
Abstract
Activation of kappa-opioid receptors (KORs) in monoamine circuits results in dysphoria-like behaviors and stress-induced reinstatement of drug seeking in both conditioned place preference (CPP) and self-administration models. Noradrenergic (NA) receptor systems have also been implicated in similar behaviors. Dynorphinergic projections terminate within the locus coeruleus (LC), a primary source of norepinephrine in the forebrain, suggesting a possible link between the NA and dynorphin/kappa opioid systems, yet the implications of these putative interactions have not been investigated. We isolated the necessity of KORs in the LC in kappa opioid agonist (U50,488)-induced reinstatement of cocaine CPP by blocking KORs in the LC with NorBNI (KOR antagonist). KOR-induced reinstatement was significantly attenuated in mice injected with NorBNI in the LC. To determine the sufficiency of KORs in the LC on U50,488-induced reinstatement of cocaine CPP, we virally re-expressed KORs in the LC of KOR knockout mice. We found that KORs expression in the LC alone was sufficient to partially rescue KOR-induced reinstatement. Next we assessed the role of NA signaling in KOR-induced reinstatement of cocaine CPP in the presence and absence of a α2-agonist (clonidine), β-adrenergic receptor antagonist (propranolol), and β1- and β2-antagonist (betaxolol and ICI-118,551 HCl). Both the blockade of postsynaptic β1-adrenergic receptors and the activation of presynaptic inhibitory adrenergic autoreceptors selectively potentiated the magnitude of KOR-induced reinstatement of cocaine CPP but not cocaine-primed CPP reinstatement. Finally, viral restoration of KORs in the LC together with β-adrenergic receptor blockade did not potentiate KOR-induced reinstatement to cocaine CPP, suggesting that adrenergic receptor interactions occur at KOR-expressing regions external to the LC. These results identify a previously unknown interaction between KORs and NA systems and suggest a NA regulation of KOR-dependent reinstatement of cocaine CPP.
Similar content being viewed by others


INTRODUCTION
Stress increases the risk of drug abuse, relapse to drug seeking, and can potentiate the rewarding properties of cocaine (Bruchas et al, 2010; Covington and Miczek, 2001; McLaughlin et al, 2003; Redila and Chavkin, 2008; Schindler et al, 2012). Numerous previous reports have implicated the dynorphin/kappa opioid system in the mediation of stress-induced behaviors. For example, stress-induced reinstatement to alcohol, cocaine, and nicotine seeking is absent following pretreatment with kappa-opioid receptor (KOR) antagonists and in KOR knockout (KOR KO) mice. However, KOR agonists induce reinstatement of cocaine conditioned place preference (CPP), and reinstatement of self-administration behavior (Beardsley et al, 2005; Jackson et al, 2012; Redila and Chavkin, 2008; Walker and Koob, 2008; Walker et al, 2011).
The KOR system is composed of prodynorphin peptides and KORs, seven transmembrane domain Gi/o-protein coupled receptors. These receptors not only have traditional Gi/o-negative regulatory effects on adenylate cyclase activity but also couple to mitogen-activated protein kinase pathways and inward rectifying potassium channels (Bruchas et al, 2010; Al-Hasani and Bruchas, 2011). Although the mechanisms of stress-induced reinstatement of drug seeking and drug reward are complex, it has been shown that stress causes dynorphin release and KOR activation within both dopaminergic and serotonergic nuclei (Carlezon et al, 2006; Ebner et al, 2010; Gehrke et al, 2008; Land et al, 2008; Zhang et al, 2005). This KOR activation results in reduced dopamine and serotonin levels, contributing to the dysphoria-like behavioral responses that drive reinstatement of drug seeking and reward behaviors. Interestingly, dynorphin and KORs are coexpressed within the locus coeruleus (LC) on both GABAergic and noradrenergic (NA) neurons (Kreibich et al, 2008; Reyes et al, 2006, 2007, 2008, 2009). However, the role of these LC KORs in the regulation of KOR-induced behaviors, including reinstatement of cocaine place preference, is unresolved.
The NA system is a primary regulator of stress-induced reinstatement and is partially mediated by activation of pre- and post-synaptic NA receptor systems. The LC is one of the primary sources of norepinephrine (NE) in the mammalian brain and has an extensive network of NA terminals that innervate numerous regions, including the extended amygdala (Van Bockstaele et al, 1996a, 1999; Moore, 1978; Phelix et al, 1992; Swanson and Hartman, 1975; Swanson, 1976). The LC–NE system is a critical stress response system in the mammalian brain in which stress neuromodulators and endogenous opioids intersect (Van Bockstaele et al, 1996b, 1998, 1999; Valentino et al, 1992, 2001). The behavioral implications of these anatomical interactions on reinstatement of cocaine preference have not been previously demonstrated.
In this report, we determined how LC KORs and NA systems influence kappa-opioid-mediated reinstatement of cocaine CPP. We demonstrate that specific adrenergic receptors influence KOR-mediated but not cocaine-primed reinstatement of cocaine place preference. We found that the expression of KORs in the LC influences the magnitude of KOR-induced reinstatement. Furthermore, we established a specific role for β1-adrenergic receptors in modulating KOR-mediated reinstatement behavior. Together, this study shows novel interactions between NA and kappa-opioid systems in the regulation of kappa-dependent reinstatement of cocaine preference.
MATERIALS AND METHODS
Subjects
Male C57BL/6 wild-type mice (20–30 g) were used. KOR (Oprk1) gene deletion on C57BL/6 background was generated and genotyped as previously described (Hough et al, 2000) and homozygous (KOR KO) and WT (KOR WT) littermate controls were generated by heterozygote crosses. All mice were group-housed within the Animal Core Facility at Washington University in St Louis, given access to food pellets and water ad libitum, and maintained in specific pathogen-free housing. Mice were transferred at least 1 week before testing into a colony room adjacent to the behavioral testing room to acclimatize to the study environment, undergo daily handling, and prevent stress during conditioning phases. Housing rooms were illuminated on a 12-h light/dark cycle with lights on at 0700 hours. All animal procedures were approved by the Animal Care and Use Committee of Washington University in St Louis, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drugs
Cocaine HCl, racemic U50,488 methansulfonate, and norbinaltorphimine-HCl (norBNI) were provided by the National Institute on Drug Abuse and Drug Supply Program and, in some instances, by Sigma Aldrich (St Louis, MO). Clonidine, Propranolol, ICI-118,551 HCl, and betoxolol were purchased from Tocris Biosciences (Mineapolis, MN). All drugs are dissolved in saline unless otherwise indicated.
Viral Vector Design and Production
The KOR construct was developed based on a lentiviral construct expressing the β2 subunit of the nicotinic acetylcholine receptor under the mouse phosphoglycerol kinase (PGK) promoter (Land et al, 2009; Maskos et al, 2005). The lenti-KOR vector is a monocistronic construct expressing KOR. Lenti-PGK-GFP was used as a control. Viral packaging was performed by the Washington University Hope Center Viral Vector Core (St Louis, MO) or the Fred Hutchinson Cancer Research Center (Seattle, WA) using EHS, and BSL2-monitored methods, equipment, and procedures. Virus was packaged at a titer ranging from 1.3 × 108 to 7.1 × 108 IU/ml.
Microinjection Surgical Procedure
After acclimatizing to the holding facility for 7–9 days, the animals were anesthetized in an induction chamber (4% Isoflurane) and mounted on a stereotaxic frame under sterile conditions (Kopf Instruments, Model 1900) where they were maintained at 1–2% isoflurane for the duration of the surgery. A craniotomy was performed and a bevel-tipped Hamilton syringe was lowered bilaterally into the LC (−5.45 AP, +/− 1.25 ML, −4.00 DV from bregma; bevel oriented towards the midline) with norBNI (2.5 μg/side, as described previously; (Knoll et al, 2011; Land et al, 2009; Shirayama et al, 2004; Smith et al, 2012) or saline in wild-type mice, and Lenti-PGK-GFP or Lenti-PGK-KOR (1 μl/side) in KOR KO mice. Both drugs and virus were injected at a rate of 100 nl/min for 10 min. The syringe remained in place for 3–5 min post infusion before being slowly removed. Animals were sutured (silk 45 cm suture, LOOK) and allowed to recover for at least 5–7 days before behavioral testing. Virally injected mice were allowed to recover for 3–4 weeks to allow adequate time for expression and distribution of protein.
Cocaine Place Preference and KOR-Reinstatement Paradigm
Mice were trained in an unbiased, balanced three-compartment conditioning apparatus as described (Bruchas et al, 2011; Land et al, 2009). On pre-conditioning day (day 1), mice were allowed free access to all the three chambers for 30 min. Time spent in each compartment was recorded with a video camera (ZR90; Canon) and analyzed using Ethovision 8.5 (Noldus). Mice were randomly assigned to saline and drug compartments and received a saline injection in the morning (10 ml/kg, s.c.) and a cocaine injection (15 mg/kg, s.c.) in the afternoon, at least 4 h after the morning training on 3 consecutive days. To test for cocaine place preference, the mice were allowed free access to the three compartments. Scores were calculated by subtracting the time spent in the drug-paired compartment, post-test minus the pre-test (Tzschentke, 2007). This was followed by 3 days of extinction training during which saline (10 ml/kg, s.c.) was injected both in the morning and afternoon before placement into isolated conditioning compartments. Mice were tested for extinction of place preference with free access to all the three chambers. Mice were considered to have extinguished cocaine preference if scores fell within 15% of their initial preference scores. Mice that did not meet these criteria were excluded from the study. Mice that extinguished continued on to the reinstatement phase.
To selectively isolate the role of KORs on reinstatement of cocaine CPP, mice were exposed to either of the following reinstatement protocols (timeline and drug treatments are also described in detail in each figure): (1) Mice were injected with KOR agonist, U50,488 (5 mg/kg, i.p.), placed in their home cage for 30 min as previously described (Redila and Chavkin 2008; Land et al, 2009), after which they were placed in the CPP apparatus and allowed free access to all the three compartments. (2) Mice were injected with either β-adrenergic receptor antagonist, propranolol (10 mg/kg, i.p.) or saline (10 ml/kg, i.p.) and returned to their home cage for 30 min. This was followed by a reinstatement test where mice were injected with KOR agonist, U50,488 (5 mg/kg, i.p.), where the mice were placed in their home cage for 30 min, after which they were placed in the CPP apparatus and allowed free access to all the three compartments. (3) Mice were injected with either β-adrenergic receptor antagonist, propranolol (10 mg/kg, i.p.), or α2-adrenergic receptor agonist, clonidine (0.03 mg/kg, i.p.) alone and placed in their home cage for 30 min. (4) Mice were injected with either α2-adrenergic receptor agonist, clonidine (0.03 mg/kg, i.p.), β1-adrenergic receptor antagonist, Betaxolol (10 mg/kg, i.p.), or β2-adrenergic receptor antagonist, ICI-118551 (5 mg/kg, i.p.) and placed in their home cage for 30 min. This was followed by an injection of U50,488 (5 mg/kg, i.p.) or cocaine (15 mg/kg, i.p.), and the mice were placed in their home cage for 30 min, after which they were placed in the CPP apparatus and allowed free access to all the three compartments for reinstatement testing.
Preference was described as time (s) in drug-paired chamber on day 5 minus time spent in drug-paired chamber following day 1 pretest. Reinstatement was measured as time (s) in drug-paired chamber on reinstatement day (varies depending on experiment, see individual figures) minus time spent in drug-paired chamber following extinction training (Bruchas et al, 2011; Land et al, 2009; Tzschentke, 2007).
Conditioned Place Aversion
Mice were trained in an unbiased, balanced three-compartment conditioning apparatus as described above (Bruchas et al, 2007a, 2011). Briefly, mice were allowed to explore all the three compartments for 30 min. Time spent in each compartment was recorded and analyzed as above. Mice were randomly assigned to saline and U50,488 (KOR agonist) compartments and received a saline injection in the morning (10 ml/kg, s.c.) and U50,488 injection (2.5 or 5 mg/kg, s.c.) in the afternoon, at least 4 h after the morning training on 2 consecutive days (days 2–3). To test for U50,488-induced aversion (day 4), the mice were either allowed immediate free access to the three chambers or were pre-treated with propranolol (10 mg/kg, s.c.) 30 min before being placed in the three chambers. Scores were calculated by subtracting the time spent in the drug-paired compartment post-test minus the pre-test.
Locomotor Activity
During the 3-day conditioning period and during reinstatement day, locomotor activity was video recorded and analyzed as distance travelled (cm) throughout the 30-min period using Ethovision 8.5 (Noldus). During the conditioning period, the total distance (cm) on conditioning days 1–3 are represented. During reinstatement post-test trials, the distance (cm) is represented as 5-min bins throughout the 30-min test period.
Immunohistochemistry
Immunohistochemistry was performed as described in Bruchas et al, (2007a), Land et al (2008), and Lemos et al (2011). Mice were anesthetized with pentobarbital and intracardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (PB). Brains were dissected, post-fixed 24 h at 4 °C and cryoprotected with a solution of 30% sucrose in 0.1 M PB at 4 °C for at least 24 h, cut into 30-μm sections and processed for immunostaining. Sections were washed three times in PBS and blocked in PBS containing 0.5% Triton X-100 (G-Biosciences) and 5% normal goat serum (Cell Signaling). To stain for KOR-agonist-induced phospho-ERK and determine functional blockade of KOR signaling by norBNI, sections were then incubated for 72 h at 4 °C with rabbit anti-phospho-ERK antibody (1 : 200, Cell Signaling) and chicken anti-tyrosine hydroxylase antibody (1 : 2000, Aves Lab). To stain for KORs, sections were then incubated for 16 h at 4 °C with a polyclonal chicken anti-KOR antibody targeted to amino acids 359–372 of rat KOR (1 : 100, Antibody Research Corporation). Following incubation, sections were washed three times in PBS and then incubated for 2 h at room temperature in Alexa fluor 488 goat anti-mouse IgG (1 : 1000, Invitrogen) and Alexa fluor 594 goat anti-rabbit IgG (1 : 500, Invitrogen). Sections were then washed three times in PBS and followed by three 10-min rinses in PB and mounted on glass slides with Vectashield (Vector Labs) and sealed with nail polish for microscopy. In some cases, a 1-h incubation with fluorescent Nissl stain (1 : 400, Neurotrace, Invitrogen) was conducted between the PBS and PB rinses. All the sections were imaged on both epifluorescent (Olympus BX61) and confocal microscopes (Olympus FV500) in the Washington University Pain Center or Bakewell Imaging Centers. Gain and exposure time were constant throughout, and all the image groups were processed at the same time.
Quantification of pERK Expression and Viral Expression of KORs
Quantification of pERK in tyrosine hydroxylase-positive LC neurons was adapted as described (Kim et al, 2013). Animals were perfused 30 min following injection with KOR agonist, U50,488 (5 mg/kg, i.p.), and immunohistochemistry was performed. Slices from the LC originated from approximately −5.45 mm caudal to bregma. For quantification, a 400 × 200 μm2 compartment was superimposed over the structure. Tyrosine hydroxylase- and pERK-labeled channels were separated, the compartment mask was applied, an exclusive threshold was set to detect light objects, and positive staining for each channel was counted in a blind-to-treatment fashion using Metamorph 7.6 (Molecular Devices). The counts from each channel were then overlaid and percentage of TH+ cells expressing pERK were reported. For quantification of virally expressed KORs in KOR KO mice used in the behavioral assays, the LC was anatomically defined using fluorescent Nissl stain and a 400 × 200 μm compartment was superimposed over the structure. Nissl- and KOR-labeled channels were separated, the compartment mask was applied, an inclusive threshold was set to detect light objects, and average signal intensity was gathered in a blind-to-treatment fashion using Metamorph 7.6 (Molecular Devices). KOR staining intensities are presented normalized to KOR KO expressing the control virus.
Data Analyses and Statistics
Data are expressed as means±SEM. All raw behavioral data are calculated via Ethovision and then analyzed as described above. Data were normally distributed, and differences between the groups were determined using the Student’s independent t-tests, one-way ANOVA, or two-way ANOVA as appropriate. ANOVAs were followed by post-hoc Bonferroni comparisons if the main effect was significant at p<0.05. Statistical analyses were conducted using the GraphPad Prism 5.0F (GraphPad).
Results
KOR Antagonism in the LC Reduces the Magnitude of KOR-Induced Reinstatement of Cocaine Place Preference
KORs are known to be expressed in the LC and are highly implicated in stress and KOR-induced reinstatement of nicotine, alcohol, and cocaine preference and self-administration (Van Bockstaele et al, 2010; Bruchas et al, 2010). In this experiment, we isolated the effects of KOR activation on reinstatement by determining the necessity of KOR expression within the LC for U50-induced reinstatement to cocaine preference. We locally injected a long acting KOR antagonist, norBNI, that stably inactivates KOR signaling in mice for >3 weeks (Bruchas et al, 2007b; Horan et al, 1992; Land et al, 2009). To demonstrate local LC KOR inactivation, we performed immunohistochemistry to observe that the infusion of norBNI into the LC blocks U50,488-induced phosphorylation of ERK (pERK), a common downstream signaling target of KORs (Bruchas et al, 2006, 2007b; Ebner et al, 2010). We found that U50,488-induced pERK expression was significantly decreased within the LC of norBNI (2.5μg/side)-injected mice, but remained intact in LC saline-injected animals, consistent with specificity of this local KOR inactivation approach (Land et al, 2009; Knoll et al, 2011) (Figure 1a and b). We then used a cocaine CPP and KOR agonist-induced reinstatement paradigm (Figure 1c) to pharmacologically isolate KOR-specific effects on reinstatement and to determine whether inactivation of KORs in the LC affect KOR reinstatement of cocaine CPP using the selective KOR agonist, U50,488 (5 mg/kg, i.p). Mice injected with norBNI directly into their LC showed robust cocaine place preference. Following extinction, however, LC norBNI-injected mice showed significantly reduced levels of U50,488-induced reinstatement of cocaine place preference (Figure 1d, n=8–13, one way ANOVA, F(3,34)=3.532, p<0.05; Bonferoni’s post-hoc test). These data suggest that KORs in the LC are, in part, necessary for KOR-induced reinstatement of cocaine place preference. In contrast, LC saline-injected controls showed both cocaine place preference and robust U50,488-induced reinstatement (Table 1: LC Sal:U50 308.6±58.7 vs LC NorBNI:U50 134.6±40.68; Figure 1d). The injection of both saline and norBNI in the LC did not alter conditioning, as both the groups formed a place preference to cocaine with a similar magnitude as the non-surgical control group (Figure 1d and e). Furthermore, injection into the LC had no effect on cocaine-induced locomotor activity in both the norBNI and saline control groups, suggesting that neither surgical procedure or LC KOR antagonism alters cocaine-induced locomotor plasticity (Figure 1d; Samaha et al, 2004). Together, these data suggest that KOR activity within the LC acts to influence the magnitude of KOR-mediated reinstatement of cocaine CPP.

Selective KOR antagonism in the LC attenuates KOR-induced reinstatement of cocaine place preference. (a, left) Image of a coronal section (plane −5.40 mm from bregma) highlighting the LC; (right) representative photomicrograph of tyrosine hydroxylase (TH, green) and phosphorylated-ERK (pERK, purple) in the LC of WT mice injected with either NorBNI (2.5 μg/side) or saline. White arrows indicate colocalization of pERK and TH. All scale bars are 100 μm. (B) NorBNI treatment decreases co-immunoreactivity of pERK and TH compared with Saline treatment (n=3 slices per brain from three brains for each group; ***p=0.0001; LC Saline vs LC NorBNI; Student’s t-test). (c) Timeline of cocaine place preference/reinstatement experiment, indicating previous injection of into the LC. (d) Cocaine preference scores, calculated as post-test minus pre-test on the cocaine-paired side and U50,488 (5 mg/kg i.p.)-induced reinstatement scores of extinguished cocaine place preference (For all results, mice were classified as extinguished if the extinction post-test was within 15% of the pre-test values). Data show a significant reduction in KOR-induced reinstatement of cocaine place preference following local norBNI infusion into the LC. Data represent the mean preference (s)±SEM, n=8–13 (*p<0.05, LC Saline/U50 vs LC NorBNI/U50 and no surgery control vs LC NorBNI/U50; one-way ANOVA followed by Bonferroni’s post-hoc test). (e) No significant difference in locomotor activity was observed, measured as distance travelled (cm) during the 3-day conditioning period in the drug-paired side. Data represent the mean distance (cm)±SEM, n=8–13.
Selective KOR Expression in the LC Partially Restores KOR-Mediated Cocaine Reinstatement
As antagonism of KORs in the LC significantly reduced the level of U50,488-induced reinstatement to cocaine place preference, we next determined whether expression of KORs in the LC is sufficient for KOR-induced reinstatement of cocaine place preference. In this series of experiments, we used a KOR knockout (KOR KO) mouse model; these mice lack KOR expression globally (Hough et al, 2000). A lentivirus (Lenti-KOR) was injected into the LC of mice to selectively restore KOR expression into the LC (Land et al, 2009) (Figure 2a). Viral expression of Lenti-KOR and control virus (Lenti-GFP) within the LC was confirmed via IHC visualization of KOR immunoreactivity and subsequent quantification or expression of the GFP control viral reporter (Figure 2a, b and c). Three-to-four weeks after the injection of lentivirus into the LC of KOR KO mice, the mice underwent cocaine conditioning followed by extinction and U50,488-induced reinstatement (Figure 2e). Viral restoration of KOR expression in the LC resulted in partial, but significant U50,488-induced reinstatement to cocaine place preference, as compared with KOR KO mice that were injected with the control Lenti-GFP virus (Table 1: Lenti-GFP:U50 −1.802±44.68 vs Lenti-KOR:U50 117.8±10.06 (Figure 2f; n=6, Student’s t-test, p< 0.05)). The absence or presence of KORs in the LC did not significantly alter basal or cocaine-induced locomotor activity, supporting the conclusion that U50,488-induced reinstatement following viral restoration in the LC is attributed solely to involvement of LC-KORs in reinstatement to cocaine place preference and not due to alterations of cocaine-induced locomotor activity during the test phases (Figure 2g). Together, these results suggest that KOR expression within the LC acts to influence the magnitude of KOR-mediated reinstatement of cocaine place preference.

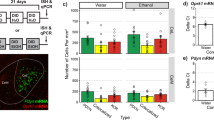
Viral restoration and activation of KORs exclusively in the LC is sufficient for KOR-induced reinstatement of CPP. (a) Schematic of the KOR lentiviral (Lenti-KOR) construct. LTR, long terminal repeats; RRE, Rev response element; FLAP, HIV-1 flap sequence; PGK, phosphoglycerate kinase promoter; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element; PPT, polypurine tract; U3, Unique 3′ region. (b) Representative photomicrograph demonstrating LC targeting (TH, red) via expression of the Lenti-GFP reporter virus (green). Dashed lines indicate the border of the fourth ventricle. Arrows indicate representative colocalization of GFP and TH. All scale bars are 100 μm. (c) Representative photomicrographs of KOR rescue in KOR KO. The LC is anatomically identified with a fluorescent Nissl stain (blue) and the presence of the fourth ventricle (dashed lines). The rescue of KORs in the LC following injection of Lenti-KOR is identified using a chicken anti-KOR antibody (red). All scale bars are 100 μm. (d) KOR immunoreactivity, measured as average intensity in the LC, is rescued in Lenti-KOR-injected mice (n=8 slices from eight brains for each group; ****p<0.0001; Lenti-GFP vs Lenti-KOR; Student’s t-test). (e) Timeline of the lentiviral injections, cocaine place preference, and KOR-induced reinstatement of cocaine place preference. (f) Cocaine preference scores, calculated as post-test minus pre-test on the cocaine-paired side and U50,488-induced (5 mg/kg i.p.) reinstatement scores of extinguished cocaine place preference in LC-injected mice. Data show that viral restoration of KORs within the LC partially recovers KOR-mediated reinstatement of cocaine place preference. Data represent mean time±SEM, n=6 (*p<0.05; Lenti-KOR/U50 vs Lenti-GFP/U50; Student’s t-test). (g) No significant difference in locomotor activity was observed, measured as distance (cm)±SEM, n=6 in Lenti-KOR/U50 vs Lenti-GFP/U50 groups during the 3-day conditioning period.
The NA System Regulates the Magnitude of KOR-Induced Reinstatement
The NA system is highly implicated in the regulation of stress-induced reinstatement; however, no studies have determined if kappa opioid-dependent reinstatement is sensitive to blockade of adrenergic receptors. It has also been reported that stress-induced reinstatement behaviors are mediated by activation of pre- and post-synaptic NA receptor systems (Mantsch et al, 2010; Shaham et al, 2000a, 2000b). Our experiments reveal a key role for LC NA KOR systems in KOR-induced reinstatement of cocaine place preference. We therefore next determined the involvement of NA signaling in KOR-induced reinstatement to cocaine place preference. Clonidine, an α2-adrenergic receptor agonist, decreases NE cell firing and release (Aghajanian and VanderMaelen, 1982) and has been shown to be involved in stress-induced relapse to heroin (Shaham et al, 2000b). We injected mice with clonidine before U50,488 during the reinstatement test. Mice underwent cocaine CPP conditioning, saline extinction, and, before the reinstatement testing phase, were systemically injected with clonidine (0.03 mg/kg i.p.) 30 min before U50,488 (5 mg/kg, i.p.; Figure 3a and b). Clonidine significantly potentiated U50,488-induced reinstatement to drug cocaine place preference (Table 1: Clonidine/Saline 22.68±18.71 vs Clonidine/U50 743.5±106.4); however, the injection of clonidine alone 30 min before reinstatement testing did not cause reinstatement of cocaine place preference, suggesting that potentiating effect of clonidine on reinstatement requires KOR activation. (Figure 3c; n=10–12; one way ANOVA, F(3,33)= 32.96, p<0.0001; Bonferoni’s post-hoc test *p<0.05, Saline/Saline vs Saline/U50; **p<0.01, Saline/U50 vs Clonidine/Saline; ****p<0.0001, Saline/U50 vs Clonidine/U50 and Clonidine/Saline vs Clonidine/U50). This suggests that NE is required to control the magnitude of KOR-induced cocaine reinstatement. To extend this finding, we determined whether propranolol (β-adrenergic receptor selective antagonist) also potentiates U50,488-induced reinstatement to cocaine place preference, thereby indicating whether the potentiation of U50,488-induced cocaine reinstatement is modulated exclusively by NA autoreceptor function or also by NE action at post-synaptic receptor sites. Consistent with a requirement of NE action at post-synaptic β-adrenergic receptors, systemic propranolol (10 mg/kg, i.p.) significantly potentiated U50,488-induced reinstatement to cocaine place preference, whereas propranolol alone did not induce reinstatement (Table 1: U50 alone 248.4±26.82 vs Propranolol/U50 425.7±52.13; Figure 3cB; n=7–13; one-way ANOVA, F(3,35)= 9.412, p<0.0005; Bonferoni’s post-hoc test *p<0.05, Saline/U50 vs Propranolol/U50; ****p<0.0001, Saline/Propranolol vs Propranolol/U50). Locomotor activity (cm) was also measured during reinstatement phase to ascertain whether clonidine or propranolol influences locomotor activity during the KOR reinstatement phase. No significant difference in locomotor activity was observed between all the groups as measured during the reinstatement test trial (Figure 3e). Taken together, these data suggest removal of NA tone via NA receptor blockade results in an increase in the magnitude of KOR-induced reinstatement of cocaine CPP.

The magnitude of KOR-induced reinstatement to cocaine preference requires noradrenergic signalling. (a) Timeline of the KOR-induced reinstatement of cocaine preference experiment, Sal, Saline, Prop, Propranolol, Clon, Clonidine. (b) Representative Ethovision behavioral traces of mouse activity post cocaine conditioning, extinction and propranolol/U50-induced reinstatement. (c) Mice injected with clonidine (α2-adrenergic receptor agonist, 0.03 mg/kg, i.p.) 30 min before treatment with U50,488 (5 mg/kg i.p.). Clonidine alone did not induce reinstatement. Data represent the mean preference (s)±SEM, n= 10–12 (*p<0.05, Saline/Saline vs Saline/U50; **p<0.001, Saline/U50 vs Clonidine/Saline; ****p<0.0001, Saline/U50 vs Clonidine/U50, Saline/Saline vs Clonidine/U50; one-way ANOVA followed by Bonferroni’s post-hoc test). (d) Mice were injected with propranolol (β-adrenergic receptor antagonist, 10 mg/kg i.p.) 30 min before treatment with U50,488 (5 mg/kg, i.p). Propanolol alone did not induce reinstatement. Data show that blockade of noradrenergic signaling via α2-adrenergic receptor agonist or β-adrenergic receptor antagonist causes a potentiation in KOR-mediated reinstatement of cocaine place preference behavior. Data represent the mean preference (s)±SEM, n=7–13 (*p<0.05, Saline/U50 vs Propranolol/U50; ****p<0.0001, Saline/Propranolol vs Propranolol/U50; one-way ANOVA followed by Bonferroni’s post-hoc test). (e) Locomotor activity measured in 5-min bins during reinstatement phase. Data represent mean distance travelled (cm)±SEM, n=7–13. To identify which β-adrenergic receptor subtype may be modulating the potentiation of KOR-induced reinstatement, betaxolol (Betax; selective β1-adrenergic receptor antagonist, 10 mg/kg i.p.) or ICI-11855 (ICI; selective β2-adrenergic receptor antagonist, 5 mg/kg i.p.) was systemically injected before U50,488-induced reinstatement. (f) Betaxolol, but not ICI-118551, significantly potentiated U50,488-induced reinstatement. Data represent the mean preference (s)±SEM, n=6–12 (*p<0.05, Saline/U50 vs Betax/U50; **p<0.01, Saline/U50 vs Propranolol/U50; one-way ANOVA, Bonferroni’s post-hoc test). (g) Locomotor activity, measured as average distance travelled, in 5-min bins during reinstatement phase. Data represent mean distance travelled (cm)±SEM, n= 6–12.
Propranolol is a non-selective β-antagonist, acting on both β1- and β2-adrenergic receptors. Here we determined which β-adrenergic receptor subtype mediates potentiation of U50,488-induced reinstatement to cocaine place preference. Following cocaine CPP and saline extinction, we injected betaxolol hydrochloride (selective β1-antagonist, 10 mg/kg, i.p.) or ICI-118,551 hydrochloride (selective β2-antagonist, 5 mg/kg, i.p.) 30 min before U50,488 (Figure 3a). Betaxolol, but not ICI-118,551, significantly potentiated U50,488-induced reinstatement to cocaine place preference (Figure 3f; n=7–12; one-way ANOVA, F(4,47)=7.816, p<0.0005; Bonferoni’s post-hoc test *p<0.05, Saline/U50 vs betaxolol/U50; **p<0.01, Saline/U50 vs Propranolol/U50). There was no significant difference in locomotor activity during the reinstatement test between the different subtype-selective β-adrenergic antagonists (Figure 3g). These data suggest that blockade of β1-adrenergic signaling causes potentiation of KOR-induced reinstatement of cocaine place preference.
β-Adrenergic Receptor Antagonism has no Effect on Cocaine-Primed Reinstatement
To ascertain whether the NA modulation of reinstatement was selective to KOR receptor activation and KOR-dependent reinstatement to cocaine place preference, we repeated the same cocaine place preference reinstatement paradigm using a cocaine prime injection to initiate reinstatement behavior (Figure 4a and b). To examine the role of β-adrenergic receptor antagonism during conditioning, propranolol was injected 30 min before cocaine during the conditioning sessions. In this experiment, propranolol had no significant effect on the cocaine conditioning phases of place preference (Figure 4b, n=6–9; one-way ANOVA, F(3,19)=18.52; Bonferroni’s post-hoc test ***p<0.001, Saline vs Cocaine and Saline vs Prop/Cocaine) or locomotor activity (Figure 4c; n=6–9; two-way ANOVA, F(4,84)=1.89, p<0.05; Bonferroni’s post-hoc test ****p<0.0001, Day 2 Saline vs Cocaine, Day 3 Saline vs Cocaine; **p<0.01, Day 2 Saline vs Propranolol/Cocaine), *p<0.05 Day 3 Saline vs Propranolol/Cocaine). Systemic injection of propranolol (10 mg/kg, i.p) 30 min before cocaine prime did not potentiate cocaine-induced reinstatement (Figure 4e). This result indicates that the potentiation of reinstatement via removal of β-adrenergic signaling is selective for KOR-mediated cocaine reinstatement of CPP. These data further support the hypothesis that KOR interactions with the NA system modulate KOR-induced, but not cocaine-induced, reinstatement of CPP behavior.

Propanolol does not potentiate cocaine prime-induced reinstatement or effect KOR-mediated place aversion. (a) Timeline of cocaine place preference, including propranolol pretreatment during the 3-day conditioning period. (b) Propanolol does not alter conditioning to cocaine. Data represent the mean preference (s)±SEM, n=6–12; one-way ANOVA, Bonferroni’s post-hoc test ***p<0.001, Saline vs Propranolol/Cocaine; ***p<0.01, Saline vs Cocaine). (c) Treatment with propranolol before cocaine conditioning did not alter locomotor activity recorded during the 30-min conditioning session (n=6–12; ****p<0.0001, Day 3 Saline vs Cocaine, Day 4 Saline vs Cocaine; **p<0.01, Day 3 Saline vs Propranolol/Cocaine; *p<0.05 Day 4 Saline vs Prop/Coc; two-way ANOVA, Bonferroni’s post-hoc test). (d) Timeline of cocaine place preference, extinction and cocaine-primed reinstatement. (e) Propanolol does not potentiate cocaine-induced reinstatement to drug seeking. Data represent the mean preference (s)±SEM, n=6–12. (f) Timeline of U50,488 conditioned place aversion. (g) The potentiating effect of propranolol is not due to enhancement of KOR-mediated aversion behavior. No significant difference was observed between the groups (n=8–16).
Expression of KOR-Mediated Aversion is not Sensitive to β-Adrenergic Modulation
It has previously been shown that U50,488 (2.5 mg/kg, i.p.) induces conditioned place aversion (Bruchas et al, 2011; Land et al, 2008) and reduction in reward seeking (Potter et al, 2011) We wanted to use this paradigm (Figure 4f) to identify whether removal of β-adrenergic tone acts to change the valence of KOR-induced place aversion behavior, indicative of β-adrenergic receptors suppressing an otherwise potent KOR-mediated aversion. Mice were conditioned to U50,488 (2.5 mg/kg or 5 mg/kg i.p.), and both doses of U50,488 showed a similar magnitude of conditioned place aversion (Figure 4g). An additional group was also conditioned to U50,488 but injected with propranolol 30 min before post-testing to observe whether the expression of KOR-mediated aversion is altered by removal of β-adrenergic tone. However, propranolol before the post-test did not alter KOR-induced aversion, suggesting that the potentiating effect of removing NA tone on KOR-mediated reinstatement is not due to an enhancement of KOR-mediated aversion-like behavior (Figure 4g).
Blockade of NA Signaling in Mice with Selective KOR Expression in the LC does not Cause Potentiation of KOR-Reinstatement
This experiment examined whether KOR expression in the LC is sufficient to induce β-adrenergic potentiation of KOR-mediated reinstatement of cocaine CPP. We again used viral re-expression of KORs in the LC of KOR-KO mice. Three-to-four weeks after the injection of lentivirus, mice underwent cocaine CPP and saline extinction. Mice were then injected with propranolol (10 mg/kg i.p.) 30 min before U50,488 (5 mg/kg i.p.) reinstatement (Figure 5a). The viral restoration of KOR expression in the LC together with β-adrenergic antagonism resulted in a similar restoration of U50,488-induced reinstatement to our initial finding in Figure 2 (Figure 5b; n=8; Student’s t-test, F(5,4)=23.68, p<0.05; Table 1: 113.3±36.51 s). The absence or presence of KORs in the LC did not significantly alter basal or cocaine-induced locomotor activity (Figure 5c), consistent with our findings in Figure 2. These results show that LC KORs are sufficient to partially restore a component of KOR-mediated reinstatement, but importantly these data also suggest that the NA component of kappa-opioid reinstatement may occur via influence over KOR-containing circuits outside the LC, as previously reported (Bruchas et al, 2011; Graziane et al, 2013; Van’t Veer et al, 2013).

Rescue of KORs in the LC and block of β-adrenergic receptors does not block potentiation of KOR-induced reinstatement. (a) Timeline of KOR-induced reinstatement experiment, including injection of lentivirus. (b) Cocaine preference scores, calculated as post-test minus pre-test on the cocaine-paired side and propranolol (10 mg/kg i.p.)/U50,488-induced (5 mg/kg i.p.) reinstatement scores in LC-injected mice. Data represent means±SEM, n=8 (*p<0.05; Lenti-KOR, Propranolol/U50 vs Lenti-GFP, Propranolol/U50; Student’s t-test). Propanolol-induced potentiation of U50,488-induced reinstatement to cocaine CPP is absent in KOR KO mice in which KORs have been virally re-expressed in the LC. (c) No significant difference in locomotor activity was observed, measured as distance (cm)±SEM, n= 8 in Lenti-KOR vs Lenti-GFP groups during the 3-day conditioning period. (d) Hypothetical model representing the interaction sites between dynorphin/KOR and noradrenergic neural systems in the modulation of KOR-mediated reinstatement of cocaine place preference. Proposed model shows that KOR activity in the LC is likely to act upstream of previously identified KOR circuits involved in KOR-mediated behavioral responses. This is confirmed in the current study where local norBNI infusion into the LC does not completely block KOR-induced reinstatement to cocaine CPP. In addition, noradrenergic circuits negativity regulate the magnitude of these responses (as identified by antagonism studies in this report). Output of KOR-induced inhibition of LC activity and modulation of noradrenergic outflow are likely mechanisms for the potentiating effect of noradrenergic blockade isolated in this report. The bed nucleus of the stria terminalis (BNST) and the basolateral amygdala (BLA) are likely output structures given their enriched noradrenergic terminal projections and expression of β1-adrenergic receptors. DA, dopamine; 5-HT, serotonin; k, kappa opioid receptor, NE, norepinephrine.
DISCUSSION
NA –KOR Interactions in Kappa Opioid-Mediated Cocaine Reinstatement
The principal findings of this report are that KORs within the LC NA nuclei are involved in kappa opioid-mediated reinstatement of cocaine CPP behavior. This report highlights the behavioral consequences of interactions between KORs and NA systems. In this study, we used KOR-induced reinstatement of a cocaine CPP as a behavioral paradigm to isolate the select role of KOR–NA interactions. We identified that KOR expression in the LC is partially required for KOR-induced reinstatement to cocaine CPP and that the magnitude of the KOR-induced reinstatement response is regulated by post-synaptic β-adrenergic activation. However, this NA-mediated reinstatement efficacy is exclusive to KOR-induced reinstatement and has no effect on the magnitude of reinstatement observed following a cocaine-primed reinstatement. The resulting evidence indicates a role for interactions between the NA system and KORs in the modulation of downstream KOR circuits. This circuitry has recently been well established to control KOR- and stress-induced reinstatement of cocaine seeking and cocaine place preference (Figure 5d; Bruchas et al, 2011; Graziane et al, 2013). The results presented here support the hypothesis that KOR function and β-adrenergic receptor modulation act together to negatively modulate KOR-mediated reinstatement behavior at known downstream circuits (ie, dopaminergic, serotonergic) reported to be critical for KOR function in behavioral affect (Figure 5d).
It is well accepted that the dynorphin/kappa opioid system has a key role in the mediation and vulnerability to stress, anxiety, dysphoria, and depression-like behaviors. Dynorphins are released during stress exposure and the dynorphin/KOR system has been shown to be critical for stress-induced depression-like and anxiety-like behaviors as well as reinstatement to drug seeking (Beardsley et al, 2005; Carey et al, 2007; Carlezon et al, 2006; Chartoff et al, 2012; Ebner et al, 2010; Land et al, 2008; Mague et al, 2003; McLaughlin et al, 2003). It has been shown that following stress, the subsequent release of dynorphin activates KORs within dopaminergic and serotonergic nuclei to produce reinstatement, aversion, and negative affective-like behavioral states (Bruchas et al, 2011; Graziane et al, 2013; Land et al, 2009; Tejeda et al, 2013; Van’t Veer et al, 2013). In addition to decreasing striatal dopamine levels and inhibiting the firing of dopamine neurons, several reports demonstrate that KOR agonists inhibit dopamine release and alter behavioral affect via action within the nucleus accumbens and/or VTA dopaminergic system (Ebner et al, 2010; Graziane et al, 2013; Shippenberg et al, 2007). Recently, we also demonstrated that activation of KORs within dorsal raphe serotoninergic circuits reduces serotonergic tone to influence cocaine place preference and reinstatement (Land et al, 2009; Bruchas et al, 2011; Schindler et al, 2012; Lemos et al, 2012). Together these reports suggest that KOR function in both serotonergic and dopaminergic circuits have a necessary role in the processing of KOR-mediated behaviors. These reports, however, also highlight the unique complexity of KOR function within various monoamine nuclei. In the current study, we found an additional role for KORs in reinstatement behavior that requires KOR expression and interaction with NA systems.
Previous reports have shown that the corticotropin-releasing factor (CRF) system is involved in drug-seeking and drug-withdrawal behaviors (Bale and Vale, 2004; Koob and Le Moal, 2008; Shaham et al, 2000a) and that both stress and CRF engage LC NA cell firing (Aston-Jones et al, 1999; Devilbiss et al, 2012; Valentino et al, 1993, 2001). Recent evidence has also shown that dynorphin/KOR signaling occurs downstream of CRF release to induce a negative affective state (Bruchas et al, 2010; Knoll et al, 2011; Land et al, 2008; Walker and Koob, 2008). The strong presence of both CRF and dynorphin within the LC NA system also suggests that these systems engage one another to affect kappa-opioid-dependent behavioral responses; however, this remains undefined. Here we report, for the first time, that kappa-opioid-induced behaviors are indeed regulated by KOR function within the LC and by KOR-dependent interactions with NA signaling, and future studies will be implemented to investigate potential CRF interactions. It will also be useful to explore the role of NA–KOR interactions on additional KOR-mediated behaviors, including aversion, intracranial self-stimulation threshold, anxiety, and fear (Bruchas et al, 2011; Ebner et al, 2010; Knoll et al, 2011; Van’t Veer et al, 2013). Given the complexity of LC NA and dynorphinergic projection sites, we focused the current study on establishing a foundational connection between dynorphin/KOR systems and NA tone in the modulation of KOR-induced reinstatement of cocaine CPP. In this study, we used viral expression, which may not completely mimic endogenous expression levels or cell type specificity of KOR in LC circuits; thus future studies with higher cellular resolution are needed to completely dissect the complexity of these dynorphin/KOR stress neuropeptide neural circuits, their cell types and the signaling pathways that mediate the effects described.
The presence of KORs within the LC NA system has been established in several reports. Recent evidence suggests that KORs are expressed both presynaptically on GABA and glutamate inputs into the LC, as well as potentially expressed on pericoerulear GABA interneurons adjacent to the LC (Kreibich et al, 2008). Furthermore, KOR agonist injection into the LC attenuates phasic LC discharge events evoked by stimuli that engage LC afferents, suggesting that KORs presynaptically inhibit afferent inputs into the LC system. KOR is also thought to be expressed directly on NA neurons to inhibit NE release, as evidenced by anatomical and physiological studies (Van Bockstaele et al, 2010; Limberger et al, 1986). However, it is important to note that few, if any, studies have examined the cell type and function of KOR signaling within the LC, and future studies in vitro and in vivo with cell type conditional KOR KO (Tejeda et al, 2013; Van’t Veer et al, 2013), cell type-selective restoration, and local circuit identification will be needed to advance our understanding of dynorphin/KOR’s role within cell types in the LC.
Interactions of the Stress, CRF, and NA Circuits in Drug-Seeking Behavior
In this report, we demonstrated, using both blockade of KORs and restoration of KORs in the LC, that KORs in the LC act to modulate KOR-induced reinstatement of cocaine place preference (Figures 1 and 2). It is important to consider that alternate brain regions and neural circuits may be mediating this KOR-dependent behavior. For instance, β-adrenergic receptor stimulation in the bed nucleus of the stria terminalis (BNST) has been shown to be critical for the aversion induced during acute opioid withdrawal and the source of NE in this behavioral response is thought to be the A1 and A2 neurons of the nucleus tractus solitarii (NTS) (Delfs et al, 2000). The BNST is also a potential site of interaction between NE and CRF as the injection of CRF into the BNST induces reinstatement, whereas the injection of a CRF antagonist in this area blocks stress-induced reinstatement (Erb and Stewart, 1999; Flavin and Winder, 2013; Nobis et al, 2011; Shaham et al, 2000b). It is thought that these behaviors are modulated by the NTS-BNST pathway, which is also thought to be necessary for stress-induced reinstatement of cocaine and opiate seeking (Shaham et al, 2000b). Importantly, a recent report has shown that activation of KOR inhibits GABA release in the BNST via a central amygdalar projection, suggesting local KOR regulation of BNST circuits (Li et al, 2012). In addition, recent evidence suggests that α2-ARs within the BNST and CeA act to inhibit fast excitatory synaptic transmission. Thus, the potentiating effects of clonidine and KOR agonist together suggest that KOR-mediated inputs may act with α2-AR modulatory regions to regulate various excitatory inputs under differing behavioral states, as has been recently suggested (for review, see Flavin and Winder, 2013). In addition, there has been some evidence to suggest that α2-ARs regulate glutamatergic transmission from the mPFC inputs, which is thought to also influence NA activity in the LC (Arnsten and Goldman-Rakic, 1984; Jodo and Aston-Jones, 1997; Jodo et al, 1998). Here we report a pronounced and important role for KOR expression within the LC in addition to the KOR-dependent NA tone seen during reinstatement of cocaine place preference. The identification of KOR-LC to BNST, other amygdalar circuits and the mPFC in stress-induced reinstatement behavior is an important future goal.
Our results also demonstrate a role for β1-adrenergic, but not β2-adrenergic, receptors in KOR-mediated cocaine reinstatement. β1-adrenergic receptors are widely expressed in the mammalian brain, including in the basolateral amygdala, BNST, hippocampus, cortex, and striatum (Correll et al, 2005; Flavin and Winder, 2013; Rosenkranz and Grace, 2002); additional studies to further isolate the critical sites and cell types that mediate the β1-adrenergic receptor’s influence on KOR reinstatement behavior will be required. Although the current findings in this study seem contrary to some recent reports showing that β-adrenergic receptor antagonism prevents stress-induced cocaine reinstatement in CPP models (Vranjkovic et al, 2012), it is important to note that our model selectively examined the reinstatement of cocaine preference to a KOR agonist. It is therefore likely that circuits expressing KORs and β1-adrenergic receptors act in opposing ways, and antagonizing β1-adrenergic receptors coupled with KOR stimulation promotes further excitatory drive of circuits that control the reinstatement response (Figure 5c). Because stress engages numerous neuropeptide receptor systems and KOR has been shown to be both necessary and sufficient for cocaine reinstatement in numerous reports (Beardsley et al, 2005; Jackson et al, 2012; Redila and Chavkin, 2008; Walker and Koob, 2008; Walker et al, 2011), we used the KOR-induced CPP reinstatement model to pharmacologically isolate KOR–NA interactions in this complex behavior. Extending these findings in stress-induced reinstatement of self-administration models is therefore warranted in future investigations.
The knowledge gained from this current study suggests that KOR pathways interact within NA systems to ultimately influence KOR-mediated behavioral responses. Our studies demonstrate that KOR interaction with the NA system modulates the magnitude of cocaine reinstatement. Together our findings also strengthen the notion that a better understanding of KOR function in neural circuits and the development of KOR antagonists may have therapeutic value in treating drug abuse disorders.
FUNDING AND DISCLOSURE
We thank the NIDA drug program for providing U50,488, cocaine HCl, and NorBNI. We also thank Selena Schrieber and Dan Messenger (Chavkin Lab, University of Washington) for providing viral vectors. We thank John Pintar (UMDMJ) for providing KOR knockout mice. These studies were supported by NIDA grant R00-DA025182 (to MRB), and McDonnell Center for Systems Neuroscience (to MRB), and WUSTL DBBS (to JGM), and NIMH F31MH101956 (to JGM), and NINDS P30NS057105. The authors declare no conflict of interest.
References
Aghajanian GK, VanderMaelen CP (1982). Alpha 2-adrenoceptor-mediated hyperpolarization of locus coeruleus neurons: intracellular studies in vivo. Science 215: 1394–1396.
Arnsten AF, Goldman-Rakic PS (1984). Selective prefrontal cortical projections to the region of the locus coeruleus and raphe nuclei in the rhesus monkey. Brain Res 306: 9–18.
Aston-Jones G, Rajkowski J, Cohen J (1999). Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry 46: 1309–1320.
Bale TL, Vale WW (2004). CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44: 525–557.
Beardsley PM, Howard JL, Shelton KL, Carroll FI (2005). Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 183: 118–126.
Bockstaele EJ, Van, Chan J, Pickel VM (1996a). Input from central nucleus of the amygdala efferents to pericoerulear dendrites, some of which contain tyrosine hydroxylase immunoreactivity. J Neurosci Res 45: 289–302.
Bockstaele EJ, Van, Colago EE, Valentino RJ (1996b). Corticotropin-releasing factor-containing axon terminals synapse onto catecholamine dendrites and may presynaptically modulate other afferents in the rostral pole of the nucleus locus coeruleus in the rat brain. J Comp Neurol 364: 523–534.
Bockstaele EJ, Van, Colago EE, Valentino RJ (1998). Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J Neuroendocrinol 10: 743–757.
Bockstaele EJ, Van, Peoples J, Valentino RJ (1999). A.E. Bennett Research Award. Anatomic basis for differential regulation of the rostrolateral peri-locus coeruleus region by limbic afferents. Biol Psychiatry 46: 1352–1363.
Bockstaele EJ, Van, Reyes BAS, Valentino RJ (2010). The locus coeruleus: a key nucleus where stress and opioids intersect to mediate vulnerability to opiate abuse. Brain Res 1314: 162–174.
Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S et al (2007a). Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27: 11614–11623.
Bruchas MR, Land BB, Chavkin C (2010). The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res 1314: 44–55.
Bruchas MR, Macey TA, Lowe JD, Chavkin C (2006). Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem 281: 18081–18089.
Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB et al (2011). Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71: 498–511.
Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S et al (2007b). Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem 282: 29803–29811.
Carey AN, Borozny K, Aldrich JV, McLaughlin JP (2007). Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol 569: 84–89.
Carlezon WA Jr, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS et al (2006). Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther 316: 440–447.
Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA (2012). Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology 62: 167–176.
Correll CM, Rosenkranz JA, Grace AA (2005). Chronic cold stress alters prefrontal cortical modulation of amygdala neuronal activity in rats. Biol Psychiatry 58: 382–391.
Covington HE 3rd, Miczek KA (2001). Repeated social-defeat stress, cocaine or morphine. Effects on behavioral sensitization and intravenous cocaine self-administration “binges.”. Psychopharmacology (Berl) 158: 388–398.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000). Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403: 430–434.
Devilbiss DM, Waterhouse BD, Berridge CW, Valentino R (2012). Corticotropin-releasing factor acting at the locus coeruleus disrupts thalamic and cortical sensory-evoked responses. Neuropsychopharmacology 37: 2020–2030.
Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH (2010). Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology (Berl) 210: 241–252.
Erb S, Stewart J (1999). A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci 19: RC35.
Flavin SA, Winder DG (2013). Noradrenergic control of the bed nucleus of the stria terminalis in stress and reward. Neuropharmacology 70: 324–330.
Gehrke BJ, Chefer VI, Shippenberg TS (2008). Effects of acute and repeated administration of salvinorin A on dopamine function in the rat dorsal striatum. Psychopharmacology (Berl) 197: 509–517.
Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA (2013). Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron 77: 942–954.
Al-Hasani R, Bruchas MR (2011). Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115: 1363–1381.
Horan P, Taylor J, Yamamura HI, Porreca F (1992). Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther 260: 1237–1243.
Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J et al (2000). Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res 880: 102–108.
Jackson KJ, McLaughlin JP, Carroll FI, Damaj MI (2012). Effects of the kappa opioid receptor antagonist, norbinaltorphimine, on stress and drug-induced reinstatement of nicotine-conditioned place preference in mice. Psychopharmacology (Berl) 226: 763–768.
Jodo E, Aston-Jones G (1997). Activation of locus coeruleus by prefrontal cortex is mediated by excitatory amino acid inputs. Brain Res 768: 327–332.
Jodo E, Chiang C, Aston-Jones G (1998). Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurons. Neuroscience 83: 63–79.
Kim T, McCall JG, Jung YH, Huang X, Siuda ER, Li Y et al (2013). Injectable, cellular-scale optoelectronics with applications for wireless optogenetics. Science 340: 211–216.
Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG et al (2011). Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry 70: 425–433.
Koob GF, Le Moal M (2008). Addiction and the brain antireward system. Annu Rev Psychol 59: 29–53.
Kreibich A, Reyes BAS, Curtis AL, Ecke L, Chavkin C, Van Bockstaele EJ et al (2008). Presynaptic inhibition of diverse afferents to the locus ceruleus by kappa-opiate receptors: a novel mechanism for regulating the central norepinephrine system. J Neurosci 28: 6516–6525.
Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci 28: 407–414.
Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D et al (2009). Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci USA 106: 19168–19173.
Lemos JC, Roth CA, Chavkin C (2011). Signaling events initiated by kappa opioid receptor activation: quantification and immunocolocalization using phospho-selective KOR, p38 MAPK, and K(IR) 3.1 antibodies. Methods Mol Biol 717: 197–219.
Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ et al (2012). Severe stress switches CRF action in the NAc from appetitive to aversive. Nature 490: 402–406.
Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB et al (2012). Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biol Psychiatry 71: 725–732.
Limberger N, Späth L, Hölting T, Starke K (1986). Mutual interaction between presynaptic alpha 2-adrenoceptors and opioid kappa-receptors at the noradrenergic axons of rabbit brain cortex. Naunyn Schmiedebergs Arch Pharmacol 334: 166–171.
Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr et al (2003). Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther 305: 323–330.
Mantsch JR, Weyer A, Vranjkovic O, Beyer CE, Baker DA, Caretta H (2010). Involvement of noradrenergic neurotransmission in the stress- but not cocaine-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: role for β-2 adrenergic receptors. Neuropsychopharmacology 35: 2165–2178.
Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux J-P et al (2005). Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436: 103–107.
McLaughlin JP, Marton-Popovici M, Chavkin C (2003). Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci 23: 5674–5683.
Moore RY (1978). Catecholamin innervation of the basal forebrain. I. The septal area. J Comp Neurol 177: 665–684.
Nobis WP, Kash TL, Silberman Y, Winder DG (2011). β-Adrenergic receptors enhance excitatory transmission in the bed nucleus of the stria terminalis through a corticotrophin-releasing factor receptor-dependent and cocaine-regulated mechanism. Biol Psychiatry 69: 1083–1090.
Phelix CF, Liposits Z, Paull WK (1992). Monoamine innervation of bed nucleus of stria terminalis: an electron microscopic investigation. Brain Res Bull 28: 949–965.
Potter DN, Damez-Werno D, Carlezon WA Jr, Cohen BM, Chartoff EH (2011). Repeated exposure to the κ-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry 70: 744–753.
Redila VA, Chavkin C (2008). Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 200: 59–70.
Reyes BAS, Chavkin C, van Bockstaele EJ (2009). Subcellular targeting of kappa-opioid receptors in the rat nucleus locus coeruleus. J Comp Neurol 512: 419–431.
Reyes BAS, Drolet G, Van Bockstaele EJ (2008). Dynorphin and stress-related peptides in rat locus coeruleus: contribution of amygdalar efferents. J Comp Neurol 508: 663–675.
Reyes BAS, Glaser JD, Magtoto R, Van Bockstaele EJ (2006). Pro-opiomelanocortin colocalizes with corticotropin- releasing factor in axon terminals of the noradrenergic nucleus locus coeruleus. Eur J Neurosci 23: 2067–2077.
Reyes BAS, Johnson AD, Glaser JD, Commons KG, Van Bockstaele EJ (2007). Dynorphin-containing axons directly innervate noradrenergic neurons in the rat nucleus locus coeruleus. Neuroscience 145: 1077–1086.
Rosenkranz JA, Grace AA (2002). Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci 22: 324–337.
Samaha A-N, Mallet N, Ferguson SM, Gonon F, Robinson TE (2004). The rate of cocaine administration alters gene regulation and behavioral plasticity: implications for addiction. J Neurosci 24: 6362–6370.
Schindler AG, Messinger DI, Smith JS, Shankar H, Gustin RM, Schattauer SS et al (2012). Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake. J Neurosci 32: 17582–17596.
Shaham Y, Erb S, Stewart J (2000a). Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev 33: 13–33.
Shaham Y, Highfield D, Delfs J, Leung S, Stewart J (2000b). Clonidine blocks stress-induced reinstatement of heroin seeking in rats: an effect independent of locus coeruleus noradrenergic neurons. Eur J Neurosci 12: 292–302.
Shippenberg TS, Zapata A, Chefer VI (2007). Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther 116: 306–321.
Shirayama Y, Ishida H, Iwata M, Hazama G-I, Kawahara R, Duman RS (2004). Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem 90: 1258–1268.
Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C (2012). Stress-induced activation of the dynorphin/κ-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci 32: 1488–1495.
Swanson LW (1976). The locus coeruleus: a cytoarchitectonic, Golgi and immunohistochemical study in the albino rat. Brain Res 110: 39–56.
Swanson LW, Hartman BK (1975). The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol 163: 467–505.
Tejeda HA, Counotte DS, Oh E, Rammamoorthy S, Schultz-Kuszak KN, Bäckman CM et al (2013). Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology. doi:10.1038/npp.2013.76.
Tzschentke TM (2007). Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol 12: 227–462.
Valentino RJ, Foote SL, Page ME (1993). The locus coeruleus as a site for integrating corticotropin-releasing factor and noradrenergic mediation of stress responses. Ann N Y Acad Sci 697: 173–188.
Valentino RJ, Page M, Bockstaele E, Van, Aston-Jones G (1992). Corticotropin-releasing factor innervation of the locus coeruleus region: distribution of fibers and sources of input. Neuroscience 48: 689–705.
Valentino RJ, Rudoy C, Saunders A, Liu XB, Van Bockstaele EJ (2001). Corticotropin-releasing factor is preferentially colocalized with excitatory rather than inhibitory amino acids in axon terminals in the peri-locus coeruleus region. Neuroscience 106: 375–384.
Van’t Veer A, Bechtholt AJ, Onvani S, Potter D, Wang Y, Liu-Chen L-Y et al (2013). Ablation of kappa-opioid receptors from brain dopamine neurons has anxiolytic-like effects and enhances cocaine-induced plasticity. Neuropsychopharmacology 38: 1585–1597.
Vranjkovic O, Hang S, Baker DA, Mantsch JR (2012). β-adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for β1 and β2 adrenergic receptors. J Pharmacol Exp Ther 342: 541–551.
Walker BM, Koob GF (2008). Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology 33: 643–652.
Walker BM, Zorrilla EP, Koob GF (2011). Systemic κ-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol 16: 116–119.
Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ (2005). Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology (Berl) 179: 551–558.
Acknowledgements
We thank the HOPE Center viral vector core, Bakewell Imaging Center. We also thank the members of the Bruchas laboratory for helpful discussion and technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al-Hasani, R., McCall, J., Foshage, A. et al. Locus Coeruleus Kappa-Opioid Receptors Modulate Reinstatement of Cocaine Place Preference Through a Noradrenergic Mechanism. Neuropsychopharmacol 38, 2484–2497 (2013). https://doi.org/10.1038/npp.2013.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.151
Keywords
This article is cited by
-
“Effects of the novel relatively short-acting kappa opioid receptor antagonist LY2444296 in behaviors observed after chronic extended-access cocaine self-administration in rats”
Psychopharmacology (2017)
-
The role of the dynorphin/κ opioid receptor system in anxiety
Acta Pharmacologica Sinica (2015)
-
Fabrication and application of flexible, multimodal light-emitting devices for wireless optogenetics
Nature Protocols (2013)
