee SH, Goddard ME, Visscher PM (2011). GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88: 76–82.
Yang J, Wray NR, Visscher PM (2010). Comparing apples and oranges: equating the power of case-control and quantitative trait association studies. Genet Epidemiol 34: 254–257.
Zaitlen N, Pasaniuc B, Sankararaman S, Bhatia G, Zhang J, Gusev A et al (2014). Leveraging population admixture to characterize the heritability of complex traits. Nat Genet 46: 1356–1362.
Zannas AS, Binder EB (2014). Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav 13: 25–37.
Zarchi O, Diamond A, Weinberger R, Abbott D, Carmel M, Frisch A et al (2014). A comparative study of the neuropsychiatric and neurocognitive phenotype in two microdeletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. Eur Psychiatry 29: 203–210.
Zhao X, Sun L, Sun YH, Ren C, Chen J, Wu ZQ et al (2014). Association of HTR2A T102C and A-1438G polymorphisms with susceptibility to major depressive disorder: a meta-analysis. Neurol Sci 35: 1857–1866.
Ziemann AE, Allen JE, Dahdaleh NS, Drebot II, Coryell MW, Wunsch AM et al (2009). The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 139: 1012–1021.
Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, Uhr M et al (2011). Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry 168: 1107–1116.
Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S et al (2014). Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 111: e455–e464.
Acknowledgements
Supported in part by NIMH grants K24MH094614 and R01MH101486 to JWS. Dr Smoller is the Tepper Family MGH Research Scholar. Dr Smoller is an unpaid member of the Scientific Advisory Board of PsyBrain, Inc. For: 2016 Neuropsychopharmacology Reviews: The Next Generation of Progress Volume IX. Impact of Stress on the Brain: Pathology, Treatment and Prevention. Editors: Kerry Ressler, MD, PhD and Jordan Smoller, MD, ScD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Smoller, J. The Genetics of Stress-Related Disorders: PTSD, Depression, and Anxiety Disorders. Neuropsychopharmacol 41, 297–319 (2016). https://doi.org/10.1038/npp.2015.266
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.266
This article is cited by
-
Updating a social cognitive model of PTSD: Implications for cross-group PTSD research
Current Psychology (2024)
-
Volumetric MRI Findings in Mild Traumatic Brain Injury (mTBI) and Neuropsychological Outcome
Neuropsychology Review (2023)
-
The role of epigenetics in anxiety disorders
Molecular Biology Reports (2023)
-
Genome-wide multi-trait analysis of irritable bowel syndrome and related mental conditions identifies 38 new independent variants
Journal of Translational Medicine (2023)
