
Abstract
Alleviating anxiety and depression is pivotal for reducing the risk of relapse in alcoholics. Currently available anxiolytic treatments are limited by side effects, including reduced efficacy in alcoholics, addiction, and sedation. We examined whether the neuropeptide S receptor (NPSR) was effective at controlling ethanol consumption and the anxiety and depression produced by forced abstinence from ethanol. We found that the anxiolytic and anti-depressant effects of NPS are enhanced in acute ethanol abstinent mice. In addition, we found that NPS reduced ethanol consumption and is not in and of itself rewarding. We also provide evidence that ethanol consumption increases the ability of NPS to modulate neuronal activity in the basolateral amygdala. Finally, we found that local injection of NPS in the basolateral amygdala promotes anxiolysis after chronic ethanol consumption, thereby providing insight into the molecular mechanism underlying the changes in behavioral response to NPS. In light of the improved anxiolytic efficacy and benign side effects of NPS in ethanol-withdrawn animals, the NPSR may prove a suitable target for reducing relapse in alcoholism.
Similar content being viewed by others


INTRODUCTION
Anxiety, stress, and abuse of ethanol (EtOH) are intricately linked through a cycle of hormonal dysregulation (Breese et al, 2005; Rose et al, 2010). Indeed, according to National Epidemiologic Survey on Alcohol and Related Conditions, alcohol-dependent subjects are three times more likely to have an anxiety disorder than the general population, and though they are more likely to seek treatment than those without co-morbid anxiety, their treatment outcome is worse. Selective serotonin reuptake inhibitors (SSRIs) and benzodiazepines (BZDs) are the two classes of drug most commonly used to treat anxiety disorders. SSRIs are generally well-tolerated; however, they take several weeks to take effect, and each SSRI is only effective in a minority of patients, who cannot be identified before initiating treatment. BZDs, in turn, are themselves habit-forming. Importantly, BZDs have also been shown to increase the palatability of EtOH and increase alcohol consumption (Soderpalm and Hansen, 1998). Thus, there is a significant need for novel targets and treatments for anxiety disorders, especially for anxiety that is co-morbid with alcoholism.
Neuropeptide S (NPS) and its receptor (NPSR) comprise a recently de-orphanized G protein-coupled receptor system that has been implicated in stress and anxiety (Xu et al, 2004; Domschke et al, 2011). Anatomically, both NPS precursor and receptor mRNAs are found predominately in the central nervous system (Xu et al, 2007). Functionally, central administration of NPS increases locomotor activity in both naïve and habituated mice, effects that are abolished in mice with a disruption of the NPSR (Duangdao et al, 2009). NPS also significantly increases wakefulness and decreases rapid eye movement and slow wave sleep. In addition, NPS suppresses food seeking, as well as anxiety-like behaviors in behavioral paradigms measuring responses to novelty or stress (for a review see, Reinscheid (2008)). This is likely due to the fact that it acts upstream of several diverse signaling systems, including corticotropin-releasing factor, orexin, and γ-aminobutyric acid (GABA), which regulate the psychological and physiological management of stressful and anxiogenic stimuli (Niimi, 2006; Meis et al, 2008; Paneda et al, 2009).
Chronic EtOH consumption induces activity in the hypothalamic pituitary adrenal axis simulating a condition of chronic stress (Rose et al, 2010). This imbalance will, over time, lead to a hormonal hypo-responsiveness to stressful stimuli, while at the same time sensitizing the central nervous system to the influences of stress (Richardson et al, 2008). These adaptations in the stress response have been linked to both enhanced consumption of EtOH and the reinstatement of drug-seeking behavior in animal models and relapse in alcoholics (Little et al, 1999; Sinha, 2001). Consequently, pharmacological management of stress and anxiety is thought to be pivotal for successful abstinence in treatment-seeking alcoholic patients.
What sets the NPSR apart from the anxiolytic targets that are currently clinically accessible is that, while NPS reduces anxiety-like behavior and stress responses, it also promotes wakefulness, reduces all stages of sleep, increases locomotion, and may reduce feeding and drug seeking (Xu et al, 2004; Badia-Elder et al, 2008; Cifani et al, 2011). Consequently, the NPSR could be a suitable target for the treatment of stress and anxiety with an improved side-effect profile compared to existing therapeutics and a reduced liability for maintaining the dependent state. Here, we examined how activation of the NPSR with its endogenous ligand, NPS, affected a spectrum of alcohol-related behaviors in mice voluntarily consuming alcohol.
MATERIALS AND METHODS
Material
Mouse NPS (SFRNGVGSGAKKTSFRRAKQ) was purchased from Anaspec (catalog no. 61246). Two hundred proof EtOH was purchased from Gold Shield Chemical (Hayward, CA). Guide cannula (catalog no. C315G), internal dummy cannula (catalog no. C315DC), and injector (catalog no. C315I) were purchased from PlasticsOne. Preservative-free saline (Hospira product no. 488-20), ketamine (ANADA no. 200-055), xylazine (ANADA no. 139-236), Tylenol (no. 3954234), and topical antibiotics Polysporin (no. 2013928) were purchased from ButlerSchein. Topical analgesic Lidocaine was from J&J sales and logistics (no. 0501-3798-01). Bupropion hydrochloride (no. B102) and desipramine hydrochloride (no. D3900) were purchased from Sigma-Aldrich.
Animals
Male C57Bl/6J mice, 8 weeks old, were purchased from Jackson. Animals that underwent surgery for cannula implantation were directly thereafter transferred into a reverse light–dark cycle room (lights out 1000 h), and were allowed a minimum of a week to recuperate. Animals that received traditional anti-depressants were delivered into the reverse light–dark cycle room and allowed at least 2 weeks of acclimation time before any behavioral test. Each animal was used only once for between-group comparisons in the anxiety tests and forced swim test, and twice for a within animal comparison in all other tests. The animals were always tested in their dark/active cycle. In the animals drinking EtOH, all behavioral tests were carried out between 18 and 24 h after their last EtOH consumption to assure clearance of EtOH. All animal experiments were performed in accordance with the Ernest Gallo Clinic and Research Center Institutional Animal Care and Use Committee guidelines in our AAALAC-approved facility.
Surgery
Animals were sedated for intracerebroventricular cannulization with ketamine/xylazine (100 and 10 mg/kg, respectively), and anesthesia was maintained by ketamine boosts (20 mg/kg) when necessary. For intracerebroventricular surgery, the skull was bared, and a hole (0.6 mm diameter) was drilled at bregma +1.1 mm lateral and −0.5 mm posterior using a stereotax. A 36-gauge guide cannula with an internal dummy cannula was inserted to −2 mm below the skull to the right side ventricle. Two screws (size no. 000, 3/32 in long) were partly inserted into the skull surrounding the guide cannula; dental cement was applied around the screws and cannula to fix the cannula in place. Animals were sedated for BLA cannulation with inhaled isoflurane. For basolateral cannulas, the skull was bared and a hole (0.6 mm diameter) was drilled at bregma ±3.2 mm lateral and −1.3 mm posterior using a stereotax. A 36-gauge guide cannula with an injector cannula was inserted to −4.2 mm below the skull to central BLA and then raised back to −4.0 mm below skull, at which point the cannula was fixed in place with dental cement. Finally, the injector was removed and replaced by a dummy cannula.
Drug Delivery
Each animal was handled multiple times before the first intracerebroventricular injection to minimize the stress from handling. Injections were carried out using an automated pump (Harvard Apparatus) delivering 2 μl of vehicle (saline) or NPS (1.5 nmol/μl) over 1 min. The dose of NPS (3 nmol per animal) was chosen based on earlier studies showing this concentration to be effective in multiple behavioral paradigms (Xu et al, 2004; Badia-Elder et al, 2008; Li et al, 2009). The animals were allowed to move freely in their home cage during the injection. For BLA injections, each mouse was lightly restrained (held by tail in home cage) and injected with 1 nmol NPS per side in 0.3 μl saline over 3 min: injector was removed 1 min later.
EtOH Consumption
Animals were allowed access to 20% EtOH by replacing the water bottle with EtOH between 1200 h (2 h into the dark cycle) and 1600 h on Monday, Wednesday, and Friday for a minimum of 4 weeks before all tests (anxiety, depression, consumption, and conditioned place preference (CPP); see Supplementary Table 1 for a list of all cohorts, what experiments were performed on each, and the average total EtOH consumption for the group at the time of the test). Animal drinking reached a peak plateau within 1 week. Drug or saline was administered immediately before 1200 h when the water bottle was replaced with EtOH (or was not replaced on the water drinking days).
Anxiety Models
All animals were naïve to the room and the anxiety apparatuses when first presented with the tests, and no habituation was performed.
Light/dark transition box
Intracerebroventricular animals were injected with vehicle or NPS (in the dark in order to avoid disturbing the animal's light cycle and reduce stress exposure before the test) and immediately placed on the light side of a light/dark transition box (Med Associates). The lux of the apparatus was 20 on the light side and 3 on the dark side, and it was kept at room temperature of 20–22°C. BLA animals were injected with vehicle or NPS (between 0700 h and noon) and placed on the light side of the light/dark transition box 7 min post-injection. The data were analyzed for the fraction of time spent on light side and total distance traveled during a 5-min test.
Elevated plus maze
Animals were injected with vehicle or NPS and immediately placed in the center of the elevated plus maze (4 arms 30 × 9 cm2 in size at right angles forming a plus; two arms open and two with 13 cm tall walls with a 9 × 9 cm2 square in the center). The apparatus was in a room with 640 lx and room temperature of 20–21°C. The animals were video-recorded over 5 min and an investigator blind to drug scored each video. Data are presented as the percent of time spent in open arms.
Open field
Animals were injected with vehicle or NPS and immediately placed in the center of a lit open field chamber (40.5 × 40.5 cm3 in size; Med Associates). The apparatus was kept at 10 lx uniform across the field and 20–21°C. Each animal was tracked for 60 min and data are presented as the percent of time spent in the center portion (inner 40% of the surface area) and the total distance traveled.
Depression Models
Forced swim test
One swim session was performed per mouse, as described in Sunal et al (1994). Animals were injected with bupropion (4 mg/kg in saline, intraperitoneally; Figure 2b and Supplementary Figure S2A) 40 min before the test, or were injected with vehicle or NPS and immediately tested. Swim sessions lasted 10 min (in ambient light of 580–680 lx), and were conducted in individual clear plastic cylinders (35.5 cm tall × 25 cm in diameter) filled with water (26.5°C, ±0.7°C) to a depth of 20 cm±1.5 cm. A camera positioned on an angle to the cylinders recorded the sessions. An experimenter blind to treatment scored animals. Manual scoring was for total immobile time during the last 6 min of the session. Immobility was defined as the absence of movement, except that necessary to keep afloat.
Tail suspension test
Tail suspension sessions (8 min) were conducted (in ambient light of 30 lx) in visually isolated chambers as per the original paper by Steru et al (1985). Animals were injected with desipramine (20 mg/kg in saline, intraperitoneally; Figure 2 and Supplementary Figure S2B) 40 min before the test, or were injected with vehicle or NPS and immediately tested. Mice were suspended 15 cm from all walls and 35 cm from the floor by tape (2–3 cm from the tip of their tails) to a length of plastic tubing 38 cm from the point of attachment. A camera positioned on an angle to the chambers recorded the sessions. Animals were scored for total immobile time during the last 6 min of the session by an investigator blind to drug treatment. Mice that climbed their tails in the initial 2-min acclimatization time were replaced in a suspended position; mice that persisted in climbing their tails were removed to their home cages and not included in the analysis.
Test of NPS on EtOH Consumption
On the test day, animals were injected intracerebroventricularly with vehicle or NPS in the dark (3 h after lights out) and allowed access to 20% EtOH only, for 1 h immediately after injection. Tests were carried out within each animal with alternating vehicle or NPS on consecutive weeks. Each bottle was weighed before and after each drinking session. Spillage was subtracted (three bottles in empty cages spread across the rack). Body weight was measured the same week. Consumption is presented as g EtOH per kg body mass. Baseline drinking in the absence of injection (EtOH basal) was measured to determine whether saline injection intracerebroventricularly and/or differences in timing for start of drinking session altered drinking behavior. Drug effect on water consumption was measured by injection of NPS or saline on a water-only day 3 h after lights out. Group size n=16.
Conditioned Place Preference
This method was carried out essentially as described recently by Berger and Whistler (2011). Each CPP box (Med Associates) was divided into two separate chambers differentiated by floor texture, wall pattern, and scent (almond or lemon, 20 μl of McCormick extract pipetted onto 2 cm squares of Whatman filter paper taped to sides of the box), resulting in an unbiased environment with equivalent light of 10–25 lx on both sides of the box. On day 1, animals were allowed 30 min non-restricted access to both sides of the box to establish baseline preference. On days 2 and 3, animals were injected (in the dark) intracerebroventricularly with vehicle or NPS (alternating within each animal on consecutive days; Figure 4) or with morphine (10 mg/kg in saline, subcutaneously, separate cohort; Figure 4) and restricted to one side of the CPP box (with a random assignment of drug-paired side) for 30 min. At 72 h after the last pairing session, animals were allowed 30 min unrestricted access to the CPP box with no drug on board. Data are presented as the percent time spent on the drug-paired side on final test day minus percent time spent on drug-paired side on day 1; n=14 water and 16 EtOH drinking animals, respectively, for the NPS cohorts and n=14 animals for the morphine (MS) cohort.
Electrophysiology
Coronal brain slices of the basolateral amygdala (BLA) were prepared from mice that either drank EtOH as above or water only. The mice were anesthetized with 5% isoflurane and immediately decapitated using a guillotine. Brain slices 220 μm thick were cut in ice-cold modified artificial cerebral spinal fluid (aCSF) solution, saturated with 95% O2–5% CO2 (carbogen). Composition of aCSF was (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 25 dextrose and 25 NaHCO3; 295–300 osmolarity. GABA currents were recorded in the presence of DNQX (10 μM) and Strychnine (10 μM) to block glutamate and glycine receptors, respectively. Whole-cell patch-clamp recordings with 3–5 MΩ electrodes were made with a Multiclamp 700B amplifier using Clampex 10.0 (Axon Instruments, Union City, CA) and Igor Pro 6.0 (Wavemetrics, Lake Oswego, OR) for data acquisition. The series resistance (Rs), input resistance (Ri), and holding current (Ihold) of all recordings were continuously monitored. Recordings with large deviations in any of these properties, indicating a poor clamp or large leak currents, were not included in the analysis. To record GABA IPSCs, we used an internal solution of the following composition (in mM): 125 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, 1 EGTA, 2 Na-ATP, 0.6 Na-GTP, 5 creatine phosphate (pH 7.2–7.4), and osmolarity 275–285. Neurons were visualized with an upright microscope equipped with infrared differential interference contrast (DIC) using the Axiovision camera and software (Carl Zeiss Microimaging, Thornwood, NY). aCSF at 30–32 °C was continuously perfused at 2–3 ml/min over brain slices. Whole-cell voltage-clamp recordings were performed at the holding potential of −70 mV in all recordings of GABA IPSCs. The IPSCs were stimulated by a stimulating electrical pulse delivered by a microelectrode placed around 100–200 μm from the cell.
Statistical Analysis
Statistical significance was tested by one-way ANOVA and Tukey's multiple comparison test or Student's t-test for anxiety and depression tests; by repeated-measures ANOVA and Dunn's multiple comparison test or one-way ANOVA and Tukey's multiple comparison test for consumption studies; by Wilcoxon's signed-rank test for conditioned placed preference tests; and Student's t-test for electrophysiology.
RESULTS
The anxiolytic effects of intracerebroventricularly administered NPS are well established for mice in the inactive/light cycle (Xu et al, 2004; Li et al, 2009; Zhu et al, 2010). Here, we demonstrated that NPS delivered intracerebroventricularly decreased anxiety-like behavior (Supplementary Figure S1A) and increased locomotion (Supplementary Figure S1B) even when administered in the active/dark cycle.
Chronic EtOH Self-Administration Enhances the Anxiolytic Effect of NPS
We next examined whether chronic voluntary consumption of EtOH altered the effects of NPS. We utilized a voluntary EtOH consumption paradigm in mice that includes limited access to 20% alcohol to induce bouts of high drinking followed by forced abstinence. All behavioral tests were carried out 18 to 24 h after the last drinking session to allow clearance of alcohol.
To examine the effects of NPS on anxiety-like behavior, we used three different behavioral assays: the elevated plus maze, the light/dark transition box, and an open field test. Using these three assays, we compared the effects of intracerebroventricular NPS on anxiety-like behavior both before and after chronic consumption of EtOH. NPS was an effective anxiolytic agent in both naïve and EtOH-drinking mice (Figure 1 and Supplementary Figure S1).

The anxiolytic effect of neuropeptide S (NPS) is enhanced after chronic ethanol consumption. (a, b) Elevated plus maze, (c, d) light/dark transition box test, or (e, f) open field test. Four groups of mice were assayed: ethanol-naïve mice treated with saline (white bar); ethanol-naïve mice treated with NPS (white hatched bar); ethanol-drinking mice treated with saline (gray bar); ethanol-drinking mice treated with NPS (gray hatched bar). (a) Fraction of total time (5 min) spent in open arms (one-way analysis of variance (ANOVA), F(3, 44)=6.5, p<0.001); (b) number of closed arm to closed arm transitions (one-way ANOVA, F(3, 44)=4.7, p<0.0065); (c) Fraction of total time (5 min) spent in the light side (one-way ANOVA, F(3, 43)=26.64, p<0.0001); (d) total distance covered over time from (c) (one-way ANOVA, F(3, 43)=7.51, p<0.0004); (e) fraction of total time (1 h) spent in the center portion of the open field box (one-way ANOVA, F(3, 26)=7.069, p<0.0013); and (f) total distance covered over time from (e) (one-way ANOVA, F(3, 26)=2.908, p<0.0536). Group size: elevated plus maze, n=15, 15, 9, and 9; light dark transition test, n=11, 11, 9, and 10; and open field, n=7, 7, 8, and 8. Comparison by one-way ANOVA and Tukey's multiple comparison test; *p<0.05; **p<0.01; ***p<0.001.
Mice who had been drinking EtOH showed increased anxiety compared to EtOH-naïve mice as assessed by the elevated plus maze, which was expressed as diminished time spent in open arms (Figure 1a; mean saline chronic EtOH is 34±8% of saline in EtOH-naïve mice; one-way ANOVA, F(3, 44)=6.5, p<0.001, post-test saline versus EtOH saline, p<0.05), an effect that has been previously reported in some other rodent drinking models (van Rijn et al, 2010). Despite this increased anxiety-like behavior on the elevated plus maze, NPS was more effective at reducing anxiety in drinking mice (Figure 1a; chronic EtOH, 242±36% increase in time spent in the open arm with NPS versus saline; one-way ANOVA, F(3, 44)=6.5, p<0.001, post-test EtOH saline versus EtOH NPS, p<0.01) compared to the EtOH naïve cohort (Figure 1a; 15±14% increase in time spent in the open arm with NPS versus saline; one-way ANOVA, F(3, 44)=6.5, p<0.001, post-test saline versus NPS, p>0.05). This apparent increased effect of NPS could reflect a reversal of the discomfort elicited by the forced EtOH abstinence, or an increased potency/efficacy of NPS per se. Regardless, this sustained effect of NPS in EtOH-drinking animals is in contrast to that of a classical anxiolytic, diazepam, which we have previously shown is less effective in mice that have been drinking (van Rijn et al, 2010).
The effect of NPS on anxiety-like behavior, as assessed by the light/dark transition box, was significantly enhanced after EtOH consumption (Figure 1c; chronic EtOH, 113±10% increase in time spent on the light side with NPS versus EtOH saline; one-way ANOVA, F(3, 43)=26.64, p<0.0001, post-test EtOH saline versus EtOH NPS, p<0.001) as compared to in EtOH-naïve animals (Figure 1c; 45±10% increase in time spent on the light side with NPS versus saline; one-way ANOVA, F(3, 43)=26.64, p<0.0001, post-test saline versus NPS, p<0.05). In fact, NPS was so effective after chronic EtOH consumption in this model as to abolish fear of the light side in mice that had been drinking EtOH (Figure 1c; NPS chronic EtOH mice spent 53±3% of total session time on the light side of the box).
In a third measure of anxiety-like behavior, time spent in the center of an open field, NPS also reduced anxiety only in EtOH-drinking mice (Figure 1e and Supplementary Figure S2; 45±11% increase in time spent in the center with NPS versus saline in EtOH-drinking mice; one-way ANOVA, F(3, 26)=7.069, p<0.0013, post-test EtOH saline versus EtOH NPS, p<0.01).
Importantly, these behavioral effects of NPS could not be attributed simply to an enhancement in the ability of NPS to stimulate locomotion in drinking animals. The locomotor effects of NPS were actually moderately decreased in the EtOH-drinking mice in both the light/dark transition test (31% increase in locomotion after NPS treatment in the water-drinking mice versus a 25% increase in the EtOH-drinking mice both compared to mice given saline instead of NPS intracerebroventricularly, Figure 1d) and the open field test (45% versus 25% increased locomotion in response to NPS in the water and EtOH-drinking mice, respectively, compared to saline-treated controls, Figure 1e). NPS-induced locomotion was moderately increased in the elevated plus maze test in the EtOH-drinking mice (34% versus 98% in water and EtOH-drinking mice, respectively, compared to saline controls) (Figure 1b).
Suppression of Depression-Like Behaviors by NPS is Unveiled after Chronic EtOH Consumption
A significant number of treatment-seeking alcoholics are diagnosed as clinically depressed and undergo treatment with conventional anti-depressants as part of their abuse management (Nunes and Levin, 2004). We, therefore, examined whether NPS affected depression-like behaviors in EtOH-naïve and EtOH-drinking mice using two depression models: the forced swim test and the tail suspension test. We first confirmed that two common anti-depressants, bupropion and desipramine, could suppress depression-like behaviors in the forced swim test and tail suspension test, respectively (Supplementary Figure S3A, two-tailed t-test bupropion versus saline in forced swim test, p<0.0296; Supplementary Figure S3B, two-tailed t-test desipramine versus saline in forced swim test, p<0.0039; Supplementary Figure S3C, two-tailed t-test desipramine versus saline in tail suspension test, p<0.0001). We found that both bupropion and desipramine were effective in the forced swim test, but only desipramine was effective in the tail suspension test (data not shown). Using these tests, we found that NPS showed an increased ability to reduce depression-like behavior after chronic EtOH consumption both in the forced swim test (Figure 2a; one-way ANOVA, F(3, 26)=3.422, p<0.0319; post-test EtOH saline versus EtOH NPS, p<0.05) and in the tail suspension test (Figure 2c; one-way ANOVA, F(3, 56)=4.972, p<0.004; post-test EtOH saline versus EtOH NPS, p<0.01). Importantly, the anti-depressant effects of both bupropion and desipramine (both of which showed a similar degree of effectiveness as NPS in naïve animals; Supplementary Figure S3A and C) were lost in mice with a history of chronic EtOH consumption (Figure 2b and d). Animals in the two cohorts used for these tests showed no differences in their depression-like behavior in either of these tests when injected with saline (Supplementary Figure S4).

An anti-depressant-like effect of neuropeptide S (NPS) is induced after chronic ethanol consumption in mice. (a, b) Forced swim test or (c, d) tail suspension test. Six groups of mice were assayed: ethanol-naïve mice treated with saline (intracerebroventricularly (i.c.v.)—NPS controls; intraperitoneally (i.p.)—antidepressant controls, white bars); ethanol-naïve mice treated with NPS (white hatched bar); ethanol-consuming mice treated with saline (gray bar); ethanol-consuming mice treated with NPS (gray hatched bar); ethanol-consuming mice treated with antidepressant (bupropion or desipramine, black bar). Data are presented as passive time (Inactivity) in seconds in both tests. Group size forced swim test, n=7, 7, 8, 8, 9, and 9, respectively; tail suspension test, n=15, 15, 16, 16, 8, and 8, respectively. Comparison by one-way analysis of variance (ANOVA) and Tukey's multiple comparison test. (a) One-way ANOVA, F(3, 26)=3.422, p<0.0319; (c) one-way ANOVA, F(3, 26)=3.422, p<0.0319; or Student's t-test: (b) unpaired t-test, p<0.3449; (d) unpaired t-test, p<0.1379; *p<0.05; **p<0.01.
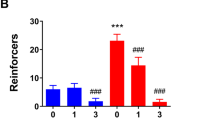
NPS Specifically Reduces EtOH Consumption
To test the effect of NPS on EtOH consumption, animals were injected with intracerebroventricular NPS, immediately returned to their home cage, and allowed access to EtOH or water for 1 h. NPS suppressed consumption of EtOH (Figure 3a and b; 40±13% reduction in consumption after 1 h; repeated-measures ANOVA, F(2, 30)=6.234, p<0.0054; post-test EtOH saline versus EtOH NPS, p<0.05). Notably, vehicle injection had no significant effect compared to consumption without any injection (basal) over the same time period (Figure 3a; repeated-measures ANOVA, F(2, 30)=6.234, p<0.0054; post-test EtOH basal versus EtOH saline p>0.05). Both saline and NPS injection somewhat reduced water consumption over 1 h, although it did not reach significance (Figure 3b; repeated-measures ANOVA, F(2, 30)=2.0219, p<0.1505). The effect of NPS on EtOH consumption was lost if drinking was recorded over 4 h, indicating a limited temporal effect of NPS, possibly due to instability of the peptide ligand (Figure 3c). After 4 h, there remained no effect of NPS on water consumption (Figure 3d). Because only one liquid is available during the test (water or EtOH), these results demonstrate a selective effect of NPS on EtOH but not water consumption, even when it is the only fluid available. Also, mice drink substantial amounts of water on the water test day because it is the only fluid available. Hence, the lack of effect of NPS on water consumption cannot be attributed to a floor effect. Importantly, using this test, we show that the same dose of NPS that reduces anxiety- and depression-like behavior in EtOH-drinking mice also decreases their EtOH consumption.

Intracerebroventricular (i.c.v.) injection of neuropeptide S (NPS) reduces ethanol consumption. (a, c) Effect of NPS on consumption of ethanol. Mice were allowed 1 h (a) or 4 h (c) access to 20% ethanol in their home cage without manipulation (Basal) or immediately after i.c.v. injection of saline (vehicle) or 3 nmol NPS (NPS). (b, d) Effect of NPS on water consumption. Mice were allowed 1 h (b) or 4 h (d) access to water in their home cage without manipulation (Basal) or immediately after i.c.v. injection of saline (vehicle) or 3 nmol NPS (NPS). (a, b) Within animal comparison of vehicle and NPS effect on consecutive weeks. Group size, n=16. Analysis of consumption data by repeated-measures analysis of variance (ANOVA) and Tukey's multiple comparison test: (a) repeated-measures ANOVA, F(2, 30)=6.234, p<0.0054; (b) repeated-measures ANOVA, F(2, 30)=2.0219, p<0.1505. (c, d) Ethanol or water consumption tests on consecutive weeks. Analysis of consumption data by one-way ANOVA and Tukey's multiple comparison test: (c) one-way ANOVA, F(2, 61)=0.1125, p<0.8938; (d) one-way ANOVA, F(2, 63)=0.1193, p<0.8878; *p<0.05; **p<0.01; NS, not significant.
Intracerebroventricular Injection of NPS Supports neither Place Preference nor Place Avoidance
The reduction in EtOH consumption after NPS injection could be explained if NPS acted to substitute for the rewarding effects of EtOH. To address this question, we used an unbiased CPP test. We used morphine, a drug known to elicit robust CPP in mice, as a positive control for our paradigm (Figure 4, morphine; Wilcoxon's signed-rank test, p<0.0012). Unlike morphine, NPS did not support CPP nor elicit conditioned place avoidance in either EtOH-naïve (Figure 4, NPS; Wilcoxon's signed-rank test, p<0.8077) or EtOH-drinking animals (Figure 4, EtOH NPS; Wilcoxon's signed-rank test, p<0.9032). In summary, our place preference data suggest that NPS is neither innately rewarding nor aversive at the dose used to reduce EtOH consumption and alleviate anxiety and depression, even after chronic EtOH consumption, which increases the response of mice to both the anxiolytic- and anti-depressant-like effects of NPS (Figures 1 and 2).

Intracerebroventricular (ICV) injection of neuropeptide S (NPS) produces neither place preference nor place avoidance. Preference to NPS in ethanol-naïve and ethanol-drinking mice 18 h after last EtOH consumption. Data are presented as the change in preference for the drug paired side as fraction of total assay time (30 min). Preference to morphine (10 mg/kg, subcutaneously (s.c.)) serves as a positive control for the CPP paradigm in ethanol-naïve animals. Within animal, comparison of preference for delta bias compared to no bias. Group size, n=14, 16, and 14, respectively. Comparison by Wilcoxon's signed-rank test: NPS, p<0.8077; EtOH NPS, p<0.9032; morphine, p<0.0012; **p<0.01.
NPS-Mediated Potentiation of GABA IPSC Amplitude in BLA is Significantly Enhanced after EtOH Consumption
We next sought a molecular mechanism that could explain the increased effectiveness of NPS in EtOH consuming mice. An NPS influence in one or several different brain regions could explain our findings. We began by examining whether the effects of NPS were altered in the BLA, a region of the brain known to modulate anxiety (for a review see, Koob (2000)). We began in this region because NPSR mRNA expression has been confirmed in the BLA (Jungling et al, 2008), and the NPSR has previously been shown to modulate activity in areas of the amygdala that converge on the BLA (Jungling et al, 2008; Meis et al, 2008; Dannlowski et al, 2011). Importantly, neuronal activity in the BLA has also been shown to influence directly the anxiety paradigms used in this study (Tye et al, 2011). We found that NPS modulation of electrically evoked GABA IPSC amplitude was significantly increased in EtOH-consuming mice (Figure 5a–b, naïve (water drinking): 102.7±21.77, n=5; chronic EtOH drinking: 215.6±33.74, n=4, unpaired t–test, p<0.0221). This constitutes a direct demonstration of enhanced NPS efficacy, brought about by ethanol consumption, in a brain area relevant for the anxiety-like behaviors monitored in this study.

Neuropeptide S (NPS)-mediated potentiation of γ-aminobutyric acid (GABA) inhibitory postsynaptic current (IPSC) amplitude in the basolateral amygdala (BLA) is significantly enhanced after ethanol consumption. (a) Effect of 1 μM NPS on GABA IPSCs in BLA neurons from water-drinking and ethanol-drinking mice. (b) NPS produces a potentiation of GABA IPSCs in BLA neurons only in ethanol-drinking mice. The gray bar in (a) represents the region from which the average GABA amplitude was computed. Naïve (water drinking): 102.7±21.77, n=5; chronic ethanol drinking: 215.6±33.74, n=4, unpaired t–test, p<0.0221; *p<0.05.
NPS Injection into BLA Produces Anxiolytic Effects in the Light/Dark Transition Box
We next examined whether NPS delivered specifically to the BLA affected anxiety-like behavior in EtOH-drinking mice. Animals with bilateral surgically implanted guide cannulas were allowed to drink EtOH for 4 weeks as above. At 18 h after forced EtOH abstinence, mice were bilaterally injected with NPS (1 nmol NPS per side, 0.3 μl volume) and anxiety-like behavior was assessed using the light–dark transition box. Intra-BLA NPS significantly reduced anxiety-like behavior measured as an increase in the percent time spent on the light side of the transition box, and also produced a small increase in locomotor activity (Figure 6a and b; unpaired t–test, p<0.0038 and p<0.0139, for Figure 6a and b NPS versus saline, respectively). Taken together, these data suggest that intra-BLA NPS is sufficient to alleviate anxiety-like behavior in EtOH-drinking mice.

Injection of neuropeptide S (NPS) in the basolateral amygdala has an anxiolytic-like effect as assessed by the light/dark transition test. (a) Fraction of total time (5 min) spent in the light side. (b) Total distance covered over total time (5 min). Comparison by unpaired t-test: (a) p<0.0038; (b) p<0.0139; *p<0.05; **p<0.01.
DISCUSSION
Here, we show an enhanced influence of the NPSR system on diverse anxiety-associated and depression-like behaviors in mice after chronic voluntary consumption of EtOH. These results, together with our findings that NPS reduces consumption of EtOH but does not support CPP at the same doses that reduce anxiety and depression, indicate that the NPSR could be a suitable target for the intervention in alcoholism and alcohol-related disorders.
The enhanced anxiolytic and antidepressant effect of NPS after chronic EtOH consumption and forced abstinence can be interpreted as an enhancement of NPSR function. This is supported by a recent study that demonstrated that chronic exposure to high levels of EtOH combined with a period of abstinence led to a significant upregulation of NPSR mRNA levels, both after acute EtOH withdrawal and after prolonged abstinence, in several brain regions—including the BLA (Ruggeri et al, 2010). However, an increased NPS effect on mood may also reflect a change in neuronal excitability or neural network function in response to chronic EtOH consumption, unrelated to NPSR levels per se, that make the neuron/system more sensitive to NPSR activation. A number of studies support this notion, showing altered activity of multiple signaling pathways in the amygdala after chronic EtOH consumption, with a direct link to anxiety, depression, and enhanced EtOH consumption (Rassnick et al, 1993; Sheline et al, 1998; Pandey et al, 2003; Lack et al, 2007; Walker et al, 2010). Our electrophysiological findings do not differentiate between these two mechanisms described above, and they need not be mutually exclusive. Nevertheless, our findings do provide a possible neural correlate to the heightened anxiolytic effect of NPS seen after chronic EtOH consumption. We report an absence of NPS effects on evoked GABA IPSCs in saline-drinking mice. This observation correlates with a recent study showing that NPS perfusion positively modulates glutamatergic but not GABAergic synaptic transmission in the amygdala (Jungling et al, 2008). We go on to further show that after chronic EtOH, NPS application now does stimulate GABA IPSCs, suggesting that EtOH consumption has resulted in a gain of function, either via an increased expression of the NPSR or a greater effect of NPS on BLA neuronal excitability.
Our finding that NPS, delivered directly to the BLA, was sufficient to reduce anxiety-like behavior in drinking animals indicates that NPSR-expressing neurons in the BLA have a role in modulating anxiety. However, it is possible that other brain regions implicated in anxiety, and/or alcohol-withdrawal-induced anxiety, including the BNST, for example (McKay et al, 2004; Funk et al, 2006), could also have an altered response to NPS after chronic drinking.
It is worth noting that we find indications that anxiety is increased in the animals in forced abstinence from EtOH (Figure 1a) findings reminiscent of those seen in the clinical data from human alcoholics, who often show comorbid anxiety and depression. It remains to be determined which specific circuits have been altered by EtOH exposure to cause these anxiety and depression-like effects. Nevertheless, the fact that NPS alleviates both of these potential side effects of EtOH abstinence, while classical anxiolytics and anti-depressants do not (see (van Rijn et al, 2010) and Figure 2b and d) suggests that either the circuits mediating these anxiety- and depression-like effects both have functional NPSRs or that these circuits are overlapping.
Conventional anxiolytic drugs available today are rewarding in and of themselves, and/or enhance the rewarding effects or the consumption of EtOH. They may, therefore, be subject to abusive use or reinforce drug-seeking behaviors in alcoholics (Soderpalm and Hansen, 1998; Schmitt et al, 2002; Isbister et al, 2004; O'Brien C, 2005). Our data indicate that NPS alleviates anxiety and depression at doses that do not elicit CPP, but nevertheless actually reduce EtOH consumption. In support of our data, a previous study has likewise shown that NPS is not itself rewarding when administered to mice, and has gone further to show that NPS can actually block the rewarding properties of morphine (Li et al, 2009). In contrast, Cao et al (2011) recently showed that NPS administration in rats can lead to both aversion and preference depending on which dose of NPS is used. These two studies used similar methodology but differed in model species, which could explain the discrepancies. With regards to the effect of NPS on EtOH consumption, Badia-Elder et al (2008) found that NPS suppressed EtOH consumption only in selectively bred EtOH-preferring rats and not non-preferring rats. The genetic make up of this selectively bred alcohol-preferring rat strain is not completely clear, but could presumably include effects on NPS, NPSR, or effector systems downstream of the receptor. In line with Badia-Elder's data in EtOH non-preferring rats, Cannella et al (2009) showed that NPS had no effect on EtOH consumption in wistar rats. Thus, genetic factors clearly influence the effect of NPS both on reward and EtOH consumption. In addition, the drinking paradigm used could also influence the results. Importantly, regardless of model species, NPS show an improved side-effect profile over conventional anxiolytic drugs by not enhancing consumption of EtOH.
A potentially attractive side effect of NPS in the treatment of alcohol abstinent patients is the suppression of all stages of sleep. Alcohol abuse produces long-lasting perturbations of night-time sleep, which leads to an increased risk of fatigue-related injuries in the recuperating patient (Williams and Rundell, 1981). NPS could possibly counteract these issues when used during daytime wake hours, thus enabling the patient to sleep when appropriate, and lead a more productive life.
In conclusion, our data support a role for a non-classical anxiolytic impact of the NPSR in the treatment of EtOH-induced depression and anxiety, and could, thus, be used to reduce relapse to addictive behaviors in treatment-seeking alcoholic patients.
References
Badia-Elder NE, Henderson AN, Bertholomey ML, Dodge NC, Stewart RB (2008). The effects of neuropeptide S on ethanol drinking and other related behaviors in alcohol-preferring and -nonpreferring rats. Alcohol Clin Exp Res 32: 1380–1387.
Berger AC, Whistler JL (2011). Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol Med 7: 385–397.
Breese GR, Chu K, Dayas CV, Funk D, Knapp DJ, Koob GF et al (2005). Stress enhancement of craving during sobriety: a risk for relapse. Alcohol Clin Exp Res 29: 185–195.
Cannella N, Economidou D, Kallupi M, Stopponi S, Heilig M, Massi M et al (2009). Persistent increase of alcohol-seeking evoked by neuropeptide S: an effect mediated by the hypothalamic hypocretin system. Neuropsychopharmacology 34: 2125–2134.
Cao J, de Lecea L, Ikemoto S (2011). Intraventricular administration of neuropeptide S has reward-like effects. Eur J Pharmacol 658: 16–21.
Cifani C, Di Bonaventura MV, Cannella N, Fedeli A, Guerrini R, Calo G et al (2011). Effect of neuropeptide S receptor antagonists and partial agonists on palatable food consumption in the rat. Peptides 32: 44–50.
Dannlowski U, Kugel H, Franke F, Stuhrmann A, Hohoff C, Zwanzger P et al (2011). Neuropeptide-S (NPS) receptor genotype modulates basolateral amygdala responsiveness to aversive stimuli. Neuropsychopharmacology 36: 1879–1885.
Domschke K, Reif A, Weber H, Richter J, Hohoff C, Ohrmann P et al (2011). Neuropeptide S receptor gene—converging evidence for a role in panic disorder. Mol Psychiatry 9: 938–948.
Duangdao DM, Clark SD, Okamura N, Reinscheid RK (2009). Behavioral phenotyping of neuropeptide S receptor knockout mice. Behav Brain Res 205: 1–9.
Funk CK, O'Dell LE, Crawford EF, Koob GF (2006). Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci 26: 11324–11332.
Isbister GK, O'Regan L, Sibbritt D, Whyte IM (2004). Alprazolam is relatively more toxic than other benzodiazepines in overdose. Br J Clin Pharmacol 58: 88–95.
Jungling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD et al (2008). Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron 59: 298–310.
Koob GF (2000). Neurobiology of addiction. Toward the development of new therapies. Ann N Y Acad Sci 909: 170–185.
Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA (2007). Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol 98: 3185–3196.
Li W, Gao YH, Chang M, Peng YL, Yao J, Han RW et al (2009). Neuropeptide S inhibits the acquisition and the expression of conditioned place preference to morphine in mice. Peptides 30: 234–240.
Little HJ, O'Callaghan MJ, Butterworth AR, Wilson J, Cole J, Watson WP (1999). Low alcohol preference among the ‘high alcohol preference’ C57 strain of mice; preference increased by saline injections. Psychopharmacology (Berl) 147: 182–189.
McKay PF, Foster KL, Mason D, Cummings R, Garcia M, Williams LS et al (2004). A high affinity ligand for GABAA-receptor containing alpha5 subunit antagonizes ethanol's neurobehavioral effects in Long–Evans rats. Psychopharmacology (Berl) 172: 455–462.
Meis S, Bergado-Acosta JR, Yanagawa Y, Obata K, Stork O, Munsch T (2008). Identification of a neuropeptide S responsive circuitry shaping amygdala activity via the endopiriform nucleus. PLoS One 3: e2695.
Niimi M (2006). Centrally administered neuropeptide S activates orexin-containing neurons in the hypothalamus and stimulates feeding in rats. Endocrine 30: 75–79.
Nunes EV, Levin FR (2004). Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA 291: 1887–1896.
O'Brien C P (2005). Benzodiazepine use, abuse, and dependence. J Clin Psychiatry 66 (Suppl 2): 28–33.
Pandey SC, Roy A, Zhang H (2003). The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res 27: 396–409.
Paneda C, Huitron-Resendiz S, Frago LM, Chowen JA, Picetti R, de Lecea L et al (2009). Neuropeptide S reinstates cocaine-seeking behavior and increases locomotor activity through corticotropin-releasing factor receptor 1 in mice. J Neurosci 29: 4155–4161.
Rassnick S, Heinrichs SC, Britton KT, Koob GF (1993). Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res 605: 25–32.
Reinscheid RK (2008). Neuropeptide S: anatomy, pharmacology, genetics and physiological functions. Results Probl Cell Differ 46: 145–158.
Richardson HN, Lee SY, O'Dell LE, Koob GF, Rivier CL (2008). Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci 28: 1641–1653.
Rose AK, Shaw SG, Prendergast MA, Little HJ (2010). The importance of glucocorticoids in alcohol dependence and neurotoxicity. Alcohol Clin Exp Res 34: 2011–2018.
Ruggeri B, Braconi S, Cannella N, Kallupi M, Soverchia L, Ciccocioppo R et al (2010). Neuropeptide S receptor gene expression in alcohol withdrawal and protracted abstinence in postdependent rats. Alcohol Clin Exp Res 34: 90–97.
Schmitt U, Waldhofer S, Weigelt T, Hiemke C (2002). Free-choice ethanol consumption under the influence of GABAergic drugs in rats. Alcohol Clin Exp Res 26: 457–462.
Sheline YI, Gado MH, Price JL (1998). Amygdala core nuclei volumes are decreased in recurrent major depression. Neuro Report 9: 2023–2028.
Sinha R (2001). How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 158: 343–359.
Soderpalm AH, Hansen S (1998). Benzodiazepines enhance the consumption and palatability of alcohol in the rat. Psychopharmacology (Berl) 137: 215–222.
Steru L, Chermat R, Thierry B, Simon P (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 85: 367–370.
Sunal R, Gumusel B, Kayaalp SO (1994). Effect of changes in swimming area on results of ‘behavioral despair test’. Pharmacol Biochem Behav 49: 891–896.
Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H et al (2011). Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471: 358–362.
van Rijn RM, Brissett DI, Whistler JL (2010). Dual efficacy of delta opioid receptor-selective ligands for ethanol drinking and anxiety. J Pharmacol Exp Ther 335: 133–139.
Walker BM, Drimmer DA, Walker JL, Liu T, Mathe AA, Ehlers CL (2010). Effects of prolonged ethanol vapor exposure on forced swim behavior, and neuropeptide Y and corticotropin-releasing factor levels in rat brains. Alcohol 44: 487–493.
Williams HL, Rundell Jr OH (1981). Altered sleep physiology in chronic alcoholics: reversal with abstinence. Alcohol Clin Exp Res 5: 318–325.
Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK (2007). Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol 500: 84–102.
Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH et al (2004). Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43: 487–497.
Zhu H, Mingler MK, McBride ML, Murphy AJ, Valenzuela DM, Yancopoulos GD et al (2010). Abnormal response to stress and impaired NPS-induced hyperlocomotion, anxiolytic effect and corticosterone increase in mice lacking NPSR1. Psychoneuroendocrinology 35: 1119–1132.
Acknowledgements
This study was supported in part by a grant from the American Asthma Foundation (AAF) and NIH Grant R01AA020401 to JL Whistler; a long-term fellowship from Svenska Stiftelsen for Medicinsk Forskning (SSMF) to J Enquist and funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco to JL Whistler. A Madhavan is supported by an individual NRSA (5F32DA027286-02) and funds from the NARSAD Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article
Cite this article
Enquist, J., Ferwerda, M., Madhavan, A. et al. Chronic Ethanol Potentiates the Effect of Neuropeptide S in the Basolateral Amygdala and Shows Increased Anxiolytic and Anti-Depressive Effects. Neuropsychopharmacol 37, 2436–2445 (2012). https://doi.org/10.1038/npp.2012.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.102

{kind=link}
{kind=link}
{kind=link}
{kind=link}