
Abstract
ΔFosB, a FosB gene product, is induced in the prefrontal cortex (PFC) by repeated exposure to several stimuli including antipsychotic drugs such as haloperidol. However, the functional consequences of increased ΔFosB expression following antipsychotic treatment have not been explored. Here, we assessed whether ΔFosB induction by haloperidol mediates the positive or negative consequences or clinical-related actions of antipsychotic treatment. We show that individuals with schizophrenia who were medicated with antipsychotic drugs at their time of death display increased ΔFosB levels in the PFC, an effect that is replicated in rats treated chronically with haloperidol. In contrast, individuals with schizophrenia who were medication-free did not exhibit this effect. Viral-mediated overexpression of ΔFosB in the PFC of rodents induced cognitive deficits as measured by inhibitory avoidance, increased startle responses in prepulse inhibition tasks, and increased MK-801-induced anxiety-like behaviors. Together, these results suggest that ΔFosB induction in the PFC by antipsychotic treatment contributes to the deleterious effects of these drugs and not to their therapeutic actions.
Similar content being viewed by others

INTRODUCTION
Schizophrenia is a debilitating disorder that affects ∼1% of the population (Sawa and Snyder, 2002). First and second generation antipsychotic drugs are the cornerstone of treatment for schizophrenia. These drugs, antagonists of dopamine D2 receptors along with variable actions at additional receptors, are generally effective against psychotic (positive) symptoms, but are largely ineffective at treating negative and cognitive symptoms (Lieberman et al, 2005; Marder et al, 2011). In fact, chronic use of antipsychotic drugs can induce or exacerbate a multitude of symptoms such as affective and cognitive decline, anhedonia, and apathy (Lader, 1994; Park et al, 2012).
It is generally thought that the clinical actions of antipsychotic drugs, which may take days or weeks to achieve maximal efficacy, are mediated in part through changes in gene expression that govern long-term neuroadaptations (Beerpoot et al, 1996; Hyman and Nestler, 1996). ΔFosB is a member of the Fos family of transcription factors, but is unique in its stable accumulation in the brain in response to a range of chronic stimuli (Nestler et al, 1999; Nestler, 2008). Both first and second generation antipsychotic drugs have been shown to increase ΔFosB expression in several limbic brain regions including the prefrontal cortex (PFC) (Atkins et al, 1999; Hiroi and Graybiel, 1996; Kontkanen et al, 2002; Perrotti et al, 2005), which is highly implicated in the cognitive deficits associated with schizophrenia.
However, despite our knowledge of ΔFosB induction by antipsychotic drugs, it remains unclear whether the accumulation of ΔFosB contributes to the clinical efficacy of these medications or instead promotes some of their negative side effects. The objective of the present study was to address this question.
MATERIALS AND METHODS
Animals
Both rats and mice were used in these studies. Male 8- to 10-week-old male C57BL/6J mice were housed four to five per cage. Male Sprague Dawley rats weighing 275–300 g were pair-housed. Colony rooms were on a 12 h light/dark cycle (lights on from 0700 to 1900 hours) at constant temperature (23 °C) with ad libitum access to water and food. All animal protocols were approved by the animal care and use committee at the Icahn School of Medicine at Mount Sinai and the State University of New York at Buffalo.
Human Tissue Samples
Human specimens were obtained from the Dallas Brain Collection (Stan et al, 2006). With next of kin permission, tissue samples were collected from cases examined by the Dallas County Medical Examiner’s Office and the Transplant Service Center at The University of Texas Southwestern Medical Center at Dallas. Blood toxicology screens were conducted in each case, and subjects with a recent or past history of drug abuse, neurological disorders, or head injury were excluded. None of the cases had sustained agonal factors at the time of death (see Table 1). Clinical records and collateral information from telephone interviews with a primary caregiver were obtained for each case. Two psychiatrists performed extensive reviews of the clinical records and made independent diagnoses followed by a consensus diagnosis using Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, criteria. Specimens of human PFC corresponding to Brodmann area 9 were obtained, and protein was extracted as described below.
Drug Treatments
Rats were injected s.c. daily for 7 days with haloperidol (Sigma) at a dose of 0.1 mg/kg and volume of 1 ml/kg. The haloperidol was dissolved in a minimal amount of acetic acid and then diluted in 0.9% saline and raised to a pH of 5.8–6.0. Control rats received equivalent injections of vehicle. Twenty-four hours following the last injection of drug, animals were killed by decapitation and 14 gauge punch dissections of medial PFC targeting the prelimbic cortex (Paxinos and Watson, 2009) were obtained from 1 mm coronal slices and rapidly frozen.
Western Blot Quantification of ΔFosB
Frozen tissue punches from rat or pulverized samples dissected from human were homogenized in 30 μl of homogenization buffer containing 320 mM sucrose, 5 mM HEPES buffer, 1% SDS, phosphatase inhibitor cocktails I and II (Sigma), and protease inhibitors (Roche) using an ultrasonic processor (Cole Parmer). Protein concentrations were determined using a DC protein assay (Bio-Rad), and 30 μg of protein were loaded onto 18% gradient Tris-HCl polyacrylamide gels for electrophoresis fractionation (Bio-Rad). Proteins were transferred to nitrocellulose membranes, blocked with Odyssey blocking buffer (Li-Cor), and incubated overnight at 4 °C with anti-rabbit ΔFosB primary antibody (1 : 500; Cell Signaling) in Odyssey blocking buffer. After thorough washing with TBS plus 0.1% Tween-20, membranes were incubated with IRDye secondary antibodies (1 : 5000 Li-Cor) dissolved in Odyssey blocking buffer for 1 h at room temperature. For analysis, the blots were imaged with the Odyssey Infrared Imaging system (Li-Cor) and quantified by densitometry using NIH ImageJ. The amount of protein blotted onto each lane was normalized to levels of actin or tubulin, which were no different between experimental and control groups in rat and human samples.
Chromatin Immunoprecipitation
Freshly dissected brain punches were prepared for chromatin immunoprecipitation (ChIP) as described previously (Maze et al, 2011; Sun et al, 2012). For each ChIP, medial PFC punches were pooled from two rats. Tissue was lightly fixed with formaldehyde to crosslink DNA with associated proteins, and the material was further sheared and immunoprecipitated using sheep anti-mouse magnetic beads (Invitrogen) conjugated to an antibody that specifically recognizes G9a (Abcam). G9a-immunoprecipitated DNA was subjected to qPCR analysis.
Viral Gene Transfer
Expression plasmids for ΔFosB, subcloned into adeno-associated viral (AAV) vectors, were used as previously published (Maze et al, 2010; Renthal et al, 2008). Mice or rats were positioned in small animal stereotaxic instruments, under ketamine (100 mg/kg)/xylazine (10 mg/kg) anesthesia, and their cranial surfaces were exposed. Thirty-three gauge syringe needles were bilaterally lowered into the prelimbic area of the PFC and infused with 0.5 μl (mice) or 1 μl (rats) of the viral or vehicle solution. Infusions occurred at a rate of 0.1 μl/min. Rat surgical coordinates were 3.0 mm A-P; 0.7 mm M-L; 3.5 mm D-V (Paxinos and Watson, 2009). Mouse coordinates were 1.9 mm A-P; 0.6 mm M-L; 2.2 mm D-V (Paxinos and Franklin, 2003). To ensure maximal ΔFosB accumulation and mimic long periods of ΔFosB induction caused by long-term antipsychotic treatment, animals were tested 8 weeks post-viral surgery.
Locomotor Assays
Locomotor activity was recorded in Accuscan Versamax locomotor activity chambers. Data were automatically scored using a 16 × 16 IR grid. Distance traveled within the central and peripheral fields, time spent in the central vs the peripheral fields, and frequency of center crossing, were all recoded. Mice were placed in locomotion chambers and their activity was recorded: in the first half hour, mice were allowed to habituate to the chambers; followed by a challenge with saline (1 ml/kg i.p.); then they were challenged with the N-methyl-D-aspartate (NMDA) receptor antagonist (+)-MK-801 hydrogen maleate (0.3 mg/kg or 0.1 mg/kg, i.p; Sigma-Aldrich) or d-amphetamine (3 mg/kg, i.p.; gifted from NIDA), and locomotion was recorded for an additional 60 min or 120 min, respectively.
Prepulse Inhibition (PPI)
Startle magnitude and sensory gating were examined in a 40-trial prepulse inhibition (PPI) assay (San Diego Instruments SR-Lab). Mice were placed in isolation chambers inside closed Plexiglas tubes outfitted with accelerometers. Following a 5-min habituation period with 74 dB background white noise, each animal received 40 randomized trials separated by 20–30 s. Trials consisted of 10 each background readings taken at 74 dB, startle trials with readings following a 40 ms 115 dB tone, PPI trials, where the 115 dB tone was preceded by a 20 ms 78 dB tone 100 ms earlier, and control trials consisting of only the 20 ms 78 dB prepulse. On all trials, maximum magnitude of the animal’s startle (or other motion) was automatically recorded by the accelerometer.
Inhibitory Avoidance
We also tested fear memory in inhibitory avoidance (IA), a task widely used in animal models relevant to the associative deficits in patients with schizophrenia (Arguello and Gogos, 2006; Savonenko et al, 2008). This test was performed in rats, in which it is better validated. IA was conducted exactly as previously described (Arguello et al, 2013). Briefly, the IA chamber consisted of a rectangular-shaped box, divided into a safe compartment that was white and illuminated. and a shock compartment that was black and dark (Model ENV-010MC, Med Associates). Foot shocks were delivered through the grid floor of the dark chamber via a constant current scrambler circuit. The IA boxes were located in a sound-attenuated, dark room. During training sessions, the rat was placed in the safe compartment. After 10 s, the door separating the two compartments was automatically opened, allowing the rat access to enter the shock compartment. Acquisition was measured, in sec, as the latency to enter the shock compartment. The automatic door closed 1 s after the rat entered the shock compartment, and a brief foot shock (0.9 mA for 2 s) was administered. Ten seconds after the foot shock, the rat was returned to the home cage. Memory retention was tested at different time points after training. During testing, the rat was placed into the safe compartment and the latency to enter the shock compartment was recorded. No foot shock was administered during retention tests, and the testing was terminated at 540 s.
Statistics
Locomotor response amphetamine and IA were analyzed using two-way repeated measures analysis of variance (ANOVA) with time as the within subject factor and virus (ΔFosB or GFP) as the between-subject factor followed by Bonferroni post hoc tests when appropriate. Western blot of ΔFosB levels from human tissue and psychostimulant-induced rearing were analyzed by one-way ANOVA followed by the Tukey’s post hoc test when appropriate. PPI, ChIP, MK-801-induced anxiety, and all other western blot data were analyzed using two-tailed Student’s t-tests, and statistical significance was set to be P<0.05 using GraphPad statistical software. Locomotor response to MK-801 was analyzed using three-factor ANOVA, with drug (MK-801 dose) and virus (ΔFosB or GFP) as between-subject factors and time as the within subject factor. Follow-up post hoc using Bonferroni corrections for multiple comparisons was performed using SPSS statistical software. All data are represented as the mean±SEM.
RESULTS
We first examined the expression of ΔFosB in the PFC of patients with schizophrenia who were either on or off antipsychotic medications at their time of death compared with the tissue from control subjects. As seen in Figure 1a, ΔFosB levels are increased in the PFC of medicated schizophrenic patients (n=9) compared with controls (n=10) (F (2,24)=5.081; P<0.05). This effect was not due to the disease state per se, as a post hoc comparison revealed that non-medicated patients showed no such increase in ΔFosB levels compared with controls (n=6; p>0.05). To confirm our hypothesis that the increased levels of ΔFosB in PFC seen in medicated patients are a result of drug exposure, we examined whether repeated administration of the standard antipsychotic drug, haloperidol, altered ΔFosB expression in a homologous region of PFC of rats. Following 7 daily injections of haloperidol (0.1 mg/kg s.c.), using a dose that is behaviorally active in rodents (Mead et al, 2008), we found increased levels of ΔFosB in haloperidol-treated rats compared with vehicle-treated controls (Figure 1b: t=1.996, df=8; p<0.05; n=5 per group).

Antipsychotic drug regulation of ΔFosB expression in humans and rats. (a) Relative ΔFosB protein expression in the PFC of diagnosed schizophrenia (SZ) patients that were either on antipsychotic drugs or non-medicated at their time of death. (b) ΔFosB expression in the PFC of rats after 7 days of repeated haloperidol administration (0.1 mg/kg; s.c.). *p<0.05 significantly different from (a) control patients or (b) vehicle-treated rats.
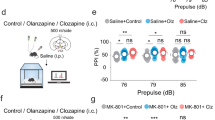
We next asked whether the expression of ΔFosB in the PFC was a key factor in regulating the clinical efficacy of haloperidol or, conversely, perhaps some of its negative side effects. In our first experiment to address this question, we injected AAV-ΔFosB or AAV-GFP into the medial PFC of wild-type C57BL/6J mice. Behavioral testing was conducted 8 weeks after viral surgery. This time point was chosen based on previous studies that have determined that long-term ΔFosB expression (>6 weeks) leads to greater AP-1-dependent gene transcription in vivo (McClung and Nestler, 2003). At this time point, mice were tested for PPI, which is widely used in animal studies of schizophrenia and antipsychotic drugs: a deficit in PPI is thought to reflect impaired sensorimotor gating that occurs in schizophrenia and in several other neuropsychiatric disorders, whereas the reversal of such deficits with antipsychotic drugs is thought to be a measure of their clinical-like efficacy (Nestler and Hyman, 2010). As seen in Figure 2a, overexpression of ΔFosB (n=9) in the medial PFC of mice decreases (impairs) PPI when compared with GFP controls (n=8) (t=2.317, df=15; P<0.05). It would be interesting in future studies to explore the action of ΔFosB in this brain region on PPI responses under a wide range of experimental conditions, including in animals with abnormal baseline PPI functioning as more accurate animal models of schizophrenia are devised.

Behavioral effects of ΔFosB overexpression in the medial PFC of mice. (a) Adeno-associated viral (AAV)-mediated overexpression of ΔFosB in this region decreases percent inhibition in the prepulse inhibition (PPI) assay compared with AAV-GFP controls. (b and c) There was no difference in locomotor activity following a saline challenge between mice overexpressing ΔFosB or GFP. MK-801 and amphetamine increased locomotor activity in both groups of mice equally. (d) ΔFosB overexpression did not alter the rearing response to MK-801 or amphetamine. (e) MK-801 (0.3 mg/kg; i.p.) induced an anxiety-like phenotype in the AAV-ΔFosB group compared with AAV-GFP controls. *p<0.05 significantly different from AAV-GFP control group; **p<0.01 significantly different from the 0.3 mg/kg MK-801 dose; ##p<0.001 significantly different from the saline baseline.
Psychostimulants such as amphetamine or NMDA receptor antagonists such as MK-801 induce psychotic symptoms in normal humans and exacerbate such symptoms in schizophrenic patients. As such, the ability of antipsychotic drugs to antagonize locomotor responses to such challenges is taken as a measure of their therapeutic-like effects in animals (Moreno et al, 2009). On the basis of our PPI findings, where ΔFosB produced an effect opposite to that of antipsychotic drugs, we hypothesized that animals with increased ΔFosB expression in the medial PFC would exhibit exaggerated locomotor responses to MK-801 and amphetamine. To this end, we injected mice overexpressing ΔFosB (n=8–9) or GFP (n=7–10) in the medial PFC with MK-801 (0.3 mg/kg or 0.1 mg/kg i.p.) or d-amphetamine (3 mg/kg; i.p) and recorded locomotor responses. Three-way ANOVA revealed main effects of time (F (8,264)=85.146; P<0.001) and drug (F (1,33)=10.319; P<0.01), and a significant interaction between time × drug (F (8,264)=19.933; P<0.001), indicating that all mice showed an increase in locomotor activity in response to MK-801 (Figure 2b). Follow-up analysis revealed that animals injected with 0.3 mg/kg MK-801 had significantly greater locomotor activity compared with the lower dose of MK-801 at the 30–60 min post injection (P<0.01). Contrary to our hypothesis, this increase in activity was not affected, up or down, by ΔFosB expression (P>0.05). As depicted in Figure 2c, amphetamine-induced locomotor activity was increased compared with a baseline saline challenge (F (17 238)=52.58; P<0.001), but this increase was also not altered by ΔFosB overexpression (P>0.05). Additionally, overexpression of ΔFosB did not affect psychostimulant-induced rearing when compared with GFP controls (P>0.05: Figure 2d); ΔFosB also had no effect on response to novelty (data not shown). However, further analysis of the data demonstrated that mice overexpressing ΔFosB in the medial PFC, when compared with AAV-GFP control mice, demonstrated a strong anxiety-like phenotype as measured by a reduction in time spent in the center of the open arena (t=2.268, df=17; P<0.05: Figure 2e). This effect was specific for MK-801 (0.3 mg/kg), as there was no difference in center time during habituation or following a saline challenge (P<0.05). This increase in anxiety-like behavior may be of clinical relevance, as several studies have demonstrated a high comorbidity of anxiety symptoms in patients with schizophrenia (Seedat et al, 2007).
We next asked whether overexpression of ΔFosB in medial PFC alters the ability to withhold a response following a learned aversion, another widely used assay in studies of schizophrenia and antipsychotic drugs action. For this assessment, we chose to use the IA paradigm in rats, in which the animal must inhibit the natural motivation to cross from a brightly lit compartment to a dark chamber that has been previously paired with a foot shock. As depicted in Figure 3, using two-way repeated measures, we found that there was a main effect of training (F (1, 15)=46.44; P<0.001) and virus (F (1, 15)=14.28; P<0.01) and a significant interaction between them (F (1, 15)=16.01; P<0.01). Further post hoc testing revealed that there was no difference in latency to enter into the dark chamber before foot shock pairing (P>0.05). However, for both the AAV-ΔFosB and control AAV-GFP rats there was an increase in latency to enter the paired chamber 24 h following a one-trial conditioning session (P<0.05). Importantly, this latency to enter the paired chamber was largely driven by the robust significant increase in latency of the AAV-GFP controls rats when directly compared with rats injected with AAV-ΔFosB (P<0.001). The latency to enter from the brightly lit start compartment to the dark chamber was similar across all groups, demonstrating that the overexpression of ΔFosB in medial PFC had no effect on baseline latency. However, following the training session, the overexpression of ΔFosB significantly reduced the latency to enter the chamber previously paired with foot shock when compared with AAV-GFP controls, with no effect on latency before training (P>0.05).

Behavioral effects of ΔFosB overexpression in medial PFC of rats. Mean latency (sec) to enter the paired chamber during baseline (before training) or 24 h after a single conditioning session in the inhibitory avoidance (IA) assay. Adeno-associated viral (AAV)-ΔFosB and AAV-GFP groups show an increase in latency to enter the paired chamber on the test day **p<0.01. On test day, ΔFosB overexpression reduced the latency to enter the paired chamber compared with GFP controls; ***p<0.001.
Finally, because the expression of ΔFosB in other brain regions has been shown to be controlled in part through an auto-regulatory feedback loop by the histone methyltransferase G9A (Maze et al, 2010; Sun et al, 2012), we determined whether haloperidol induction of ΔFosB in medial PFC was associated with reduced binding of this enzyme, which represses ΔFosB expression in other systems. Using a previously validated G9a antibody, we performed a ChIP on the medial PFC from haloperidol- and vehicle-treated rats. As depicted in Figure 4, we found that haloperidol treatment (n=3) markedly decreases G9a binding at the FosB gene promoter when compared with vehicle-treated control rats (n=4) (t=3.101, df=3; p<0.05), consistent with ΔFosB induction under these conditions.

Regulation of G9a binding to the FosB gene promoter by haloperidol. Chromatin immunoprecipitation (ChIP) analysis showed that 7 days of repeated haloperidol administration (0.1 mg/kg; s.c.) reduced G9a binding in the medial PFC compared with vehicle-treated rats; *p<0.05.
DISCUSSION
The current studies reveal a novel role for ΔFosB induction by antipsychotic medications. Here, consistent with previous findings from our group (Atkins et al, 1999), we demonstrate that the first-generation antipsychotic drug, haloperidol, increases ΔFosB expression in the medial PFC of rats. We then added to these findings by demonstrating that this increase in ΔFosB is associated with epigenetic modifications at the FosB gene, specifically, reduced binding of G9a, which catalyzes repressive histone methylation at bound genes. Importantly, we show that schizophrenic patients medicated with antipsychotic drugs at their time of death exhibit increased ΔFosB protein levels in PFC, and that this abnormality is likely not due to the disease state itself, as no such induction was observed in non-medicated patients. Finally, we found that viral-mediated overexpression of ΔFosB in the rodent PFC leads to behavioral changes that are associated with negative behavioral outcomes, including possible worsening of psychotic symptoms (eg, including impaired PPI and IA) as best as can be inferred from animal models (Nestler and Hyman, 2010), which could be part of the molecular mechanisms underlying the so-called ‘neuroleptic-induced deficit syndrome,’ a common side effect observed with antipsychotic treatment (Lader, 1993, 1994; Park et al, 2012). These results suggest that ΔFosB induction in PFC in response to antipsychotic treatment contributes to the deleterious effects of these drugs, which are believed to limit their therapeutic efficacy (Hill et al, 2009; Miyamoto et al, 2012). However, to fully determine a causal relationship between ΔFosB and these negative outcomes, future testing will be necessary to determine whether the blockade of ΔFosB activity in PFC during antipsychotic treatment prevents the adverse behavioral responses.
ΔFosB induction in the mouse medial PFC has recently been implicated in mediating certain deleterious effects of chronic social defeat stress, including increased social withdrawal and anhedonia-like symptoms (Vialou et al, 2012). Given that epidemiological data suggest that social stress may trigger the onset or worsen the severity of psychotic episodes in humans (Cantor-Graae, 2007; Selten and Cantor-Graae, 2005), these findings further support the notion that ΔFosB induction in medial PFC by antipsychotic drugs would worsen clinical outcomes.
However, the exact mechanism by which ΔFosB in medial PFC mediates these behavioral effects remains unknown. Further studies are required to determine the cell type that shows ΔFosB induction after antipsychotic drug treatment, for example, whether this response occurs in glutamatergic pyramidal neurons or GABAergic interneurons, information that will be key in understanding the underlying circuit level mechanisms involved (O’Donnell, 2012). Furthermore, as ΔFosB is a transcription factor, further studies will be necessary to identify the exact target genes that are regulated by ΔFosB following antipsychotic drug treatment and delineating which genes are responsible for the various negative behavioral outcomes seen. One established gene target for ΔFosB in medial PFC is that which encodes the cholecystokinin B receptor (Vialou et al, 2012), which is highly implicated in anxiogenic responses consistent with observations of our studies (Ang et al, 2001; Bowers et al, 2012; Christoffel et al, 2012; McClung and Nestler, 2003). It will now be interesting to use more open-ended approaches to identify many more relevant ΔFosB target genes.
Finally, clinical evidence has demonstrated that a relatively large proportion of patients with schizophrenia do not respond adequately to existing antipsychotic medications (Conley and Kelly, 2001; Suzuki et al, 2012). Although the molecular mechanisms that are responsible for this lack of efficacy remain poorly understood, results of the present study raise the possibility that ΔFosB induction may contribute. These findings thus suggest that suppressing ΔFosB activity or that of its various target genes is a novel path toward boosting the therapeutic actions of available medications.
FUNDING AND DISCLOSURE
Dr Tamminga reports consulting income from Astellas, Eli Lilly, IntraCelluar Therapies, Lundbeck, and PureTech Ventures. The remaining authors declare no conflict of interest.
References
Ang E, Chen J, Zagouras P, Magna H, Holland J, Schaeffer E et al (2001). Induction of nuclear factor-κB in nucleus accumbens by chronic cocaine administration. J Neurochem 79: 221–224.
Arguello AA, Ye X, Bozdagi O, Pollonini G, Tronel S, Bambah-Mukku D et al (2013). CCAAT enhancer binding protein Δ Plays an essential role in memory consolidation and reconsolidation. J Neurosci 33: 3646–3658.
Arguello PA, Gogos JA (2006). Modeling madness in mice: one piece at a time. Neuron 52: 179–196.
Atkins JB, Chlan-Fourney J, Nye HE, Hiroi N, Carlezon WA, Nestler EJ (1999). Region-specific induction of ΔFosB by repeated administration of typical versus atypical antipsychotic drugs. Synapse 33: 118–128.
Beerpoot LJ, Lipska BK, Weinberger DR (1996). Neurobiology of treatment-resistant schizophrenia: new insights and new models. Eu Neuropsychopharmacol 6 (Suppl 2): S27–S34.
Bowers ME, Choi DC, Ressler KJ (2012). Neuropeptide regulation of fear and anxiety: Implications of cholecystokinin, endogenous opioids, and neuropeptide Y. Physiol Behavior 107: 699–710.
Cantor-Graae E (2007). The contribution of social factors to the development of schizophrenia: a review of recent findings. Can J Psychiatry 52: 277–286.
Christoffel DJ, Golden SA, Heshmati M, Graham A, Birnbaum S, Neve RL et al (2012). Effects of inhibitor of [kappa]B kinase activity in the nucleus accumbens on emotional behavior. Neuropsychopharmacology 37: 2615–2623.
Conley RR, Kelly DL (2001). Management of treatment resistance in schizophrenia. Biol Psychiatry 50: 898–911.
Hill SK, Bishop JR, Palumbo D, Sweeney JA (2009). Effect of second-generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother 10: 43–57.
Hiroi N, Graybiel AM (1996). Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J Comp Neurol 374: 70–83.
Hyman SE, Nestler EJ (1996). Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry 153: 151–162.
Kontkanen O, Törönen P, Lakso M, Wong G, Castrén E (2002). Antipsychotic drug treatment induces differential gene expression in the rat cortex. J Neurochem 83: 1043–1053.
Lader M (1993). Neuroleptic-induced deficit syndrome: old problem, new challenge. J Psychopharmacol (Oxford, England) 7: 392–393.
Lader M (1994). Neuroleptic-induced deficit syndrome. Historical introduction. Acta Psychiatr Scand Suppl 380: 6–7.
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO et al (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353: 1209–1223.
Marder SR, Roth B, Sullivan PF, Scolnick EM, Nestler EJ, Geyer MA et al (2011). Advancing drug discovery for schizophrenia. Ann N Y Acad Sci 1236: 30–43.
Maze I, Covington HE, Dietz DM, LaPlant Q, Renthal W, Russo SJ et al (2010). Essential role of the histone methyltransferase g9a in cocaine-induced plasticity. Science 327: 213–216.
Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ (2011). Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci USA 108: 3035–3040.
McClung CA, Nestler EJ (2003). Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nature Neurosci 6: 1208–1215.
Mead A, Li M, Kapur S (2008). Clozapine and olanzapine exhibit an intrinsic anxiolytic property in two conditioned fear paradigms: contrast with haloperidol and chlordiazepoxide. Pharmacol Biochem Behav 90: 551–562.
Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA (2012). Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry 17: 1206–1227.
Moreno J, Sealfon S, González-Maeso J (2009). Group II metabotropic glutamate receptors and schizophrenia. Cell Mol Life Sci 66: 3777–3785.
Nestler EJ (2008). Transcriptional mechanisms of addiction: role of deltaFosB. Philos Trans R Soc London B Biol Sci 363: 3245–3255.
Nestler EJ, Hyman SE (2010). Animal models of neuropsychiatric disorders. Nature Neurosci 13: 1161–1169.
Nestler EJ, Kelz MB, Chen JS (1999). ΔFosB: A molecular mediator of long-term neural and behavioral plasticity. Brain Res 835: 10–17.
O’Donnell P (2012). Cortical interneurons, immune factors and oxidative stress as early targets for schizophrenia. Eur J Neurosci 35: 1866–1870.
Park CH, Park TW, Yang JC, Lee KH, Huang GB, Tong Z et al (2012). No negative symptoms in healthy volunteers after single doses of amisulpride, aripiprazole, and haloperidol: a double-blind placebo-controlled trial. Int Clin Psychopharmacol 27: 114–120.
Paxinos G, Franklin K (2003) The Mouse Brain in Stereotaxic Coordinates. Elsevier Publisher: London, UK.
Paxinos G, Watson C (2009) The Rat Brain in Stereotaxic Coordinates. Elsevier Publisher: London, UK.
Perrotti LI, Bolaños CA, Choi K-H, Russo SJ, Edwards S, Ulery PG et al (2005). ΔFosB accumulates in a GABAergic cell population in the posterior tail of the ventral tegmental area after psychostimulant treatment. Eur J Neurosci 21: 2817–2824.
Renthal W, Carle TL, Maze I, Covington HE, Truong H-T, Alibhai I et al (2008). ΔFosB Mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neuroscience 28: 7344–7349.
Savonenko AV, Melnikova T, Laird FM, Stewart K-A, Price DL, Wong PC (2008). Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA 105: 5585–5590.
Sawa A, Snyder SH (2002). Schizophrenia: diverse approaches to a complex disease. Science 296: 692–695.
Seedat S, Fritelli V, Oosthuizen P, Emsley RA, Stein DJ (2007). Measuring anxiety in patients with schizophrenia. J Nerv Ment Dis 195: 320–324.
Selten J-P, Cantor-Graae E (2005). Social defeat: risk factor for schizophrenia? Br J Psychiatry 187: 101–102.
Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ et al (2006). Human postmortem tissue: what quality markers matter? Brain Res 1123: 1–11.
Sun H, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D et al (2012). Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci 32: 17454–17464.
Suzuki T, Remington G, Mulsant BH, Uchida H, Rajji TK, Graff-Guerrero A et al (2012). Defining treatment-resistant schizophrenia and response to antipsychotics: A review and recommendation. Psychiatry Res 197: 1–6.
Vialou V, Bagot RC, Ferguson D, Feng J, Ku SM, Nestler EJ (2012). Role of deltaFosB in the prefrontal cortex in CCK responses and vulnerability to stress. Neuroscience Meeting Planner New Orleans, LA: Society for Neuroscience 2012.
Acknowledgements
This work was supported by grants from the National Institute of Mental Health (EJN) and National Institute on Alcohol Abuse and Alcoholism (AMG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dietz, D., Kennedy, P., Sun, H. et al. ΔFosB Induction in Prefrontal Cortex by Antipsychotic Drugs is Associated with Negative Behavioral Outcomes. Neuropsychopharmacol 39, 538–544 (2014). https://doi.org/10.1038/npp.2013.255
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.255
