
Abstract
The potential contribution of chronic inflammation to the development of neuropsychiatric disorders such as major depression has received increasing attention. Elevated biomarkers of inflammation, including inflammatory cytokines and acute-phase proteins, have been found in depressed patients, and administration of inflammatory stimuli has been associated with the development of depressive symptoms. Data also have demonstrated that inflammatory cytokines can interact with multiple pathways known to be involved in the development of depression, including monoamine metabolism, neuroendocrine function, synaptic plasticity, and neurocircuits relevant to mood regulation. Further understanding of mechanisms by which cytokines alter behavior have revealed a host of pharmacologic targets that may be unique to the impact of inflammation on behavior and may be especially relevant to the treatment and prevention of depression in patients with evidence of increased inflammation. Such targets include the inflammatory signaling pathways cyclooxygenase, p38 mitogen-activated protein kinase, and nuclear factor-κB, as well as the metabolic enzyme, indoleamine-2,3-dioxygenase, which breaks down tryptophan into kynurenine. Other targets include the cytokines themselves in addition to chemokines, which attract inflammatory cells from the periphery to the brain. Psychosocial stress, diet, obesity, a leaky gut, and an imbalance between regulatory and pro-inflammatory T cells also contribute to inflammation and may serve as a focus for preventative strategies relevant to both the development of depression and its recurrence. Taken together, identification of mechanisms by which cytokines influence behavior may reveal a panoply of personalized treatment options that target the unique contributions of the immune system to depression.
Similar content being viewed by others

INTRODUCTION
According to a special issue of the journal Science, the recognition that inflammation is a primary pathophysiologic mechanism in chronic illness is one of the major scientific insights of the decade (Science, Volume 330, issue 6011, page 1621, 2010). Although much of the attention on the role of inflammation in disease has focused on cardiovascular disease, diabetes, cancer, and neurodegenerative disorders, there is increasing appreciation that neuropsychiatric diseases such as major depression should appropriately be added to the list of disorders where inflammation is meaningfully involved. Indeed, there has been a virtual explosion of information implicating inflammation as a pivotal pathway to depression, bringing psychiatry and medicine in lock step for the search for targets unique to inflammation for the treatment and prevention of disease development and progression. Given data suggesting that depression is associated with inflammation has also begun to shed new light on the well-established impact of co-morbid depression on the outcome of diseases associated with depression and inflammation, including heart disease and cancer. The purpose of this review is to present data that support the notion that major depression is a psychiatric illness in which inflammation has a role in at least some significant sub-population of depressed patients. Given this evidence, we will then proceed to elaborate those pathways that may be unique to the contribution of inflammation to depression, thereby identifying a series of pharmacologic targets that may be of primary relevance to those depressed individuals who show increased inflammatory biomarkers. A primary goal of this exercise is to further establish a personalized approach to the treatment and prevention of neuropsychiatric disease through the identification of specific pathophysiologic pathways that warrant specific treatment interventions.
INFLAMMATION AND DEPRESSION: MAKING THE CASE
Patients with Major Depression Show Increased Biomarkers of Inflammation
Some of the earliest evidence that major depression was associated with increased inflammation came from the work of Maes et al (1991, 1992a, 1992b) who identified increased inflammatory biomarkers in depressed patients, including increases in acute-phase proteins and the production of inflammatory cytokines, as well as increased cellular markers of immune activation. These data came as somewhat of a surprise, because they occurred in the context of a pre-existing literature on the immune system in depression, which was dominated by reports of decreased cellular (lymphocyte) responses and reduced natural killer-cell activity (Irwin and Miller, 2007). Although not entirely resolved, preclinical data suggest that the inflammatory response itself may be implicated in the impaired lymphocyte function in major depression, including direct effects of inflammatory cytokines on signaling through the T-cell receptor (Clark et al, 2005; Blume et al, 2011). Since the early reports of immune activation in major depression, a vast literature has reproduced these findings, and meta-analyses have revealed that peripheral blood elevations in the cytokines, interleukin (IL)-6, tumor necrosis factor (TNF)-α, and the acute-phase reactant, c-reactive protein (CRP) are some of the most reliable biomarkers of increased inflammation in depressed patients (Zorrilla et al, 2001; Dowlati et al, 2010). Increases in peripheral blood chemokines and cellular adhesion molecules, as well as increased stress-induced nuclear factor-κB (NF-κB), a lynchpin-signaling molecule in the inflammatory response (see below), have also been described in patients with major depression. Of note, not only have mean differences in inflammatory markers between depressives and controls been reported, but also studies have found significant associations between blood concentrations of inflammatory factors and the severity of depressive symptoms, including impaired sleep, cognitive dysfunction, and fatigue (Bower et al, 2002; Meyers et al, 2005; Motivala et al, 2005).
Although not entirely consistent, increased inflammation in depression appears to be a state-dependent phenomenon, with a number of studies demonstrating that increased inflammatory markers in depressed patients return to control levels following successful antidepressant treatment (Miller et al, 2009). A corollary to these findings is that depressed patients who fail to respond to antidepressant therapy show increased inflammatory markers (Sluzewska et al, 1997; Lanquillon et al, 2000; Fitzgerald et al, 2006). Moreover, patients with increased inflammatory markers at baseline are less likely to show a response to treatment, suggesting a relationship between inflammation and treatment resistance (Sluzewska et al, 1997; Lanquillon et al, 2000; Fitzgerald et al, 2006). Polymorphisms in the IL-1β gene have also been associated with treatment response as well as alterations in emotional processing as measured by functional magnetic resonance imaging (fMRI) (Yu et al, 2003; Baune et al, 2010). Finally, epidemiologic data have indicated that increased inflammation in major depression may be especially associated with patients who have experienced childhood maltreatment (Danese et al, 2007). For example, in a representative birth cohort of 1000 individuals followed to age 32, patients with a current major depression and a documented history of childhood maltreatment were over two times more likely to show high peripheral blood concentrations of high-sensitivity (hs) CRP (>3 mg/l) compared with depressed individuals without a history of childhood abuse (Danese et al, 2008). It should be noted that, based on the relevance of inflammation to disease outcome, especially cardiovascular disease, guidelines regarding quantification of inflammation by using peripheral blood hsCRP have been suggested, including the designation of low (hsCRP<1 mg/l), medium (hsCRP 1–3 mg/l), and high (hsCRP >3 mg/l) inflammation (Ridker, 2003). Given the potential targeting of inflammatory pathways for depression treatment, such guidelines can provide useful starting points for the identification of sub-groups of depressed patients who may be most appropriate for immune-targeted therapies.
Cytokine Administration Induces Depressive Symptoms
A second major body of data that has supported the notion that inflammation may have a role in depression is the findings that administration of inflammatory cytokines or cytokine inducers can induce a depressive-like behavior in both laboratory animals and humans. For example, acute administration of endotoxin and typhoid vaccination to humans leads to a host of behavioral changes, including depressed mood, fatigue, and cognitive dysfunction (Reichenberg et al, 2001; Brydon et al, 2008; Harrison et al, 2009a, 2009b; Eisenberger et al, 2010; Hannestad et al, 2011). Chronic administration of the inflammatory cytokine, interferon-α (IFN-α) has also been found to induce depressive symptoms, with as many as 30–50% of IFN-α-treated patients meeting the symptom criteria for major depression (according to the Diagnostic and Statistical Manual IV) depending on the dose (Musselman et al, 2001; Raison et al, 2005a, 2005b). Interestingly, a comparison of depressive symptoms in patients with IFN-α-induced depression vs ostensibly medically healthy depressed patients revealed a large degree of overlap in both symptom expression and symptom severity (Capuron et al, 2009). Differences were related primarily to increased psychomotor slowing and anorexia, and decreased feelings of guilt and self-reproach in IFN-α treated patients vs medically healthy depressed controls. Further supporting the similarity between depression associated with IFN-α and depression in other populations of depressed individuals is the capacity of IFN-α-induced depression to be prevented by the pre-administration of conventional antidepressant medications, including paroxetine. Indeed, randomized, placebo-controlled clinical trials in both patients with malignant melanoma and hepatitis-C have shown that pretreatment with paroxetine can significantly reduce the development of depressive symptoms, especially in patients with elevated symptoms of depression at baseline (Musselman et al, 2001; Raison et al, 2007). Baseline levels of depression are one of the strongest predictors of the development of depression during IFN-α therapy (Raison et al, 2005a, 2005b). Of note, endotoxin-induced behavioral changes can also be reduced by antidepressant pretreatment (Hannestad et al, 2011).
Stress Increases Inflammation
Psychosocial stressors are well-established precipitants of episodes of major depression (Kendler et al, 1999). Thus, data that acute and chronic psychosocial stress can activate the inflammatory response represent a major breakthrough in the link between stress and depression as it relates to the immune system. One of the important early findings in this area was that exposure to the Trier Social Stress Test (TSST), a public speaking and mental arithmetic stressor, was associated with a significant increase in the DNA binding of the inflammatory transcription factor NF-κB in peripheral blood mononuclear cells (PBMCs) as measured by electromobility shift assay and compared with individuals who were spectators of the task (Bierhaus et al, 2003). Interestingly, major depression appears to augment the inflammatory response to stress as demonstrated by exaggerated increases in NF-κB DNA binding in PBMCs as well as increased plasma IL-6 in response to the TSST in patients with major depression as compared with healthy controls (Pace et al, 2006). Of note, patients with major depression in this latter study were also victims of early-life stress, and similar heightened IL-6 responses to acute psychosocial stress have recently been described in non-depressed individuals exposed to early-life stress (Carpenter et al, 2010). These data further support the notion that there is an interaction between early-life stress (and/or childhood maltreatment) and depression that may predispose to increased inflammation both under baseline conditions and following stress. This vulnerability to increased inflammatory responses may also contribute to the medical consequences of both depression and early-life adversity (Brown et al, 2009).
In terms of the mechanisms by which stress activates the inflammatory response, attention has been focused on the sympathetic nervous system (SNS). For example, catecholamines acting through α- and β-adrenergic receptors have been shown to increase cytokine expression in both the brain and the periphery of rats (Johnson et al, 2005), and α- adrenergic antagonists were noted to block the increased peripheral blood concentrations of IL-6 induced by altitude stress in humans (Mazzeo et al, 2001). In addition, α- and β-adrenergic agonists have been shown to directly activate NF-κB in vitro (Bierhaus et al, 2003). Nevertheless, the impact of the SNS on the immune system is complex and involves both stimulatory and inhibitory aspects (Nance and Sanders, 2007).
It should be noted that there is an emerging literature that indicates that the parasympathetic nervous system is also involved in immune regulation (Tracey, 2009). Stimulation of efferent parasympathetic nervous system fibers, including the motor vagus, has been shown to reduce mortality secondary to endotoxin administration in laboratory rats, while also reducing the endotoxin-induced activation of NF-κB as well as TNF-α (Tracey, 2009). These inhibitory effects on the inflammatory response have been shown to be mediated by the vagal release of acetylcholine, which in turn activates the α7 subunit of the nicotinic acetylcholine receptor (nAChR), which can regulate both cytokine transcription and translation (often referred to as the ‘cholinergic anti-inflammatory pathway’) (Tracey, 2009). In addition, recent data suggest that there may be a cellular component to this inhibitory cholinergic reflex. Indeed, adoptive transfer of T effector cells from vagotomized mice was shown to aggravate colitis in association with increased inflammatory scores and reduced regulatory T cells (T regs) (O’Mahony et al, 2009). Consistent with the Yin-Yang influence of the SNS and parasympathetic nervous systems on inflammation are studies showing a relationship between reduced heart rate variability, a reflection of reduced parasympathetic tone, and increased inflammatory biomarkers, including IL-6 and CRP, in medically healthy individuals as well as patients with major depression and heart disease (Frasure-Smith et al, 2009; Thayer and Fischer, 2009).
MECHANISMS OF CYTOKINE EFFECTS ON THE BRAIN
Given the association of depression with increased inflammatory biomarkers and the capacity of inflammatory stimuli and cytokines to induce depressive symptoms, there has been considerable attention paid to the mechanisms by which cytokines influence behavior.
Cytokine Signals Access the Brain
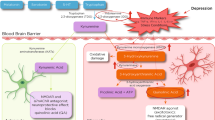
Cytokines are relatively large proteins (ranging from 6–70 kDa) and therefore do not readily pass through the blood–brain barrier (BBB). Research has therefore focused on how cytokine signals reach the brain (for review see Quan and Banks, 2007). At least five potential pathways have been described, including (1) passage through leaky regions in the BBB such as the circumventricular organs, which are highly vascularized structures that have permeable, fenestrated capillaries located around the third and fourth ventricles (Breder et al, 1988; Komaki et al, 1992; Ericsson et al, 1994); (2) active transport through saturable transport molecules (Banks et al, 1989); (3) activation of cells lining the cerebral vasculature (including endothelial cells and perivascular macrophages), which then release cytokines and other inflammatory mediators into the brain parenchyma (the pathway responsible for the febrile response to endotoxin) (Fabry et al, 1993; Cao et al, 1997); (4) binding to cytokine receptors associated with peripheral afferent nerve fibers (eg, the vagus nerve), which then relay cytokine signals to relevant brain regions, including the nucleus of the solitary tract and hypothalamus (the so called ‘neural route’) (Bluthe et al, 1994; Ericsson et al, 1994; Watkins et al, 1994); and (5) recruitment of activated cells such as monocytes/macrophages from the periphery to the brain, where these cells can in turn produce cytokines (D’Mello et al, 2009) (Figure 1). It should be mentioned, however, that these pathways by which cytokine signals access the brain have been elucidated through the use of experimental strategies, which yield peripheral blood cytokine concentrations well above what have been described in depression and/or stress. Therefore, it remains unclear which pathways may be most relevant for transmission of cytokine signals to the brain under these conditions. Of note, studies have indicated that NF-κB at the blood–brain interface is an important molecule in transmitting cytokine signals to the brain. Indeed, central blockade of NF-κB in rodents was shown to inhibit c-fos activation in multiple brain regions following peripheral administration of IL-1β, while also inhibiting IL-1β and endotoxin-induced behavioral changes (Godbout et al, 2005; Nadjar et al, 2005).

Immune cell trafficking in CNS resilience and pathology. Immune cell trafficking to the brain can have important roles in mental health and illness, including (a) the delivery of anti-inflammatory molecules that can promote neuronal integrity and resilience, and (b) the communication of inflammatory signals, which may contribute to pathology. For example, (a) during stress, the production of glucocorticoids increases the expression of ICAM-1 by choroid plexus cells, which then attracts CD4+ T cells to the brain. Production of IL-4 by these T cells has been shown to shift the phenotype of meningeal myeloid cells (eg, macrophages) from an M1 (or proinflammatory) phenotype to an M2 (or anti-inflammatory) phenotype. In addition, IL-4, which enters the circulation or the CSF, can diffuse into brain parenchyma and induce glial elements, including astrocytes, to produce BDNF, which promotes neurogenesis and synaptic plasticity. By contrast, (b) peripherally elaborated TNF-α can lead to the activation of microglia, which in turn produce the chemokine, MCP-1. MCP-1 attracts monocytes to the brain where they enter the brain parenchyma as activated macrophages, capable of producing TNF-α as well as additional inflammatory mediators such as other inflammatory cytokines and reactive nitrogen and oxygen species.
Once cytokine signals reach the brain, there is a rich cytokine network throughout the brain, including glial elements (astrocytes and microglia) and neurons, which produce cytokines and express cytokine receptors (Miller et al, 2009). The most active cytokine producing cells in the brain are microglia, the equivalent of macrophages in the brain. Of relevance to the discussions above regarding the stress-induced activation of inflammatory responses, microglia can be activated by stress and have been found to be an important substrate of stress-induced inflammatory responses in the brain of laboratory animals (Frank et al, 2007). In addition, increased microglial density has been found in several brain regions, including the dorsolateral prefrontal cortex (PFC), the anterior cingulate cortex, and the mediodorsal thalamic nucleus of suicide victims with affective disorders (major depression and bipolar disorder, depressed) as well as schizophrenia (Steiner et al, 2008).
Cytokines Alter Neurotransmitter Metabolism
Probably best studied of the mechanisms by which cytokines influence behavior is their effects on neurotransmitter metabolism. Numerous human and laboratory animal studies have demonstrated that multiple neurotransmitter systems are affected by acute and chronic administration of cytokines, including monoamines, serotonin, and dopamine, as well as glutamate (Dunn and Wang, 1995; Anisman et al, 2008; Miller et al, 2009).
Cytokine effects on monoamine metabolism The role of monoamines, especially serotonin, in the development of depressive symptoms during cytokine exposure in humans has been the focus of a number of studies. Early demonstration that serotonin reuptake inhibitors could prevent and/or treat depressive symptoms during chronic exposure to IFN-α in patients with infectious diseases and cancer provided strong evidence that serotonin pathways are likely involved in the cytokine effects on behavior (Musselman et al, 2001; Raison et al, 2007). Genetic studies have complemented this work indicating that polymorphisms in the promoter region of the serotonin transporter (SERT) gene (5-HTTLPR) are associated with IFN-α-induced behavioral changes in patients with hepatitis-C. More specifically, in two independent studies, the L(A) allele was associated with a decreased likelihood of developing major depression or depressive symptoms during IFN-α administration (Bull et al, 2008; Lotrich et al, 2009). Further implicating alterations in serotonin metabolism in cytokine-induced behavioral changes, data have shown that IFN-α-associated increases in the cerebrospinal fluid (CSF) concentrations of IL-6 were negatively correlated with the serotonin metabolite, 5-hydroxyindoleacetic acid (5-HIAA), which was in turn negatively correlated with IFN-α-induced depression severity (Raison et al, 2009).
In conjunction with these findings, data have suggested that cytokines have a significant impact on dopamine pathways as well. As discussed below, neuroimaging studies have indicated altered blood flow and metabolic activity in basal ganglia nuclei during exposure to inflammatory stimuli (Juengling et al, 2000; Capuron et al, 2007; Brydon et al, 2008), and in studies in non-human primates, reduced CSF concentrations of the dopamine metabolite, homovanillic acid, were associated with depressive-like huddling behavior secondary to chronic IFN-α administration (Felger et al, 2007). Of note, early observations of huddling behavior in non-human primates were made in animals following chronic administration of the monoamine-depleting drug, reserpine (McKinney et al, 1971). Rodent studies have also indicated that cytokines target the basal ganglia and dopamine pathways. For example, acute administration of a high dose of endotoxin (5 mg/kg) to adult mice led to an almost 50% decrease in tyrosine hydroxylase-expressing neurons in the substantia nigra after 10 months (Qin et al, 2007), and chronic administration of high-dose endotoxin (2 weeks at 5 ng/h) into the brain was associated with a 70% reduction of nigral dopaminergic neurons within 10 weeks (Gao et al, 2002). These effects appear to be mediated in part by the endotoxin-induced production of TNF-α and the related activation of oxidative stress (Gao et al, 2002; Qin et al, 2007).
Mechanisms by which cytokines affect monoamine metabolism. Several mechanisms have received special attention regarding the impact of cytokines on monoamine metabolism that may ultimately serve as targets for pharmacologic intervention (discussed below).
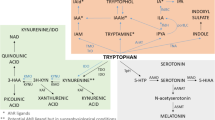
Indoleamine-2,3-dioxygenase. Indoleamine-2,3-dioxygenase (IDO) is an enzyme expressed in multiple cell types, including macrophages, dendritic cells, microglia, astrocytes, and neurons (Guillemin et al, 2005a, 2005b, 2005c; Huang et al, 2010). IDO is activated by a number of cytokines alone or in combination, including IFN-γ, TNF-α, IL-1, and IL-6, through stimulation of cytokine-signaling pathways such as signal transducer and activator of transcription (STAT)-1, IFN-regulatory factor-1, p38 mitogen-activated protein kinase (MAPK), and NF-κB (Pemberton et al, 1997; Fujigaki et al, 2006). Relevant to serotonin metabolism, IDO catabolizes tryptophan, the primary amino-acid precursor of serotonin, into kynurenine (Figure 2). Depletion of tryptophan inhibits effector T-cell responses and thereby contributes to immune tolerance (Huang et al, 2010). Evidence of a role of IDO in cytokine-induced depression comes from a number of studies, which have demonstrated correlations between IFN-α-induced depression and decreases in tryptophan, and increases in kynurenine and/or the kynurenine-to-tryptophan ratio (Bonaccorso et al, 2002; Capuron et al, 2003a, 2003b). In addition, studies have shown that increases in plasma kynurenine are manifested as increased kynurenine in the CSF of IFN-α-treated patients (Raison et al, 2010a, 2010b, 2010c).

IDO and the kynurenine pathway in inflammation-induced CNS pathology. Cytokine-induced activation of IDO in peripheral immune cells (eg, macrophages and dendritic cells) or cells in the brain (eg, microglia, astrocytes, and neurons) leads to the production of kynurenine, which is converted to KA by KAT-II in astrocytes or quinolinic acid by kynurenine-3-monooxygenase (KMO) and 3-hydroxy-anthranilic acid oxygenase (3-HAO) in microglia or infiltrating macrophages. Through blockade of the α7nAChR, KA can reduce glutamate release as well as the release of dopamine, both of which can contribute to cognitive dysfunction. By contrast, quinolinic acid through activation of the NMDA receptor can increase glutamate release as well as lead to lipid peroxidation, thus contributing to excitotoxicity, oxidative stress, and ultimately neurodegeneration.
Although much of the attention regarding IDO was initially focused on the depletion of tryptophan and in turn serotonin, data from laboratory animals have demonstrated that intraperitoneal administration of kynurenine can induce a depressive-like behavior as reflected by increased immobility in the forced swim test (FST) and the tail suspension test (TST) (O’Connor et al, 2008). In the CNS, kynurenine can be further catabolized into the neuroactive metabolites kynurenic acid (KA) and quinolinic acid, both of which have also been found to be increased in the CSF of IFN-α-treated patients (Schwarcz and Pellicciari, 2002; Raison et al, 2010a, 2010b, 2010c) (Figure 2). Quinolinic acid, which is produced primarily in microglia and infiltrating macrophages, can directly bind to the N-Methyl-D-aspartate (NMDA) receptor, leading to the release of glutamate (Schwarcz and Pellicciari, 2002). Quinolinic acid is also associated with lipid peroxidation and oxidative stress (Rios and Santamaria, 1991; Schwarcz and Pellicciari, 2002). In combination, these activities of quinolinic acid can lead to excitotoxicity in the brain, and therefore, excessive quinolinic acid has been implicated in a number of neurodegenerative disorders, including Huntington's disease, Amyotrophic Lateral Sclerosis, Alzheimer's disease, and dementia secondary to infection with HIV (Schwarcz and Pellicciari, 2002; Guidetti and Schwarcz, 2003; Guillemin et al, 2005a, 2005b, 2005c). Of, note increased concentrations of CSF quinolinic acid were significantly correlated with depressive symptoms in IFN-α-treated patients (Raison et al, 2010a, 2010b, 2010c).
In contrast to quinolinic acid, KA, which is produced primarily in astrocytes, reduces glutamate release and thereby has downstream effects on the release of dopamine (which is in part under glutamatergic control) (Schwarcz and Pellicciari, 2002) (Figure 2). For example, intra-striatal administration of KA to rodents leads to marked reductions in extracellular dopamine concentrations as determined by in vivo microdialysis (Wu et al, 2007). In addition, in mice with targeted deletion of the gene for kynurenine aminotransferase-II (KAT-II), a major biosynthetic enzyme of brain KA, brain KA concentrations were reduced and the mice showed significant increases in cognitive performance (object recognition, spatial discrimination, passive avoidance) compared with wild-type animals (Potter et al, 2010).
Mitogen-activated protein kinase. Another pathway that can influence monoamine metabolism is the cytokine-signaling pathway, MAPK. Both in vitro and in vivo data have established that stimulation of p38 MAPK pathways can increase the expression and function of the SERT. For example, treatment of mouse midbrain and striatal synaptosomes with IL-1β and TNF-α was shown to lead to a dose- and time-dependent increase in serotonin reuptake, which was reversed by the p38 antagonist, SB203580 (Zhu et al, 2006). These results were also associated with direct effects of cytokine-stimulated p38 MAPK on both the Vmax and Km of the SERT (Zhu et al, 2005, 2006). Interestingly, in vivo treatment with endotoxin also led to increased SERT activity paralleled by increased immobility in the FST and TST (Zhu et al, 2010). Endotoxin-associated increases in SERT activity as well as depressive-like behaviors were reversed by p38 MAPK inhibition by using the pharmacologic p38 antagonist, SB203580. Consistent with an in vivo role of p38 MAPK in serotonin metabolism is the correlation found between increased phosphorylated (activated) p38 in peripheral blood monocytes as determined by flow cytometry and decreased CSF concentrations of 5-HIAA in Rhesus monkeys exposed to early maternal neglect and abuse (Sanchez et al, 2007). In addition, increased phosphorylation of p38 MAPK following the first injection of IFN-α was associated both the acute cortisol response to IFN-α as well as the subsequent development of IFN-α-induced depression and fatigue (Felger et al, 2011).
It should be noted that MAPK pathways have also been found to influence the dopamine transporter. For example, hDAT-expressing cells transfected with a constitutively activate MAPK kinase (MEK) show increased DA reuptake (Vmax), whereas treatment of rat striatal synaptosomes with MEK inhibitors was associated with decreased DA reuptake in a concentration and time-dependent manner (Moron et al, 2003).
Tetrahydrobiopterin. Tetrahydrobiopterin (BH4) is an essential enzyme cofactor for tryptophan hydroxylase and tyrosine hydroxylase, which are the rate-limiting enzymes in the synthesis of serotonin and dopamine/norepinephrine, respectively (Neurauter et al, 2008) (Figure 3). BH4 is also a cofactor for nitric oxide (NO) synthase, which converts arginine to NO (Neurauter et al, 2008). BH4 is highly labile and highly sensitive to non-enzymatic oxidation, which leads to an irreversible degradation of BH4 to dihydroxyanthopterin (XPH2) (Neurauter et al, 2008). Of relevance to inflammation, intramuscular injection of IFN-α to rats has been shown to decrease the CNS concentrations of BH4 through stimulation of NO (Kitagami et al, 2003). Indeed, treatment with an inhibitor of NO was found to reverse IFN-α's inhibitory effects on the brain concentrations of both BH4 and DA (Kitagami et al, 2003). IL-6 also has been shown to reduce BH4 content in sympathetic neurons (Li et al, 2003). Of note, activation of an inflammatory response and microglia within the brain is associated with increased NO production (Zielasek and Hartung, 1996), suggesting that cytokine influences on BH4 through NO and oxidative stress may be a common mechanism for cytokines and inflammation to reduce monoamine availability in relevant brain regions. Because BH4 also is a cofactor for phenylalanine-4-hydroxylase (PAH), which converts phenylalanine (phe) to tyrosine (tyr), investigators have used peripheral blood measures of phe, tyr, and/or the phe/tyr ratio as a measure of PAH activity and indirectly the availability of BH4 (Neurauter et al, 2008). A number of patient populations with increased inflammation have been found to show increased peripheral blood phe concentrations, including patients with trauma, sepsis, cancer, and HIV (Neurauter et al, 2008). In addition, correlations have been found between peripheral blood phe concentrations and IL-6, IL-2 receptor, soluble TNF-α receptor-2, as well as peripheral blood markers of oxidative stress such as isoprostane-8 in patients with cancer (Neurauter et al, 2008). Moreover, in a recent study of healthy elderly persons with low-grade inflammation, the peripheral blood concentrations of phe, tyr, and the phe/tyr ratio were associated with depressive symptoms such as anhedonia and altered sleep (Capuron et al, 2011).

Tetrahydrobiopterin: Target for inflammatory effects on neurotransmitter metabolism. Tetrahydrobiopterin (BH4) is a critical cofactor for the rate-limiting enzymes involved in the synthesis of the monoamine neurotransmitters, including (1) the synthesis of tyrosine (tyr) from phenylalanine (phe) by PAH; (2) the synthesis of L-3,4-dihydroxyphenylalanine (L-DOPA) from tyrosine (tyr) by tyrosine hydroxylase (TH) leading to the production of dopamine and norepinephrine; and (3) the synthesis of 5-hydoxy-L-tryptophan (5-HTP) from tryptophan (tryp), leading to the production of serotonin. In addition, BH4 is a cofactor for the enzyme NOS, which converts arginine (arg) to NO. During these enzymatic reactions, BH4 is degraded to BH2, which can be regenerated to BH4 through pathways supported by folic acid, L-methylfolate, and SAMe. BH4 is relatively unstable and in the context of inflammation and oxidative stress can undergo non-enzymatic oxidation leading to the irreversible degradation of BH4 to XPH2.
Cytokine effects on glutamate metabolism Aside from effects on monoamines, another neurotransmitter target of inflammation is glutamate. Cytokines have been shown to have profound effects on glutamate metabolism, including a rich literature demonstrating that cytokines can (1) decrease the expression of glutamate transporters on relevant glial elements and (2) increase the release of glutamate from astrocytes (Bezzi et al, 2001; Pitt et al, 2003; Volterra and Meldolesi, 2005; Tilleux and Hermans, 2007; Ida et al, 2008). Of note, glutamate released by astrocytes has preferential access to extrasynaptic NMDA receptors, which can mediate excitotoxicity and lead to decreased production of trophic factors, including brain-derived neurotrophic factor (BDNF) (Hardingham et al, 2002; Haydon and Carmignoto, 2006). Relevant to depression, increased glutamate has been found in the frontal cortex of patients with mood disorders (Hashimoto et al, 2007). As noted above, quinolinic acid can lead to glutamate release through direct activation of the NMDA receptor, while contributing to oxidative stress. Cytokines including TNF-α and IL-1 can also induce both astrocytes and microglia to release reactive oxygen and nitrogen species that can amplify oxidative stress and impair glutamate reuptake, while stimulating glutamate release and endangering relevant cell types, including oligodendrocytes, which are especially vulnerable to oxidative damage and over-activation of calcium-permeable glutamate receptors (Matute et al, 2006; Ida et al, 2008; Matute, 2011). Interestingly, loss of glial elements such as oligodendrocytes in multiple mood-relevant brain regions, including the subgenual PFC and amygdala, has emerged as a fundamental morphologic abnormality in major depression (Ongur et al, 1998; Hamidi et al, 2004; Rajkowska and Miguel-Hidalgo, 2007).
Cytokine Effects on Neurogenesis
Neurogenesis has emerged as an important process in the development of depression and the activity of antidepressant medications (Duman and Monteggia, 2006). A number of antidepressants have been shown to increase neurogenesis in the brain, and some of the behavioral effects of these drugs are dependent on the stimulation of neurogenesis (Duman and Monteggia, 2006; David et al, 2009). Chronic stress has been shown to inhibit neurogenesis, which in turn has been associated with the development of depressive-like behavior in laboratory animals (Duman and Monteggia, 2006). In terms of the role of cytokines in these findings, data have shown that stress-induced decreases in neurogenesis as well as the expression of relevant nerve growth factors, including BDNF, which support neurogenesis, can be reversed by the administration of an IL-1 receptor antagonist (IL-1ra) or transplantation of IL-1ra-secreting neural precursor cells into the hippocampus, or by the use of IL-1 receptor-knockout (KO) mice (Barrientos et al, 2003; Ben Menachem-Zidon et al, 2008; Koo and Duman, 2008). Cytokine antagonism or the use of cytokine-KO animals has also been shown to reverse the stress-induced effects on depressive-like behavior in these studies. These data suggest that stress-induced activation of inflammatory pathways in the brain lead to the release of inflammatory cytokines such as IL-1, which in turn influence fundamental aspects of neural plasticity such as neurogenesis and ultimately behavior. Of note, in vitro studies indicate that the inhibitory effect of IL-1 on neurogenesis is mediated by the activation of NF-κB (Koo and Duman, 2008).
Cytokine Effects on Neuroendocrine Function
Alterations in the hypothalamic–pituitary–adrenal (HPA) axis are some of the most reproducible findings in patients with major depression. Patients with major depression have been found to show increased concentrations of the HPA axis hormones, ACTH and cortisol, as well as increases in the CSF measures of the HPA axis-regulatory neuropeptide, corticotrophin-releasing hormone (CRH) (Pariante and Miller, 2001; Pariante and Lightman, 2008). Of relevance to inflammation, administration of inflammatory cytokines to laboratory animals has been shown to profoundly stimulate not only ACTH and cortisol but also the expression and release of CRH (Besedovsky and del Rey, 1996). Human studies also have demonstrated that the acute ACTH and cortisol response to the first injection of IFN-α (presumably due to activation of CRH pathways) is robust and correlates with the subsequent development of depressive symptoms during IFN-α therapy in patients with cancer (Capuron et al, 2003a, 2003b). In contrast to acute administration of IFN-α, chronic IFN-α administration is associated with flattening of the diurnal curve and increased evening cortisol concentrations, both of which correlate with the development of depression and fatigue (Raison et al, 2010a, 2010b, 2010c). Flattening of the diurnal cortisol rhythm has been seen in a number of medical disorders associated with inflammation, including cardiovascular disease and cancer, where it has been associated with a worse outcome in these diseases (Raison et al, 2010a, 2010b, 2010c). Indeed, patients with metastatic breast cancer with a flattened cortisol slope were found to show decreased survival as well as decreased natural killer-cell number and activity (Sephton et al, 2000). Of note, previous studies have shown that the flattened cortisol rhythm is associated with the non-suppression of cortisol by dexamethasone (DEX) in the DEX suppression test (DST) (Spiegel et al, 2006). DST non-suppression is a common finding in major depression both in vivo and in vitro in PBMCs (Pariante and Miller, 2001; Pariante, 2004; Pariante and Lightman, 2008). DEX non-suppression, also referred to as glucocorticoid resistance, has additionally been correlated with the stimulated production of IL-1β by PBMCs in patients with major depression (Maes et al, 1993). Moreover, increased TNF-α has been associated with glucocorticoid resistance in the skin as measured by reduced cutaneous blanching in response to topically applied glucocorticoids (Fitzgerald et al, 2006).
It is generally believed that glucocorticoid resistance is a result of decreased expression and/or function of the receptor for glucocorticoids. Of relevance in this regard, there is a rich literature indicating that inflammatory cytokines can disrupt glucocorticoid receptor (GR) function while decreasing GR expression (Pace et al, 2007). For example, IFN-α has been shown to inhibit GR function by activating STAT-5, which in turns binds to the activated GR in the nucleus, thus disrupting GR–DNA binding (Hu et al, 2009). A similar protein–protein interaction between the GR and NF-κB in the nucleus has also been described (Smoak and Cidlowski, 2004). On the other hand, IL-1α and β have been shown both in vitro and in vivo to inhibit GR translocation from the cytoplasm to the nucleus through activation of p38 MAPK (Pariante et al, 1999; Wang et al, 2004; Engler et al, 2008). Of note, stress-induced alterations in GR translocation leading to glucocorticoid resistance in laboratory mice have been found to be mediated by IL-1 through the use of IL-1-KO mice, who fail to show impaired GR translocation following social disruption stress (Engler et al, 2008). Given the potent anti-inflammatory effects of glucocorticoids, it is not surprising that glucocorticoid resistance secondary to stress in mice is associated with increased lethality in response to endotoxin administration (Quan et al, 2001). Thus the effects of cytokines on GR function may lead to a feed-forward cascade, whereby increased inflammation through its effects of the GR undercuts the well-known ability of glucocorticoids to restrain inflammatory responses (Rhen and Cidlowski, 2005), leading to further increases in inflammation and reduced GR function. Finally, it should be noted that chronic exposure to inflammatory cytokines can lead to decreased GR expression as well as increased expression of GR-β, a GR isoform that has a distinct hormone-binding domain (that is unable to bind to known glucocorticoids) and a unique pattern of gene regulation (Pace et al, 2007; Kino et al, 2009). Of note, GR-β expression has been shown to be increased in patients with inflammatory disorders, including patients with bronchial asthma and rheumatoid arthritis (Kino et al, 2009).
Impact of Cytokines on the Neurocircuitry
As interest in the impact of inflammation on the brain has grown, there has been an emerging neuroimaging literature examining the brain regions that are most affected by the administration of inflammatory stimuli. Brain regions that have been most reliably identified include the basal ganglia and the dorsal anterior cingulate cortex (dACC).
Basal ganglia Using positron emission tomography, early studies of patients undergoing IFN-α therapy for cancer or hepatitis-C revealed marked increases in glucose metabolic activity in the basal ganglia, which correlated with symptoms of fatigue (Juengling et al, 2000; Capuron et al, 2007). These increases are consistent with the metabolic changes in the basal ganglia found in patients with Parkinson's disease, and are believed to reflect increased oscillatory burst activity in relevant basal ganglia nuclei secondary to DA depletion (Eidelberg et al, 1994; Spetsieris et al, 1995; Wichmann and DeLong, 1999; Mentis et al, 2002). Increased metabolic activity in the basal ganglia of Parkinson's disease patients can be reversed by the administration of levodopa (Feigin et al, 2001). Of note, IFN-α has been associated with the development of Parkinson-like symptoms that were relieved by levodopa administration (Bersano et al, 2008). Using fMRI, typhoid vaccination was also found to alter activation in the basal ganglia. In particular, vaccinated volunteers (compared with a control condition) showed increased evoked activity in the substantia nigra, which was associated with both prolonged reaction times and increased peripheral blood concentrations of IL-6 (Brydon et al, 2008). Finally, administration of endotoxin to healthy volunteers led to a reduced activation in the ventral striatum during a monetary reward task (Eisenberger et al, 2010). Between-group differences in striatal activation were in turn associated with increases in depressed mood.
The impact of cytokines on the basal ganglia and ultimately motor activity and motivation has been suggested to be an evolutionarily derived process, which may serve to reduce exploratory behavior and motor activity in order to facilitate the reallocation of energy resources to fighting infection, mounting a fever and wound-healing.
Anterior cingulate cortex Another brain region that has been found to be influenced by cytokine administration and inflammatory stimuli is the dACC. The dACC has an important role in error detection and conflict monitoring (Carter et al, 1998). Patients with hepatitis-C treated with IFN-α were found to have a significantly greater activation of the dACC (Broadman's Area 24) using fMRI and a task of visuospatial attention (Capuron et al, 2005). Interestingly a strong correlation was found in this study between activation of the dACC in IFN-α-treated patients and the number of errors made during the task. The error rate was quite low for the task, and no such correlation was found in control subjects. Of note, patients with high-trait anxiety have also been shown to show increased dACC activation during fMRI in the context of low error rates (Paulus et al, 2004). In addition, increased activation of the dACC has been observed in subjects with neuroticism and obsessive compulsive disorder, both of which are associated with increases in anxiety as well as arousal (Ursu et al, 2003; Eisenberger et al, 2005). Aside from IFN-α, increased activation of the dACC has been found using fMRI after a high-demand color word Stroop task in patients administered typhoid vaccination (Harrison et al, 2009a, 2009b). Moreover, increased activation of the dACC after a psychosocial stressor (TSST) was correlated with the degree of activation of peripheral blood IL-6, suggesting a role for stress-induced inflammation in the findings (Slavich et al, 2010).
Based on fMRI studies and a social rejection task, it has been suggested by Eisenberger and Lieberman (2004) that the dACC not only has a role in error detection and conflict monitoring, but also processes social pain. Given the connection of the dACC with downstream autonomic nervous system arousal pathways, these investigators have further hypothesized that the dACC may serve as a ‘neural alarm system’ that can both detect and respond (with arousal and distress) to threatening environmental stimuli in the social domain (Eisenberger and Lieberman, 2004). Thus, cytokines may sensitize the responsivity of the dACC, thereby contributing to the anxiety, arousal, and alarm that often accompany chronic exposure to inflammatory stimuli such as IFN-α. From an evolutionary perspective, this heightened dACC activity (and heightened sensitivity to social threat) may subserve the survival priority of vigilance against attack in an animal that is otherwise vulnerable because of infection and/or wounding (Miller, 2008a, 2008b). Taken together with the effects of inflammation on the basal ganglia, the effects of inflammation on the neurocircuitry in the brain appear to subserve two competing evolutionary survival priorities: to lay low to conserve energy resources for fighting infection and wound-healing, and to be vigilant for protection against future attack (Miller, 2008a, 2008b). Finally, it should be noted that administration of an inflammatory stimulus (typhoid vaccination) has been associated with activation of the subgenual ACC, the target of deep brain stimulation strategies in patients with treatment-resistant depression (Lozano et al, 2008).
SOURCES OF PERIPHERAL INFLAMMATION IN DEPRESSION
In looking forward to the translational implications of the impact of inflammatory processes on the brain as they relate to depression, a question that is often asked is whether pharmacologic treatments must enter the brain to be effective, or whether strategies that solely target peripheral inflammation will suffice. There is no clear answer to this question as yet, and although it is very likely that drugs that act both in the periphery and the brain may have some advantage, in the absence of frank evidence of an active inflammatory response in postmortem brain samples from patients with major depression, there are not sufficient data to suggest that drugs must enter the brain in order to impart benefit. Findings from patients peripherally administered inflammatory cytokines (IFN-α) indicate that peripheral cytokines have ready access to the brain and can induce a central inflammatory response, including the release of chemokines such as monocyte chemoattractant protein (MCP)-1 and other cytokines such as IL-6 (Raison et al, 2009). Moreover, biologic therapies such as the TNF-α antagonist, etanercept, which is not believed to appreciably enter the brain, was shown to be effective in reducing depressive symptoms in patients with the peripheral immune disorder, psoriasis (see below) (Tyring et al, 2006). Given this notion that peripheral inflammatory responses can spread readily to the brain and that drugs that target peripheral immune responses can improve depressive symptoms, there has been interest in the potential peripheral sources of chronic, non-resolving inflammation that may serve as targets for treatment and prevention strategies (see below) (Nathan and Ding, 2010) (Figure 4).

Factors contributing to chronic, non-resolving inflammation and disease. Numerous environmental and biological factors can conspire to contribute to chronic inflammation, including stress, adiposity, dietary intake, the bacterial composition of the gut microbiota, and the relative balance of T-cell subpopulations, including proinflammatory Th-17 cells and anti-inflammatory T regs. Through both epidemiologic and mechanistic studies, chronic inflammation, in turn, is now recognized to be a common pathway to pathology, having a role in a diverse set of illnesses, including cardiovascular disease, diabetes, cancer and depression. Strategies targeting these contributors to chronic inflammation represent an important approach to the prevention and treatment of these diseases.
Stress
A primary source of non-resolving inflammation in patients with major depression is likely to be psychosocial stress. Both acute and chronic stress have been shown in a multitude of studies to be important predictors of depression (Kendler et al, 1999), and as indicated above, both acute and chronic psychosocial stress, as well as early-life stress, have been associated with increased inflammatory markers in both laboratory animals and humans. Activation of fundamental inflammatory pathways, including NF-κB as well as impaired HPA axis regulation of inflammatory signaling pathways, is believed to be involved in the impact of chronic stress on inflammation. For example, in a study of familial caregivers for patients with brain cancer, microarray analyses of peripheral blood monocytes revealed that, compared with controls, caregivers showed a heightened expression of gene transcripts with response elements for NF-κB in conjunction with diminished expression of transcripts bearing response elements for the GR (Miller et al, 2008). Interestingly, whereas caregivers also showed significant elevations in the biomarkers of inflammation such as CRP, there were no differences between the groups in diurnal cortisol secretion, suggesting that peripheral glucocorticoid resistance at the level of the GR (in the absence of alterations in cortisol secretion) was ultimately related to increased inflammatory signaling. Of note, similar microarray data have been found in lonely vs non-lonely individuals, consistent with the notion that increased inflammation may be a mediator of the negative impact of social isolation on both mental and physical health (Cole et al, 2007).
Diet and Obesity
Another pathway to inflammation that has received increasing attention is obesity. Obesity is an epidemic in Western societies, and data exist that there is a bidirectional relationship between obesity and depression (Shelton and Miller, 2010). Increased body mass index (BMI), a measure of obesity, is associated with an increased likelihood of developing depression, and depression in turn is predictive of the development of an increased BMI (Vogelzangs et al, 2008; Luppino et al, 2010). Relevant to inflammation, when adipocytes expand they can compress the local vasculature, leading to hypoxia and mitochondrial dysfunction, ultimately contributing to the induction of oxidative stress and adipocyte necrosis (Nathan, 2008). These processes in addition to the production of MCP-1 by adipocytes can attract macrophages to accumulate in adipose tissue where they produce inflammatory mediators, including the inflammatory cytokines, TNF-α, IL-1β, and IL-6 (Nathan, 2008; Shelton and Miller, 2010). Adipose tissue, white adipose tissue (WAT) in particular, can contain large numbers of macrophages, and abdominal WAT (especially intra-abdominal WAT) produces a greater effect on systemic inflammation than other sites of WAT accumulation, possibly related to its proximity to the portal circulation (Shoelson et al, 2007). Of note, in a sample of women with severe or morbid obesity, BMI was positively correlated with inflammatory markers (eg, IL-6 and hsCRP), which were in turn correlated with depression (Capuron et al, 2010). Abdominal obesity in women also has been associated with increased IL-1ra responses to mental stress, and women with higher basal plasma concentrations of leptin, a hormone produced by adipose tissue that is elevated in obesity, showed greater stress-induced increases in IL-6 (Brydon, 2011). These data indicate that stress and obesity can interact to exaggerate inflammatory responses.
Related to the obesity epidemic, is the change in the Western diet, whereby in the last century there has been a dramatic shift toward a greater representation of n-6 (or omega 6) polyunsaturated fatty acids (PUFAs) in relation to n-3 fatty acids (Kiecolt-Glaser, 2010). n-3 fatty acids are found in large quantities in fish and fish oil extracts, and there is evidence that increased consumption of fish is associated with a reduced prevalence of depressive disorders (Shelton and Miller, 2010). Moreover, in a recent meta-analytic review of 14 studies comparing PUFAs in depressed individuals vs controls, depressed patients showed significantly lower docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), the major bioactive components of n-3 PUFAs, as well as decreased total n-3 PUFAs (Lin et al, 2010). Of note, postmortem studies of the frontal cortex of patients with major depression have found decreased expression of fatty acid biosynthesis genes that are responsible for converting n-3 fatty acid precursors into DHA (McNamara and Liu, 2011). In addition, a polymorphism in the phospholipase-A2 (PLA2), a key enzyme in the metabolism of PUFAs, was found to predict the development of IFN-α-induced depression, which occurred in association with lower EPA levels in the ‘at risk’ PLA2 phenotype (Su et al, 2010). Relevant to inflammation, n-6 PUFAs are metabolized into arachidonic acid and the inflammatory mediators prostaglandins and leukotrienes. Prostaglandins have been found to be reliably elevated in depressed patients (Zorrilla et al, 2001).
T-Cell Dysregulation
One of the earliest and most reproducible immunologic findings in major depression was evidence of impaired proliferative responses of lymphocytes to the T-cell mitogens concanavalin-A and phytohemagglutinin (Irwin and Miller, 2007). As noted above, there has been much speculation regarding the mechanisms of these T-cell alterations, including the impact of cytokines on T-cell receptor signaling as well as spontaneous apoptosis related to increased T-cell expression of Fas, the receptor of Fas ligand, which induces cell death through the caspase pathway (Szuster-Ciesielska et al, 2008). Nevertheless, despite both functional and genetic data indicating that alterations in T cells may be involved in depression (Wong et al, 2008), few studies have directly examined the relative expression and function of relevant T-cell subsets beyond the characterization of CD4+ and CD8+ T-cell phenotypes and mitogen-induced T-cell proliferation (Zorrilla et al, 2001; Irwin and Miller, 2007). Given recent developments in immunology regarding the role of T cells in the regulation of inflammation, the relative expression and function of T-cell subsets, including T regs and certain effector T (T eff) cells, including Th17 cells, has attracted increasing attention. T regs produce the anti-inflammatory cytokines IL-10 and transforming growth factor-β (TGF-β), and are believed to be of central importance in restraining inflammatory responses. Reduced IL-10 and TGF-β, as well as an increased IL-6/IL-10 ratio, have been found in depressed patients and are believed to be consistent with reduced T reg expression and/or function (Sutcigil et al, 2007; Dhabhar et al, 2009). In addition, two studies directly examining CD4+CD25+ T reg expression in depressed patients have found decreased T reg percentage in association with reduced IL-10 and TGF-β in depressed patients vs controls (Li et al, 2010), and increased CD4+CD25+ T reg percentage associated with decreased IL-1β following antidepressant treatment (Himmerich et al, 2010). These results support the hypothesis that CD4+CD25+ regulatory T cells may be decreased in depressed patients, and given the role of T regs in reducing inflammatory responses, may contribute to increased inflammation in certain depressed individuals.
Another T-cell subset that may be relevant to inflammation in depression are T eff cells that produce IL-17 (Th17 cells). Th17 cells have been recently recognized as a distinct, highly pro-inflammatory T-cell subset that may have a fundamental pathophysiologic role in inflammatory disorders (Acosta-Rodriguez et al, 2007; Annunziato et al, 2007; Wilson et al, 2007; Tesmer et al, 2008; Damsker et al, 2010). In conjunction with IL-6 and IL-23, IL-1β is the most effective inducer of IL-17 expression in naive T cells in humans (Boissier et al, 2008). To date, no study has examined Th17-cell expression or function in major depression. Nevertheless, based on developments regarding the regulation of inflammation by T regs and Th17-cell subsets, high inflammation in patients with major depression may be a result of T-cell dysregulation characterized by reduced T reg function in conjunction with increased Th17 activity.
Intestinal Microbiota and the Leaky Gut
It has been estimated that microbes in our bodies make up to 100 trillion cells, 10-fold the number of human cells (Lee and Mazmanian, 2010). The majority of microbes reside in the gut. Endogenous gut flora coats the small and large intestines, with a density of bacteria that peaks at 1011–1012 per gram of colonic content in the large intestine (Fleshner, 2011). The aggregate human microbiota (including bacteria from the skin, mouth, and airways) contains over 1000 bacterial species, with approximately 160 species per individual, allowing for relatively distinct bacterial composition among individuals that can be influenced by genetic and environmental factors (Lee and Mazmanian, 2010), although studies also suggest that a common bacterial species ‘core’ is shared among at least 50% of individuals (Qin et al, 2010). Recent developments in our understanding regarding the impact of these commensural microorganisms indicate that aside from assisting in digestive processes, bacteria in the gut may have an important role in the immune response, including inflammation (Lee and Mazmanian, 2010). Indeed, the presence of certain bacterial populations can influence the relative development of T regs and Th17 cells, with anti-inflammatory bacteria such as Bacteroides fragilis supporting the elaboration of T regs, and proinflammatory microbes such as segmented filamentous bacteria supporting the development of Th17 cells (Lee and Mazmanian, 2010). Interestingly, in the context of stress, bacteria have been shown to translocate from the gut to lymphoid organs in laboratory mice (Bailey et al, 2006), and treatment of mice with a cocktail of antibiotics was found to both reduce intestinal microflora while eliminating the capacity of a social disruption stressor to increase peripheral blood cytokines, including IL-6 and MCP-1 (Bailey et al, 2011). Of note, relevant to depression, patients with major depression have been found to show increased peripheral blood antibody titers to LPS derived from gram negative enterobacteria as compared with normal volunteers (Maes et al, 2008). Finally, administration of the probiotic Bifidobacterium infantis to maternally separated rats was found to reverse depressive-like behavior in the FST, reduce peripheral IL-6 concentrations, and normalize basal norepinephrine concentrations in the brainstem (Desbonnet et al, 2010). Taken together, these data suggest that the composition of intestinal microbiota as well as their translocation from the gut into the body (leaky gut) in the context of stress may contribute to inflammation as it relates to depression and depressive-like behavior.
TRANSLATIONAL IMPLICATIONS: TREATMENT TARGETS UNIQUE TO IMMUNOLOGICAL CONTRIBUTIONS
Immunological Targets
Cytokines. Cytokines are one of the most obvious targets for treating the impact of peripheral inflammation on the brain. Early case reports indicated that several of the currently available biological response modifiers (or simply ‘biologics’) that target cytokines were effective in reducing symptoms of depression and fatigue, as well as quality of life (Soczynska et al, 2009). Biologics are compounds derived from biological processes vs medicinal chemistry. Most of the biologics approved by the US Food and Drug Administration (FDA) for treatment for autoimmune or inflammatory diseases use monoclonal antibodies, soluble cytokine receptors, or other antagonists which combine cytokine receptors with other fusion proteins. Therefore, there are limited drug–drug interactions between biologics and psychotropic medications, although the side-effect profile for these agents, especially as it relates to the risk of infection, is a cause for concern. Drugs currently approved by FDA target both TNF-α and IL-1 (Table 1).
Larger scale studies have confirmed early observations that cytokine antagonists may have antidepressant properties. For example, in a large (∼300 patients per group), double-blind, randomized, placebo-controlled trial of patients with psoriasis, patients who received the TNF-α antagonist, etanercept (a fusion protein that joins TNFR2 to IgG1), showed significant improvement in depressive symptoms as measured by the Beck Depression Inventory (Tyring et al, 2006). Improved symptoms of depression occurred independently of improvement in disease activity, and were sustained for up to 96 weeks in a open-label phase of the study (Krishnan et al, 2007). In addition, administration of a single dose of etanercept was found to normalize rapid eye movement sleep in patients with alcohol dependence (Irwin et al, 2009). Moreover, in a small trial of 12 patients with advanced cancer randomly assigned to chemotherapy alone or chemotherapy in combination with etanercept, patients randomized to etanercept reported significantly less fatigue (Monk et al, 2006). Of note, the chemotherapy/etanercept combination was also associated with greater antitumor activity, consistent with the role of inflammation in cancer development and progression (Monk et al, 2006). These data are consistent with a rich literature in laboratory animals indicating that cytokine antagonism using biologics such as IL-1ra or anti-inflammatory cytokines such as IL-10 (which is also being developed for clinical use) can inhibit the induction of depressive-like behavior following exposure to inflammatory stimuli (Dantzer et al, 2008). TNF-α receptor- and IL-1-KO mice have yielded similar results, showing reduced anxiety and an antidepressant phenotype as measured in standard animal testing protocols such as the FST, sucrose consumption, elevated plus maze, and fear conditioning (Simen et al, 2006; Koo and Duman, 2009). Finally, there has been interest in small molecules that inhibit the TNF-α-converting enzyme (so-called TACE inhibitors), which may have significant advantages in oral bioavailability (the currently available biologics must be administered by subcutaneous injection or intravenous infusion) and brain penetrance, which may be especially relevant to neuroimmunologic disorders, which show manifest pathology (tissue damage and destruction) within the brain (Murumkar et al, 2010).
Inflammatory/cytokine-signaling pathways
Cyclooxygenase. Aside from studies inhibiting the cytokines themselves, probably the best studied anti-inflammatory strategy for the treatment of depression is the targeting of inflammatory signaling pathways. The most common approach in this regard has been the administration of pharmacologic agents that inhibit cyclooxygenase (COX), the enzyme that converts arachidonic acid into prostaglandin. As noted above, prostaglandins have been shown to be increased in depression and are known to have an important role in the inflammatory response, including the mediation of fever and sensitivity to pain. Moreover, inhibition of COX-1 and COX-2 selectively and in combination have been shown in laboratory animals to inhibit depressive-like behavior following administration of the inflammatory stimulus, LPS, without influencing cytokine responses (de Paiva et al, 2010; Teeling et al, 2010). Of note, there has been some controversy as to whether COX-1 vs COX-2 is the most relevant target, with some recent animal data suggesting that COX-1- but not COX-2-selective inhibitors were able to inhibit low-dose LPS-induced behavioral responses. As might be expected, indomethacin and ibuprofen, non-selective COX inhibitors, have reliably shown efficacy in animal models (Teeling et al, 2010).
In human studies, addition of acetylsalicylic acid (which blocks COX-1 and COX-2) to fluoxetine led to increased remission rates in an open-label study of 24 depressed patients previously non-responsive to fluoxetine alone (Mendlewicz et al, 2006). Similarly, in a study of 20 subjects per group, medically healthy depressed patients who received the COX-2 inhibitor, celecoxib, in combination with reboxetine showed greater symptomatic improvement compared with patients randomized to reboxetine plus placebo (Muller et al, 2006). Nevertheless, both studies involved small sample sizes, and in the celecoxib study, 50% or more of the sample in each group dropped out before conclusion of the study (Muller et al, 2006). Interestingly, the largest trial to date examining the antidepressant efficacy of a COX-2 inhibitor (cimicoxib) in combination with sertraline vs sertraline plus placebo (conducted in Austria, the Czech Republic, and Germany) has completed enrollment, and results are pending (ClinicalTrials.gov: NCT00510822).
p38 MAPK. As previously noted, inhibition of p38 MAPK was able to reverse the development of LPS-induced behavioral changes in laboratory animals, in part, through reversing the effects of p38 MAPK on the SERT (Zhu et al, 2010). A number of p38 MAPK inhibitors are in development for the treatment of a range of diseases, including autoimmune and inflammatory disorders, cardiovascular disease, pulmonary disorders, including asthma and COPD, as well as pain (Kumar et al, 2003). According to a recent market analysis, about 10 next-generation p38 MAPK inhibitors (which show fewer side effects such as liver toxicity) are in phase-I or II clinical trials. One drug, GW856553, manufactured by GlaxoSmithKline, has just completed phase-II testing in patients with major depression (ClinicalTrials.gov: NCT00976560). However, no results are available at this time. Of note, minocycline, a tetracycline derivative, which inhibits the activation of microglia as well as the microglial production of IL-1 and NO, has been shown to inhibit p38 MAPK as well as poly(ADP-ribose) polymerase-1, which along with inhibition of NF-κB (see below), may serve as the basis for its anti-inflammatory efficacy (Tikka et al, 2001; Tikka and Koistinaho, 2001; Si et al, 2004; Alano et al, 2006). Interestingly minocycline has also been found to show antidepressant properties in laboratory animals by using the FST both alone and in combination with sub-threshold doses of the tricyclic antidepressant, desipramine, and several glutamate antagonists (Molina-Hernandez et al, 2008). Minocycline has also been shown to inhibit the development of neuroinflammation and behavioral changes after LPS administration (Henry et al, 2008), and has been found to attenuate the development of opioid tolerance, which may in part be related to the capacity of opioids to induce an inflammatory response (Hutchinson et al, 2008a, 2008b; Habibi-Asl et al, 2009).
NF- κ B. NF-κB has a pivotal role in the inflammatory response in general, and is relevant to the behavioral effects of inflammation; has been shown to transmit inflammatory signals from the periphery to the brain; and mediate the inflammation-induced inhibition of neurogenesis in vitro (Nadjar et al, 2005; Koo et al, 2010). Moreover, intracerebroventricular administration of NF-κB antagonists to rats subjected to acute or chronic stress has been found to reverse the stress-induced suppression of hippocampal neurogenesis as well as the effects of chronic unpredictable stress on behavior, including sucrose consumption (Koo et al, 2010). There are hundreds of compound that have been shown to inhibit NF-κB (see www.nf-kb.org for a complete and updated listing), including compounds that block the phosphorylation of Iκ kinase, which maintains NF-κB in an inactive state in the cytosol; compounds that block NF-κB translocation to the nucleus; and compounds that inhibit NF-κB DNA binding.
Special interest has been recently paid to natural compounds such as curcumin (a derivative of the curry spice, turmeric), which has been shown to decrease NF-κB activation in the PBMCs of patients with pancreatic cancer (Dhillon et al, 2008); α tocopherol (a form of Vitamin E), which has been shown to block NF-κB and the associated neuroinflammatory and behavioral response to LPS (Godbout et al, 2005); and resveratrol (found in the skin of red grapes), which has been shown to inhibit the LPS-induced induction of inflammatory cytokines, chemokines, and inducible NO synthase (iNOS) in murine microglia and astrocytes in association with inhibition of NF-κB (Lu et al, 2010). These natural compounds are currently in a number of phase-I–III clinical trials for multiple indications, including independent examination of effects on inflammation and behavior.
Another compound of interest is salicylate, which has also been shown to be protective against neurotoxicity induced by glutamate in primary neuronal cultures and hippocampal slices through inhibition of NF-κB (Grilli et al, 1996). Of note, salsalate, a dimeric prodrug comprising two esterified salicylate moieties, was recently found to significantly improve diabetic control while showing minimal adverse reactions (Goldfine et al, 2010). Finally, there has been interest in the potential role of angiotensin-II AT1 receptor blockers in ameliorating the neuroinflammatory response. Angiotensin-II has been shown to directly activate NF-κB in cultured neurons (Mitra et al, 2010), and candesartan, a blocker of the angiotensin-II AT1 receptor, was found to inhibit the peripheral and central cytokine response to LPS, as well as the effects of LPS on iNOS and COX-2 expression, microglial activation, and anxiety-like behavior (Benicky et al, 2011).
Chemokines Activation of microglia by peripherally elaborated TNF-α in the context of chronic immune stimulation (secondary to bile duct ligation and subsequent liver inflammation) leads to the release of the chemokine, MCP-1, which in turn attracts peripheral blood monocytes to the brain, whereupon they accumulate as activated macrophages in brain parenchyma (D’Mello et al, 2009) (Figure 1). The elucidation of this cellular pathway by which peripheral inflammatory signals can reach the brain has raised the possibility that chemokine and chemokine receptor antagonists may serve as additional immunologic targets for therapeutic intervention in neuropsychiatric disease. Much of the interest in the development of chemokine and chemokine receptor antagonists has been focused on the chemokines receptors, CCR5 and CXCR4, which are the two major co-receptors for HIV entry into CD4+ T cells (Proudfoot et al, 2010). Maraviroc is the first chemokine co-receptor antagonist approved by the FDA for use in the treatment of HIV (Proudfoot et al, 2010). Given the role of chemokines and chemokine receptors in immune trafficking and cellular adherence, these molecules also have an important role in cancer cell metastasis as well as autoimmune and inflammatory disorders, including rheumatoid arthritis, asthma, psoriasis, and inflammatory bowel disease (Proudfoot et al, 2010). Plerixafor is a CXCR4 antagonist that has been approved by the FDA and is being used in combination with granulocyte colony-stimulating factor to facilitate the mobilization of bone marrow stem cells to the peripheral blood for subsequent autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma (Proudfoot et al, 2010). The remainder of the chemokine and chemokine receptor antagonists are in phase-I–III development. In general, however, the results in clinical trials for inflammatory disorders to date have been somewhat disappointing, despite promising results in animal models (Proudfoot et al, 2010).
Another potential chemokine target is fractalkine (CX3CL1) and its receptor (CX3CR1). CX3CL1, which is produced by neurons, has been shown to inhibit microglial activation in vitro, and the expression of CX3CR1 on microglia suggests that neurons may have a role in regulating microglial activation through complementary CX3CL1–CX3CR1 interactions (Corona et al, 2010). Interestingly, CXCR1-KO mice show evidence of prolonged activation of microglia after LPS administration, with associated prolonged microglial expression of IL-1β and prolonged expression of depressive-like behavior compared with wild-type mice (Corona et al, 2010). Of note, chronic administration of CX3CL1 into the striatum of rats administered 6-hydroxydopamine to induce a Parkinson's-like syndrome led to suppressed microglial activation and a fewer loss of neurons, suggesting a neuroprotective role for CX3CL1 (Pabon et al, 2011).
T-cell targets
T regs and Th17 cells. Given the recognition of the role of T regs and Th17 cells in the pathophysiology of autoimmune and inflammatory disorders, as well as other disorders now recognized to have an inflammatory component, including cardiovascular disease, diabetes, neurodegenerative disease, and cancer, there has been great interest in targeting molecules and pathways that regulate the relative balance of these T-cell subsets. Although it has yet to be established that there is increased Th17-cell activation in depression, if such evidence is obtained, there is a multitude of pharmacologic agents approved or in development that target the fundamental molecules that regulate the differentiation of naïve T cells into Th17 cells, including IL-1, IL-6, IL-21, IL-23, RORc, and Stat3 (Kato and Fox, 2010). In concert, each of these molecules has a role in the expression and activation of relative cytokine receptors, which guide Th17-cell differentiation and ultimately pathogenicity (Kato and Fox, 2010).
As noted above, depressed patients have been shown to exhibit decreased T reg-cell numbers as well as evidence of decreased T reg-cell function as manifested by decreased IL-10 and TGF-β, the primary anti-inflammatory cytokines produced by this T-cell subset. Relative to the role of T regs in mediating self-tolerance and inhibiting T eff-cell proliferation, increasing T reg function may have a therapeutic advantage in inflammatory disease (Sakaguchi et al, 2010). For example, in a murine model of HIV, adoptive transfer of CD3-activated T regs attenuated astrogliosis and microglial activation, with concomitant decreased inflammatory cytokines and increased BDNF and glial cell-derived neurotrophic factor (Liu et al, 2009). Moreover, in a rat stroke model, T regs were found to enhance the survival of progenitor cells in the sub-ventricular zone (Ishibashi et al, 2009). To date, there are few strategies to increase T reg expression and function, aside from directly infusing T regs through adoptive transfer. Nevertheless, T regs, particularly their expression of the defining molecule, Foxp3, appear to be unstable, and this Foxp3 instability can lead to the generation of pathogenic effector–memory cells that promote autoimmunity (Zhou et al, 2009a, 2009b). Thus, further understanding of the regulation of T regs and Foxp3 expression is required before more definitive therapeutic targets can be elaborated (Sakaguchi et al, 2010).
An alternative mechanism to induce T reg expression and treat inflammatory disorders is the use of probiotics, typically administered orally as lactic acid bacteria (Fink, 2010). For example, in a randomized, placebo-controlled trial, administration of Lactobacillus rhamnosus GG to pregnant mothers at risk for having atopic children was shown to significantly reduce the development of atopic disorders (primarily eczema) in their offspring who subsequently took Lactobacillus GG for the first 6 months of life (Kalliomaki et al, 2001). However, subsequent controlled studies using Lactobacillus GG and other bacterial strains have failed to show such a protective effect against atopic and allergic disorders (de Roock et al, 2010; Fink, 2010), and follow-up of the children in the original Lactobacillus GG study revealed a trend toward increased allergic rhinitis and asthma, although rates of eczema continued to be significantly decreased (Kalliomaki et al, 2007). To address the inconsistencies in the literature, human PBMCs were treated in vitro with a range of bacteria, and the expression and function of Foxp3+ T cells were measured. Interestingly, there was a range of induction of T reg expression, with Lactobacillus acidophilus W55 showing the most dramatic effect (de Roock et al, 2010). These data suggest that the type of probiotic may dictate in part the relative induction of T regs, and may explain the differential results in relevant clinical trials. Of note, administration of other microbial agents, including killed preparations of Mycobacterium vaccae, have also been shown to induce regulatory T cells associated with increased IL-10 and TGF-β, which in turn have been found to protect against inflammatory reactions in laboratory animals (Zuany-Amorim et al, 2002). Taken together, these data suggest that exposure to non-pathogenic microbes may induce T regs and promote immunologic tolerance. This capacity of bacteria and other infectious agents to invoke immune tolerance (and T regs) is the basis for the ‘hygiene hypothesis’ regarding the increased prevalence of inflammatory disorders in modern societies where exposure to pathogenic stimuli has been dramatically reduced as a function of a variety of lifestyle factors, including increased sanitary methods, reduced consumption of spoiled foods, and minimal exposure to domesticated animals (Raison et al, 2010a, 2010b, 2010c).
T eff. Another potential therapeutic approach involving T-cell subpopulations is the use of immunization strategies against CNS antigens to target T eff cells to the brain (Figure 1). For example, immunization of rats with a modified myelin basic protein (which does not induce experimental allergic encephalomyelitis, but induces weakly autoreactive T cells) was found to significantly reduce the development of anhedonia (reduced sucrose preference) and immobility in the FST in response to 4 weeks of chronic mild stress (Lewitus et al, 2009). Immunization was also found to reverse stress-related decreases in hippocampal BDNF while increasing the generation of newly formed neurons as identified by BrdU and the early neuronal differentiation marker, doublecortin, in the dentate gyrus of the hippocampus (Lewitus et al, 2009). T-cell recruitment to the CNS has also been shown to reduce anxiety-like responses (as measured by the acoustic startle response and the elevated plus maze) to predator odor in mice (Cohen et al, 2006; Lewitus et al, 2008). Interestingly, stressor (odor) exposure was associated with increased T-cell infiltration to the choroid plexus, which was associated with a significant increase in the expression of the adhesion molecule, intercellular adhesion molecule (ICAM)-1, by choroid plexus cells (Lewitus et al, 2008) (Figure 1). The role of glucocorticoids in this latter effect was established by systemic administration of corticosterone, the natural glucocorticoid of the mouse, which also significantly increased ICAM-1 expression in the choroid plexus (Lewitus et al, 2008). These data demonstrating a novel role for T cells in resilience against stress are consistent with intriguing data demonstrating that T cells may have a fundamental role in neuronal integrity (Kipnis et al, 2004; Brynskikh et al, 2008; Wolf et al, 2009). For example, using the Morris Water Maze task, hippocampus-dependent visuo-spatial learning was measured in nude mice (which are devoid of T cells) with and without T-cell replenishment (Kipnis et al, 2004). Compared with nude mice without T-cell replenishment, nude mice replenished with T cells showed significantly greater learning capacity in terms of the acquisition, extinction, and reversal phases of the task (Kipnis et al, 2004). T cells have also been shown to facilitate the expression of BDNF and hippocampal neurogenesis, including having a role in the increased neurogenesis secondary to an enriched environment (Ziv et al, 2006; Wolf et al, 2009). As in the case of stressed animals, T cells are believed to mediate their effects in healthy animals through actions in the meningeal spaces (choroid plexus and subarachnoid spaces) where they release IL-4, which both skews meningeal myeloid cells (eg, monocytes/macrophages) to an M2 or an anti-inflammatory phenotype, and has been shown to promote the production of BDNF by astrocytes in vitro (Kipnis et al, 2008; Derecki et al, 2010) (Figure 1). Taken together, these data indicate that T eff cells expressing IL-4 may subserve not only a protective effect against stress, but may also have a role in the maintenance of neuronal integrity. Thus, strategies to augment T eff-cell trafficking to the brain (eg, through immunization with CNS antigens, which induce low levels of CNS autoreactivity) may be a highly innovative strategy to treat neuropsychiatric diseases, including depression (Schwartz and Shechter, 2010).
Neurotransmitter Targets
Indoleamine-2,3-dioxygenase The importance of IDO as a potential target for the unique contributions of the immune system to depression is evidenced by studies that have administered an IDO antagonist or used IDO-KO mice in the context of immune activation or infection. For example, treatment of mice with the IDO antagonist, 1-methyl tryptophan (1-MT), has been shown to abrogate the impact of LPS as well as infection with bacille Calmette–Guerin (BCG), an attenuated form of Mycobacterium bovis, on depressive-like behavior, including increased immobility in the FST and TST (O’Connor et al, 2008, 2009). In addition, IDO-deficient mice are resistant to BCG-induced behavioral changes, while showing a normal inflammatory cytokine response to BCG administration (O’Connor et al, 2009). Targeted deletion of KAT-II (the enzyme that converts kynurenine to KA) has also been shown to increase cognitive performance in association with an increase in the amplitude of long-term potentiation in vitro, while reducing extracellular KA as measured by hippocampal in vivo microdialysis (Potter et al, 2010).
In terms of pharmacologic agents in clinical trials, 1-MT is currently being tested in patients with advanced-stage cancers, administered alone or in combination with vaccine therapies (eg, ClinicalTrials.gov: NCT00617422 and NCT01302821). These studies are based on the inhibitory effect of IDO-mediated TRP depletion on T-cell function (Katz et al, 2008). No studies are currently underway examining the impact of 1-MT on behavioral alterations in cancer patients or other patient populations, although efficacy in regard to behavior would be an important proof of concept for clinical studies of IDO inhibitors in depressive disorders. Until recently, the majority of IDO inhibitors showed enzyme affinities in the micromolar range and were derived from metabolites of tryptophan (eg, 1-MT) and β-carbolines (Rohrig et al, 2010). Nevertheless, more potent (nanomolar range) IDO inhibitors have been identified in extracts from marine invertebrates and screening of the NCI Diversity Set Library (Volgraf et al, 2008; Rohrig et al, 2010; Di Pucchio et al, 2010). In addition, elaboration of the crystal structure of human IDO has paved the way for the in silico design of new IDO inhibitors (Rohrig et al, 2010). Thus, there is great promise regarding the development of more potent IDO inhibitors, although these compounds are still somewhat removed from clinical trials (Di Pucchio et al, 2010). In a similar manner, given the elaboration of the kynurenine pathway as well as data indicating the role of KA and quinolinic acid in neuropsychiatric disease, especially neurodegenerative disorders, a number of compounds, which interfere with multiple steps in the kynurenine pathway, have been identified, including inhibitors of 3-hydroxyanthranilic acid oxygenase, which converts 3-hydroxyanthranilic acid to quinolinic acid. Nevertheless, these compounds have yet to advance to phase-I–III testing for neuropsychiatric disorders (Stone, 2001; Schwarcz and Pellicciari, 2002).
BH4 Given the primary role of BH4 as a cofactor in the synthesis of monoamines as well as the inactivation and/or depletion of BH4 as a function of inflammatory processes and their induction of NO and oxidative stress (discussed above), pharmacologic strategies that restore or maintain the availability of BH4 may have special relevance for immune-based depression. Several strategies are currently available that address deficiencies in BH4, including administration of BH4 itself as well as the use of folic acid, L-methylfolate, and S-adenosyl-methionine (SAMe), all of which have a role in the synthesis and/or regeneration of BH4 (Stahl, 2007; Miller, 2008a, 2008b) (Figure 3). As noted above, BH4 is the enzyme cofactor for PAH, which converts phe to tyr. Deficiency of BH4 can lead to impaired PAH activity and in rare cases can lead to excessive phe and phenylketonuria (PKU), which can be associated with significant brain damage, mental retardation, seizures, and behavioral alterations, similar to that found in patients with deficiency of PAH, the more common cause of PKU. Sapropterin (Kuvan) is the first non-dietary, FDA-approved, BH4 replacement for patients with PKU that has been shown in randomized, double-blind trials to be effective in lowering blood phe levels (Burton et al, 2010). Aside from its use in PKU, BH4 is also currently being tested for cardiovascular disorders, particularly as they relate to hypertension (eg, ClinicalTrials.gov: NCT00208780). These studies are based on the role of NO in vascular tone. BH4 is an essential cofactor for endothelial NOS (eNOS); however, in the face of BH4 deficiency, eNOS switches from producing NO to generating oxygen free radicals that increase oxidative stress, which as noted above can further deplete BH4 while also contributing to endothelial dysfunction and vascular damage (Alp and Channon, 2004). Interestingly, BH4 administration has not been studied in the context of depression. Nevertheless, studies examining folic acid, L-methylfolate, and SAMe have been conducted. For example, in a number of studies, low levels of serum folate have been associated with an increased risk of depression as well as non-response to antidepressant treatment and an increased likelihood of depression relapse (Fava et al, 1997; Papakostas et al, 2004a, 2004b; Gilbody et al, 2007a, 2007b). In addition, a common genetic polymorphism in the enzyme, methylene tetrahydrofolate reductase that catalyzes the conversion of methylene tetrahydrofolate to L-methylfolate, the active form of folate, which crosses the BBB and can regenerate oxidized BH4 (Antoniades et al, 2006), has been positively linked to low serum folate as well as a 36% increase in the chance of developing depression in individuals with the TT enzyme variant, which is associated with reduced enzyme activity (Gilbody et al, 2007a, 2007b). Administration of L-methylfolate (marketed as Deplin and Zervalx) to depressed patients has been shown to augment the efficacy of standard antidepressant therapy (Godfrey et al, 1990; Ginsberg et al, 2011). In a recent double-blind, placebo-controlled trial of 73 SRI non-responders, adjunctive treatment with SAMe (+SRI) was also found to lead to significantly higher rates of remission and a 50% or greater decrease in depressive symptoms compared with placebo (Papakostas et al, 2010). Of note, similar augmentation of antidepressant efficacy has been found with folic acid and folinic acid (Leucovorin), a metabolically active form of folic acid that is readily converted to L-methylfolate and is used to supplement folic acid in patients undergoing treatment with methotrexate (Coppen and Bailey, 2000; Alpert et al, 2002). Interestingly, although the impact of inflammation and associated oxidative stress on BH4 and monoamine metabolism is well recognized, studies have yet to directly test whether strategies to augment BH4 using folic acid, L-methylfolate, or SAMe might be most useful in depressed patients who show evidence of increased inflammation.
Neuroendocrine Targets
Glucocorticoid receptor Through inhibition of NF-κB and other inflammatory mediators, endogenous glucocorticoids are primary regulators of the inflammatory response (Rhen and Cidlowski, 2005). Synthetic glucocorticoids are a mainstay in the treatment of inflammatory disorders, although glucocorticoid resistance, in part related to the effects of cytokines and cytokine-signaling pathways on GR expression and function, remains a significant problem. The targeting of cytokines and their signaling pathways has been previously discussed and therefore the focus in this section will be on alternative strategies to enhance glucocorticoid sensitivity. Several strategies have been suggested regarding the enhancement of GR function, including use of pharmacologic agents that target protein kinase-A (PKA).
Protein kinase-A. A large body of data indicates that the PKA-signaling pathways have a prominent role in GR function. For example, GR associates both in vivo and in vitro with the catalytic subunit of PKA in a ligand-independent manner (Doucas et al, 2000). Moreover, PKA has been found to phosphorylate GR independent of the presence of hsp90 and activate GR function in a ligand-independent manner (Haske et al, 1994; Eickelberg et al, 1999). In addition, PKA agonists, including forskolin and 8-Br-cAMP, can increase GR mRNA stability and GR mRNA levels, while enhancing GR transcription and function (Dong et al, 1989; Penuelas et al, 1998). The β2-adrenergic receptor agonists, salbutamol or salmeterol, have been found to translocate the GR from cytoplasm to nucleus, increase GR–GRE binding, and increase GRE-driven luciferase reporter gene activity in primary human lung fibroblasts and vascular smooth muscle cells (Eickelberg et al, 1999). The phosphodiesterase type-IV inhibitor, rolipram, which antagonizes the breakdown of cAMP, has also been shown to enhance GR-mediated gene transcription, both in the presence and absence of dexamethasone, in LMCAT mouse fibroblast cells and rat C6 glioma cells (Miller et al, 2002). PKA activity has also been found to have a role in GR resistance. For example, cAMP-resistant cell lines give rise to a significantly higher frequency of glucocorticoid-resistant cell variants (10−7 vs 10−10 in wild type) (Gruol et al, 1986). Conversely, when GR-resistant lymphoid cells were co-treated with forskolin, dexamethasone caused 90% cell death (Medh et al, 1998), an effect that was reversed by the GR antagonist, RU486, indicating the GR specificity of the effect.
Relevant to the impact of cytokines on GR function, PKA-signaling pathways also interact with both NF-κB- and MAPK-signaling pathways (Saxena et al, 1999; Takahashi et al, 2002). Increased PKA activity has been shown to inhibit NF-κB transcription through interaction of the catalytic subunit of PKA with p65, blocking p65 transactivation (Takahashi et al, 2002). In addition, in a number of cell types, elevated PKA inhibits MAPK pathways by phosphorylation of serine residues on raf-1, which leads to a reduced affinity of raf for Ras (Hafner et al, 1994). In addition, forskolin, a PKA activator, has been reported to inhibit MAPK-induced raf-1 translocation (Melck et al, 1999). Tamir et al (1996) reported that forskolin additionally inhibits T-cell activation through downregulation of MAPK pathways. Taken together, these data suggest that drugs that activate cAMP–PKA pathways may represent an intriguing therapeutic strategy in reversing glucocorticoid resistance. As PKA pathways can both enhance GR function as well inhibit inflammatory signaling, pharmacologic therapies targeting PKA may represent a ‘double hit’ on relevant mechanisms driving glucocorticoid resistance (Miller et al, 2002).
Of note, interactions of PKA-signaling pathways with (a) GR signaling and (b) signaling pathways of proinflammatory and immunoregulatory cytokines are especially relevant in view of the fact that depressed patients have been found to show reduced G-protein function in mononuclear cells (Avissar et al, 1997) and reduced cAMP-dependent protein kinase activity in cultured fibroblasts (Shelton et al, 1996). cAMP/PKA signal transduction pathways have also been shown to be reduced in postmortem brain tissue from depressed patients (Shelton et al, 2009).
Autonomic nervous system targets The relevance of the cholinergic anti-inflammatory reflex activated by stimulation of the vagus nerve has been demonstrated to inhibit inflammation and limit pathology in a number of animal models of inflammatory insult, including endotoxic shock, sepsis induced by peritonitis, hemorrhagic shock, and collagen-induced arthritis (Tracey, 2009). The exquisite detail with which this pathway has been elucidated has also led to the testing of α7nAChR agonists, which have shown efficacy in animal models of arthritis, post-operative ileus, intestinal inflammation, pancreatitis, and colitis (Tracey, 2009). These data raise the possibility that activating the anti-inflammatory cholinergic reflex may not only have relevance for inflammatory diseases such as rheumatoid arthritis and inflammatory bowel disease (especially given the rich representation of cholinergic fibers in the gut), but also inflammation in the context of depression (Tracey, 2009). Prototype vagus nerve stimulation devices have already been tested in laboratory animals, and currently approved vagus nerve stimulators (which send stimulation toward the brain as opposed to the periphery) have proven to be safe in humans treated for seizure disorders and depression (Tracey, 2009). Moreover, clinical trials examining the efficacy of α7nAChR agonists in humans have already begun. Indeed, in a double-blind, placebo-controlled trial of the α7nAChR agonist, GTS-21, volunteers were treated with either GTS-21 (150 mg t.i.d. for 3 days) or placebo and then administered a low dose (2 ng/kg) of LPS intravenously (Kox et al, 2011). Although there were no differences in the LPS-induced cytokine response between groups, the blood concentrations of GTS-21 and its metabolite were highly variable between subjects, and when taken into consideration, higher GTS-21 plasma concentrations were associated with significantly lower plasma concentrations of TNF-α, IL-6, and IL-1ra (Kox et al, 2011). These data are of great interest and suggest that further testing with higher doses or more potent agonists are warranted in future studies.
COMPLEMENTARY TREATMENT AND PREVENTION STRATEGIES FOR POPULATIONS AT RISK
Weight Reduction and Dietary Manipulations
One strategy to reduce the potential impact of obesity and its association with inflammation on mood is intentional weight loss. A number of studies have examined the impact of weight loss on depressive symptoms, and a recent meta-analysis on the studies conducted to date examined the efficacy of this strategy (Fabricatore et al, 2011). Thirty-one studies were included, and treatments were categorized as lifestyle modification, non-dieting, dietary counseling, diet-alone, exercise-alone, pharmacotherapy, placebo, or control interventions. The meta-analysis revealed that all non-pharmacologic weight loss approaches were associated with reductions in depressive symptoms. However, within-group analyses failed to show a relationship between actual weight loss and decreased depressive symptoms. The study also indicated that strategies with a supervised exercise component were positively related to mood changes, consistent with previous reports of a direct link between physical activity and mood, including the capacity of exercise to induce an antidepressant response (Barbour and Blumenthal, 2005; Mead et al, 2009). Pharmacologic strategies for weight reduction did not separate from placebo in depressive symptom reduction. None of the studies examined whether changes in inflammatory markers were associated with either weight loss and/or reduction in depressive symptoms. Interestingly, in a recent study by Capuron et al (2010), gastric surgery-induced weight loss was associated with reduced inflammatory markers, notably hsCRP, as well as decreases in anxiety.
As noted above, exercise interventions have been reliably found to reduce depressive symptoms in patients with major depression (Barbour and Blumenthal, 2005; Mead et al, 2009). Exercise has also been found to reduce inflammatory markers (Woods et al, 2009). However, the link between improvement in depressive symptoms and reductions in inflammatory measures has yet to be established. For example, although a 16-week exercise programme was shown to reduce the peripheral blood concentrations of CRP in a sample of 319 sedentary women, changes in inflammation, which were especially pronounced in obese exercisers, were not associated with improvements in mood (Arikawa et al, 2010). Nevertheless, subjects in this and other studies on the effects of exercise on the immune system have not included patients with major depression or patients at risk for depression. Thus, the relationship between weight loss, exercise, and inflammation as it relates to the treatment and prevention of depressive symptoms remains to be determined.
Dietary changes are an additional strategy to potentially reduce inflammation and improve depressive symptoms. For example, adherence to the so-called Mediterranean diet pattern (MDP) in a sample of over 10 000 individuals prospectively followed for incident episodes of depression (median follow-up of 4.4 years) found that greater adherence to the MDP was associated with a decreased likelihood of developing depression (Sanchez-Villegas et al, 2009). The MDP is a diet with a high ratio of monounsaturated to saturated fats and n-3 to n-6 PUFAs, as well as an abundance of fruits, vegetables, legumes, and grains. Of relevance to inflammation, greater adherence to the MDP has also been shown to be associated with reduced peripheral blood concentrations of IL-6 and other inflammatory markers, including markers of oxidative stress (Dai et al, 2008a, 2008b). Interestingly, in a longitudinal study of diet and depressive symptoms in older adults, a greater increase in IL-6 over time was associated with higher depressive symptoms (Milaneschi et al, 2010). However, when individuals were dichotomized into those with a higher vs a lower Mediterranean diet score, higher depressive symptoms were associated with a greater increase in IL-6 only in those who were non-adherent to the MDP. These data suggest that dietary pattern may have a protective effect relative to the relationship between inflammation and depression.
Regarding more specific dietary supplementation, a number of studies have directly examined the antidepressant efficacy of n-3 PUFAs as represented by EPA, DHA, polyunsaturated fatty acid, or fish oil. In a meta-analysis of 10 double-blind, placebo-controlled trials in patients with mood disorders comprising primarily of patients with unipolar or bipolar depression, pooling of the results indicated that n-3 PUFAs significantly improved depressive symptoms with an effect size of 0.061 (p<0.003) (Lin and Su, 2007). However, significant heterogeneity was observed among studies. One possibility is the relative ratio of EPA to DHA. Another recent meta-analysis of the literature suggests that supplements containing a 3 : 2 or greater ratio of EPA to DHA were significantly more likely to show antidepressant efficacy.
Meditation/Yoga/Tai Chi and the Autonomic Nervous System
There is a small literature examining the impact of various alternative and integrative medicine approaches to both the treatment and the prevention of depression, and the reduction of inflammation. For example, mindfulness-based cognitive therapy, a meditation programme derived from cognitive behavioral therapy and mindfulness-based stress reduction, has been shown in a number of studies to reduce residual depressive symptoms in patients with major depression and reduce depression relapse (Bondolfi et al, 2010; Chiesa and Serretti, 2010). In addition, compassion meditation, a meditation technique that includes a mindfulness component, has been shown to decrease inflammatory (IL-6) responses to a psychosocial laboratory stressor (Pace et al, 2009). However, whether the impact of meditation on the inflammatory response is related to its impact on depression remains unclear. Moreover, the mechanism by which meditation may reduce inflammation has yet to be determined. One possibility is the effect of meditation and other integrative and alternative medicine practices on the autonomic nervous system. Short-term meditation has been shown to increase heart rate variability and decrease skin conductance, both evidence of increased parasympathetic tone (Tang et al, 2009). Moreover, in separate studies, Tai Chi has been shown to decrease SNS activity (Motivala et al, 2006), while also augmenting the antidepressant response of escitalopram in elderly patients with major depression (Lavretsky et al, 2011). As noted above, the SNS responses can drive inflammation, whereas parasympathetic nervous system activation through release of acetylcholine can inhibit NF-κB and inflammatory responses. Similar effects have been proposed for various yoga techniques (Brown and Gerbarg, 2005), which has also been shown in some studies to improve depressed mood (Saeed et al, 2010). Although there is much promise in the potential impact of alternative therapies on autonomic nervous system pathways and their effects on inflammation and mood, there are limited data directly linking these phenomena, and thus, there is need for further research.
FUTURE DIRECTIONS
Given the mounting data that the immune system may contribute both to the development of neuropsychiatric disease as well as the maintenance of neuronal integrity, there is an increasing need to (1) test neuropharmacologic strategies that target the immune system to treat neuropsychiatric disorders such as depression and (2) develop better biomarkers to measure the relative status of the immunologic response. In some ways, the immune hypothesis of depression has some advantages in this regard, because of the general recognition that inflammation is a central pathophysiologic mechanism in a host of disorders. Thus, there are numerous partners in the search for new anti-inflammatory treatments as well as new biomarkers relevant to both diagnosis and treatment response. As noted previously, the acute-phase protein, CRP, has already become a standard inflammatory marker for cardiovascular risk in clinical practice, with suggested guidelines regarding CRP concentrations and degree of risk. Data demonstrating that increased peripheral blood CRP concentrations can also predict the development of symptoms of depression further support the potential consistency that may exist across disease states relevant to the role of inflammation (Gimeno et al, 2009). Taken together, the fact that inflammation may contribute to the development of depression in a significant proportion of depressed patients while also contributing to the development and progression of multiple other illnesses, places psychiatry squarely in line with other medical disciplines in the search for new strategies to treat and prevent immunologic contributions to disease development.
References
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007). Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8: 942–949.
Alano CC, Kauppinen TM, Valls AV, Swanson RA (2006). Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci USA 103: 9685–9690.
Alp NJ, Channon KM (2004). Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 24: 413–420.
Alpert JE, Mischoulon D, Rubenstein GE, Bottonari K, Nierenberg AA, Fava M (2002). Folinic acid (Leucovorin) as an adjunctive treatment for SSRI-refractory depression. Ann Clin Psychiatry 14: 33–38.
Anisman H, Merali Z, Hayley S (2008). Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol 85: 1–74.
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B et al (2007). Phenotypic and functional features of human Th17 cells. J Exp Med 204: 1849–1861.
Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J et al (2006). 5-Methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 114: 1193–1201.
Arikawa AY, Thomas W, Schmitz KH, Kurzer MS (2010). Sixteen weeks of exercise reduces C-reactive protein levels in young women. Med Sci Sports Exerc 43: 1002–1009.
Avissar S, Nechamkin Y, Roitman G, Schreiber G (1997). Reduced G protein functions and immunoreactive levels in mononuclear leukocytes of patients with depression. Am J Psychiatry 154: 211–217.
Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M (2011). Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun 25: 397–407. Provocative demonstration that antibiotic treatment can eliminate stress-induced increases in peripheral blood inflammatory cytokines.
Bailey MT, Engler H, Sheridan JF (2006). Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol 171: 29–37.
Banks WA, Kastin AJ, Durham DA (1989). Bidirectional transport of interleukin-1 alpha across the blood–brain barrier. Brain Res Bull 23: 433–437.
Barbour KA, Blumenthal JA (2005). Exercise training and depression in older adults. Neurobiol Aging 26 (Suppl 1): 119–123.
Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW et al (2003). Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 121: 847–853.
Baune BT, Dannlowski U, Domschke K, Janssen DG, Jordan MA, Ohrmann P et al (2010). The interleukin 1 beta (IL1B) gene is associated with failure to achieve remission and impaired emotion processing in major depression. Biol Psychiatry 67: 543–549.
Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J et al (2011). Angiotensin II AT(1) receptor blockade ameliorates brain inflammation. Neuropsychopharmacology 36: 857–870.
Ben Menachem-Zidon O, Goshen I, Kreisel T, Ben Menahem Y, Reinhartz E, Ben Hur T et al (2008). Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 33: 2251–2262. Elegant demonstration of the role of IL-1 in mediating the impact of stress on neurogenesis and behavior.
Bersano A, Aghemo A, Rumi MG, Ballabio E, Candelise L, Colombo M (2008). Recovery after L-DOPA treatment in peginterferon and ribavirin induced parkinsonism. Eur J Intern Med 19: 370–371.
Besedovsky HO, del Rey A (1996). Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev 17: 64–102.
Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E et al (2001). CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702–710.
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D et al (2003). A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA 100: 1920–1925. One of the first demonstrations that stress can activate fundamental inflammatory signaling pathways in humans, an effect that appears to be mediated by catecholamines.
Blume J, Douglas SD, Evans DL (2011). Immune suppression and immune activation in depression. Brain Behav Immun 25: 221–229.
Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D et al (1994). Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III 317: 499–503. One of the early papers demonstrating the role of the vagus nerve in relaying cytokine signals to the brain.
Boissier MC, Assier E, Falgarone G, Bessis N (2008). Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine 75: 373–375.
Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M et al (2002). Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol 22: 86–90.
Bondolfi G, Jermann F, der Linden MV, Gex-Fabry M, Bizzini L, Rouget BW et al (2010). Depression relapse prophylaxis with Mindfulness-Based Cognitive Therapy: replication and extension in the Swiss health care system. J Affect Disord 122: 224–231.
Bower JE, Ganz PA, Aziz N, Fahey JL (2002). Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psychosom Med 64: 604–611.
Breder CD, Dinarello CA, Saper CB (1988). Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 240: 321–324.
Brown DW, Anda RF, Tiemeier H, Felitti VJ, Edwards VJ, Croft JB et al (2009). Adverse childhood experiences and the risk of premature mortality. Am J Prev Med 37: 389–396.
Brown RP, Gerbarg PL (2005). Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med 11: 711–717.
Brydon L (2011). Adiposity, leptin and stress reactivity in humans. Biol Psychol 86: 114–120.
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD (2008). Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry 63: 1022–1029.
Brynskikh A, Warren T, Zhu J, Kipnis J (2008). Adaptive immunity affects learning behavior in mice. Brain Behav Immun 22: 861–869.
Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C et al (2008). Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry 14: 1095–1104.
Burton BK, Bausell H, Katz R, Laduca H, Sullivan C (2010). Sapropterin therapy increases stability of blood phenylalanine levels in patients with BH4-responsive phenylketonuria (PKU). Mol Genet Metab 101: 110–114.
Cao C, Matsumura K, Yamagata K, Watanabe Y (1997). Involvement of cyclooxygenase-2 in LPS-induced fever and regulation of its mRNA by LPS in the rat brain. Am J Physiol 272: R1712–R1725.
Capuron L, Fornwalt FB, Knight BT, Harvey PD, Ninan PT, Miller AH (2009). Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord 119: 181–185.
Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D et al (2003a). Interferon-alpha-induced changes in tryptophan metabolism. Relationship to depression and paroxetine treatment. Biol Psychiatry 54: 906–914.
Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS et al (2005). Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry 58: 190–196. One of the early reports of the impact of chronic cytokine exposure on neurocircuits relevant to anxiety, arousal, and alarm.
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B et al (2007). Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 32: 2384–2392.
Capuron L, Poitou C, Machaux-Tholliez D, Frochot V, Bouillot JL, Basdevant A et al (2010). Relationship between adiposity, emotional status and eating behaviour in obese women: role of inflammation. Psychol Med 41: 1517–1528.
Capuron L, Raison CL, Musselman DL, Lawson DH, Nemeroff CB, Miller AH (2003b). Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry 160: 1342–1345.
Capuron L, Schroecksnadel S, Feart C, Aubert A, Higueret D, Barberger-Gateau P et al (2011). Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry 70: 175–182.
Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH (2010). Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology 35: 2617–2623. A clear demonstration that early-life stress represents a vulnerability factor for increased inflammatory responses following exposure to stress.
Carter CS, Braver TS, Barch DM, Botvinick MM, Noll D, Cohen JD (1998). Anterior cingulate cortex, error detection, and the online monitoring of performance. Science 280: 747–749.
Chiesa A, Serretti A (2010). Mindfulness based cognitive therapy for psychiatric disorders: a systematic review and meta-analysis. Psychiatry Res 187: 441–453.
Clark J, Vagenas P, Panesar M, Cope AP (2005). What does tumour necrosis factor excess do to the immune system long term? Ann Rheum Dis 64 (Suppl 4): iv70–iv76.
Cohen H, Ziv Y, Cardon M, Kaplan Z, Matar MA, Gidron Y et al (2006). Maladaptation to mental stress mitigated by the adaptive immune system via depletion of naturally occurring regulatory CD4+CD25+ cells. J Neurobiol 66: 552–563.
Cole SW, Hawkley LC, Arevalo JM, Sung CY, Rose RM, Cacioppo JT (2007). Social regulation of gene expression in human leukocytes. Genome Biol 8: R189.
Coppen A, Bailey J (2000). Enhancement of the antidepressant action of fluoxetine by folic acid: a randomised, placebo controlled trial. J Affect Disord 60: 121–130.
Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG et al (2010). Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation 7: 93.
Dai J, Jones DP, Goldberg J, Ziegler TR, Bostick RM, Wilson PW et al (2008a). Association between adherence to the Mediterranean diet and oxidative stress. Am J Clin Nutr 88: 1364–1370.
Dai J, Miller AH, Bremner JD, Goldberg J, Jones L, Shallenberger L et al (2008b). Adherence to the Mediterranean diet is inversely associated with circulating interleukin-6 among middle-aged men: a twin study. Circulation 117: 169–175.
Damsker JM, Hansen AM, Caspi RR (2010). Th1 and Th17 cells: adversaries and collaborators. Ann NY Acad Sci 1183: 211–221.
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008). Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65: 409–415.
Danese A, Pariante CM, Caspi A, Taylor A, Poulton R (2007). Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci USA 104: 1319–1324. One of the first epidemiological studies demonstrating an association between early-life stress and increased inflammation in adulthood.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56. An outstanding review of the role of the immune system in behavioral change as demonstrated by studies in laboratory animals.
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I et al (2009). Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62: 479–493.
de Paiva VN, Lima SN, Fernandes MM, Soncini R, Andrade CA, Giusti-Paiva A (2010). Prostaglandins mediate depressive-like behaviour induced by endotoxin in mice. Behav Brain Res 215: 146–151.
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR et al (2010). Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 207: 1067–1080. One of several provocative studies from the Kipnis laboratory demonstrating the importance of T cells in the maintenance of neuronal integrity and cognitive function.
de Roock S, van Elk M, van Dijk ME, Timmerman HM, Rijkers GT, Prakken BJ et al (2010). Lactic acid bacteria differ in their ability to induce functional regulatory T cells in humans. Clin Exp Allergy 40: 103–110.
Desbonnet L, Garrett L, Clarke G, Kiely B, Cryan JF, Dinan TG (2010). Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 170: 1179–1188.
Dhabhar FS, Burke HM, Epel ES, Mellon SH, Rosser R, Reus VI et al (2009). Low serum IL-10 concentrations and loss of regulatory association between IL-6 and IL-10 in adults with major depression. J Psychiatr Res 43: 962–969.
Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL et al (2008). Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res 14: 4491–4499.
Di Pucchio T, Danese S, De Cristofaro R, Rutella S (2010). Inhibitors of indoleamine 2,3-dioxygenase: a review of novel patented lead compounds. Expert Opin Ther Pat 20: 229–250.
D’Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102. Comprehensive elucidation of the ‘cellular pathway’ by which peripheral elaboration of TNF- α communicates with the brain through activation of microglia, which in turn produce MCP-1, which attracts monocytes to the brain parenchyma.
Dong Y, Aronsson M, Gustafsson JA, Okret S (1989). The mechanism of cAMP-induced glucocorticoid receptor expression. Correlation to cellular glucocorticoid response. J Biol Chem 264: 13679–13683.
Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM (2000). Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA 97: 11893–11898.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK et al (2010). A meta-analysis of cytokines in major depression. Biol Psychiatry 67: 446–457.
Duman RS, Monteggia LM (2006). A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59: 1116–1127.
Dunn AJ, Wang J (1995). Cytokine effects on CNS biogenic amines. Neuroimmunomodulation 2: 319–328.
Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M et al (1999). Ligand-independent activation of the glucocorticoid receptor by beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 274: 1005–1010.
Eidelberg D, Moeller JR, Dhawan V, Spetsieris P, Takikawa S, Ishikawa T et al (1994). The metabolic topography of parkinsonism. J Cereb Blood Flow Metab 14: 783–801.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010). Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68: 748–754.
Eisenberger NI, Lieberman MD (2004). Why rejection hurts: a common neural alarm system for physical and social pain. Trends Cogn Sci 8: 294–300.
Eisenberger NI, Lieberman MD, Satpute AB (2005). Personality from a controlled processing perspective: an fMRI study of neuroticism, extraversion, and self-consciousness. Cogn Affect Behav Neurosci 5: 169–181.
Engler H, Bailey MT, Engler A, Stiner-Jones LM, Quan N, Sheridan JF (2008). Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology 33: 108–117. An in vivo demonstration of the capacity of cytokines to inhibit glucocorticoid receptor function leading to glucocorticoid resistance.
Ericsson A, Kovacs KJ, Sawchenko PE (1994). A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J Neurosci 14: 897–913. An elegant anatomical elucidation of the neural pathway by which cytokines can signal key brain regions to affect neuroendocrine responses.
Fabricatore AN, Wadden TA, Higginbotham AJ, Faulconbridge LF, Nguyen AM, Heymsfield SB et al (2011). Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond) (e-pub ahead of print 22 February 2011).
Fabry Z, Fitzsimmons KM, Herlein JA, Moninger TO, Dobbs MB, Hart MN (1993). Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J Neuroimmunol 47: 23–34.
Fava M, Borus JS, Alpert JE, Nierenberg AA, Rosenbaum JF, Bottiglieri T (1997). Folate, vitamin B12, and homocysteine in major depressive disorder. Am J Psychiatry 154: 426–428.
Feigin A, Fukuda M, Dhawan V, Przedborski S, Jackson-Lewis V, Mentis MJ et al (2001). Metabolic correlates of levodopa response in Parkinson's disease. Neurology 57: 2083–2088.
Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM et al (2007). Effects of interferon-alpha on Rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry 62: 1324–1333.
Felger JC, Alagbe O, Pace TW, Woolwine BJ, Hu F, Raison CL et al (2011). Early activation of p38 mitogen activated protein kinase is associated with interferon-alpha-induced depression and fatigue. Brain Behav Immun 25: 1094–1098.
Fink LN (2010). Induction of regulatory T cells by probiotics: potential for treatment of allergy? Clin Exp Allergy 40: 5–8.
Fitzgerald P, O’Brien SM, Scully P, Rijkers K, Scott LV, Dinan TG (2006). Cutaneous glucocorticoid receptor sensitivity and pro-inflammatory cytokine levels in antidepressant-resistant depression. Psychol Med 36: 37–43.
Fleshner M (2011). The gut microbiota: a new player in the innate immune stress response? Brain Behav Immun 25: 395–396.
Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF (2007). Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21: 47–59. The first demonstration that stress can directly activate microglia in the brain, leading to a CNS inflammatory response.
Frasure-Smith N, Lesperance F, Irwin MR, Talajic M, Pollock BG (2009). The relationships among heart rate variability, inflammatory markers and depression in coronary heart disease patients. Brain Behav Immun 23: 1140–1147. Pivotal data supporting the potential role of the anti-inflammatory cholinergic reflex in humans as assessed by heart rate variability and peripheral blood inflammatory markers.
Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H et al (2006). The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem 139: 655–662.
Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B (2002). Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 81: 1285–1297.
Gilbody S, Lewis S, Lightfoot T (2007a). Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol 165: 1–13.
Gilbody S, Lightfoot T, Sheldon T (2007b). Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health 61: 631–637.
Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A et al (2009). Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med 39: 413–423.
Ginsberg LD, Oubre AY, Daoud YA (2011). L-methylfolate plus SSRI or SNRI from treatment initiation compared to SSRI or SNRI monotherapy in a major depressive episode. Innov Clin Neurosci 8: 19–28.
Godbout JP, Berg BM, Krzyszton C, Johnson RW (2005). Alpha-tocopherol attenuates NFkappaB activation and pro-inflammatory cytokine production in brain and improves recovery from lipopolysaccharide-induced sickness behavior. J Neuroimmunol 169: 97–105.
Godfrey PS, Toone BK, Carney MW, Flynn TG, Bottiglieri T, Laundy M et al (1990). Enhancement of recovery from psychiatric illness by methylfolate. Lancet 336: 392–395.
Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE (2010). The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346–357.
Grilli M, Pizzi M, Memo M, Spano P (1996). Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science 274: 1383–1385. Early demonstration that salicylates can provide neuroprotection through inhibition of NF- κ B and thereby may serve as a potential treatment strategy for depression in the context of increased inflammation.
Gruol DJ, Campbell NF, Bourgeois S (1986). Cyclic AMP-dependent protein kinase promotes glucocorticoid receptor function. J Biol Chem 261: 4909–4914.
Guidetti P, Schwarcz R (2003). 3-Hydroxykynurenine and quinolinate: pathogenic synergism in early grade Huntington's disease? Adv Exp Med Biol 527: 137–145.
Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM (2005a). Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol 31: 395–404.
Guillemin GJ, Meininger V, Brew BJ (2005b). Implications for the kynurenine pathway and quinolinic acid in amyotrophic lateral sclerosis. Neurodegener Dis 2: 166–176.
Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005c). Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49: 15–23. Elaboration of the spectrum of cellular elements involved in the expression of IDO and the production of quinolinic acid in the brain.
Habibi-Asl B, Hassanzadeh K, Charkhpour M (2009). Central administration of minocycline and riluzole prevents morphine-induced tolerance in rats. Anesth Analg 109: 936–942.
Hafner S, Adler HS, Mischak H, Janosch P, Heidecker G, Wolfman A et al (1994). Mechanism of inhibition of Raf-1 by protein kinase A. Mol Cell Biol 14: 6696–6703.
Hamidi M, Drevets WC, Price JL (2004). Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry 55: 563–569.
Hannestad J, DellaGioia N, Ortiz N, Pittman B, Bhagwagar Z (2011). Citalopram reduces endotoxin-induced fatigue. Brain Behav Immun 25: 256–259.
Hardingham GE, Fukunaga Y, Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009a). Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66: 407–414. The first demonstration that inflammatory stimuli can interact with neurocircuits known to be involved in depression, especially treatment-resistant depression.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ et al (2009b). Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry 66: 415–422.
Hashimoto K, Sawa A, Iyo M (2007). Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry 62: 1310–1316.
Haske T, Nakao M, Moudgil VK (1994). Phosphorylation of immunopurified rat liver glucocorticoid receptor by the catalytic subunit of cAMP-dependent protein kinase. Mol Cell Biochem 132: 163–171.
Haydon PG, Carmignoto G (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86: 1009–1031.
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT et al (2008). Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation 5: 15.
Himmerich H, Milenovic S, Fulda S, Plumakers B, Sheldrick AJ, Michel TM et al (2010). Regulatory T cells increased while IL-1beta decreased during antidepressant therapy. J Psychiatr Res 44: 1052–1057.
Hu F, Pace TW, Miller AH (2009). Interferon-alpha inhibits glucocorticoid receptor-mediated gene transcription via STAT5 activation in mouse HT22 cells. Brain Behav Immun 23: 455–463.
Huang L, Baban B, Johnson III BA, Mellor AL (2010). Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. Int Rev Immunol 29: 133–155.
Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N et al (2008a). Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun 22: 1178–1189.
Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL et al (2008b). Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun 22: 1248–1256.
Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H (2008). Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett 432: 232–236.
Irwin MR, Miller AH (2007). Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun 21: 374–383.
Irwin MR, Olmstead R, Valladares EM, Breen EC, Ehlers CL (2009). Tumor necrosis factor antagonism normalizes rapid eye movement sleep in alcohol dependence. Biol Psychiatry 66: 191–195.
Ishibashi S, Maric D, Mou Y, Ohtani R, Ruetzler C, Hallenbeck JM (2009). Mucosal tolerance to E-selectin promotes the survival of newly generated neuroblasts via regulatory T-cell induction after stroke in spontaneously hypertensive rats. J Cereb Blood Flow Metab 29: 606–620.
Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN et al (2005). Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 135: 1295–1307.
Juengling FD, Ebert D, Gut O, Engelbrecht MA, Rasenack J, Nitzsche EU et al (2000). Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology (Berl) 152: 383–389.
Kalliomaki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E (2001). Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 357: 1076–1079.
Kalliomaki M, Salminen S, Poussa T, Isolauri E (2007). Probiotics during the first 7 years of life: a cumulative risk reduction of eczema in a randomized, placebo-controlled trial. J Allergy Clin Immunol 119: 1019–1021.
Kato H, Fox DA (2010). Are Th17 cells an appropriate new target in the treatment of rheumatoid arthritis? Clin Transl Sci 3: 319–326.
Katz JB, Muller AJ, Prendergast GC (2008). Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev 222: 206–221.
Kendler KS, Karkowski LM, Prescott CA (1999). Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156: 837–841.
Kiecolt-Glaser JK (2010). Stress, food, and inflammation: psychoneuroimmunology and nutrition at the cutting edge. Psychosom Med 72: 365–369.
Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP (2009). Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun 381: 671–675.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004). T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA 101: 8180–8185.
Kipnis J, Derecki NC, Yang C, Scrable H (2008). Immunity and cognition: what do age-related dementia, HIV-dementia and ‘chemo-brain’ have in common? Trends Immunol 29: 455–463.
Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T (2003). Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood–brain barrier. Brain Res 978: 104–114.
Komaki G, Arimura A, Koves K (1992). Effect of intravenous injection of IL-1 beta on PGE2 levels in several brain areas as determined by microdialysis. Am J Physiol 262: E246–E251.
Koo JW, Duman RS (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA 105: 751–756.
Koo JW, Duman RS (2009). Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett 456: 39–43.
Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS (2010). Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA 107: 2669–2674. Demonstration of the pivotal role played by NF- κ B in the impact of stress on behavior and emphasizing the relevance of NF- κ B as a neuropharmacologic target.
Kox M, Pompe JC, Gordinou de Gouberville MC, van der Hoeven JG, Hoedemaekers CW, Pickkers P (2011). Effects of the alphaalpha7nAChR agonist GTS-21 on the innate immune response in humans. Shock 36: 5–11.
Krishnan R, Cella D, Leonardi C, Papp K, Gottlieb AB, Dunn M et al (2007). Effects of etanercept therapy on fatigue and symptoms of depression in subjects treated for moderate to severe plaque psoriasis for up to 96 weeks. Br J Dermatol 157: 1275–1277.
Kumar S, Boehm J, Lee JC (2003). p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2: 717–726.
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H (2000). Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology 22: 370–379. One of the first studies demonstrating the relationship between inflammatory markers and treatment resistance in major depression.
Lavretsky H, Alstein LL, Olmstead RE, Ercoli LM, Riparetti-Brown M, Cyr NS et al (2011). Complementary use of Tai Chi Chih augments escitalopram treatment of geriatric depression: a randomized controlled trial. Am J Geriatr Psychiatry (e-pub ahead of print).
Lee YK, Mazmanian SK (2010). Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330: 1768–1773.
Lewitus GM, Cohen H, Schwartz M (2008). Reducing post-traumatic anxiety by immunization. Brain Behav Immun 22: 1108–1114.
Lewitus GM, Wilf-Yarkoni A, Ziv Y, Shabat-Simon M, Gersner R, Zangen A et al (2009). Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry 65: 283–288.
Li W, Knowlton D, Woodward WR, Habecker BA (2003). Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem 86: 774–783.
Li Y, Xiao B, Qiu W, Yang L, Hu B, Tian X et al (2010). Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J Affect Disord 124: 68–75.
Lin PY, Huang SY, Su KP (2010). A meta-analytic review of polyunsaturated fatty acid compositions in patients with depression. Biol Psychiatry 68: 140–147.
Lin PY, Su KP (2007). A meta-analytic review of double-blind, placebo-controlled trials of antidepressant efficacy of omega-3 fatty acids. J Clin Psychiatry 68: 1056–1061. An excellent review of the role of fatty acids in depression and their potential use as therapeutic agents in the treatment of major depression.
Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE (2009). Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol 182: 3855–3865.
Lotrich FE, Ferrell RE, Rabinovitz M, Pollock BG (2009). Risk for depression during interferon-alpha treatment is affected by the serotonin transporter polymorphism. Biol Psychiatry 65: 344–348.
Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH (2008). Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol Psychiatry 64: 461–467.
Lu X, Ma L, Ruan L, Kong Y, Mou H, Zhang Z et al (2010). Resveratrol differentially modulates inflammatory responses of microglia and astrocytes. J Neuroinflammation 7: 46.
Luppino FS, de Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BW et al (2010). Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry 67: 220–229.
Maes M, Bosmans E, Meltzer HY, Scharpe S, Suy E (1993). Interleukin-1 beta: a putative mediator of HPA axis hyperactivity in major depression? Am J Psychiatry 150: 1189–1193.
Maes M, Bosmans E, Suy E, Vandervorst C, DeJonckheere C, Raus J (1991). Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1 beta and soluble interleukin-2 receptor production. Acta Psychiatr Scand 84: 379–386. One of the first papers describing evidence of increased immune activation in major depression.
Maes M, Kubera M, Leunis JC (2008). The gut–brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett 29: 117–124.
Maes M, Lambrechts J, Bosmans E, Jacobs J, Suy E, Vandervorst C et al (1992a). Evidence for a systemic immune activation during depression: results of leukocyte enumeration by flow cytometry in conjunction with monoclonal antibody staining. Psychol Med 22: 45–53.
Maes M, Scharpe S, Van Grootel L, Uyttenbroeck W, Cooreman W, Cosyns P et al (1992b). Higher alpha 1-antitrypsin, haptoglobin, ceruloplasmin and lower retinol binding protein plasma levels during depression: further evidence for the existence of an inflammatory response during that illness. J Affect Disord 24: 183–192.
Matute C (2011). Glutamate and ATP signalling in white matter pathology. J Anat 219: 53–64.
Matute C, Domercq M, Sanchez-Gomez MV (2006). Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 53: 212–224.
Mazzeo RS, Donovan D, Fleshner M, Butterfield GE, Zamudio S, Wolfel EE et al (2001). Interleukin-6 response to exercise and high-altitude exposure: influence of alpha-adrenergic blockade. J Appl Physiol 91: 2143–2149.
McKinney Jr WT, Eising RG, Moran EC, Suomi SJ, Harlow HF (1971). Effects of reserpine on the social behavior of Rhesus monkeys. Dis Nerv Syst 32: 735–741.
McNamara RK, Liu Y (2011). Reduced expression of fatty acid biosynthesis genes in the prefrontal cortex of patients with major depressive disorder. J Affect Disord 129: 359–363.
Mead GE, Morley W, Campbell P, Greig CA, McMurdo M, Lawlor DA (2009). Exercise for depression. Cochrane Database Syst Rev CD004366.
Medh RD, Saeed MF, Johnson BH, Thompson EB (1998). Resistance of human leukemic CEM-C1 cells is overcome by synergism between glucocorticoid and protein kinase A pathways: correlation with c-Myc suppression. Cancer Res 58: 3684–3693.
Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzman M, Di Marzo V (1999). Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett 463: 235–240.
Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N (2006). Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol 21: 227–231.
Mentis MJ, McIntosh AR, Perrine K, Dhawan V, Berlin B, Feigin A et al (2002). Relationships among the metabolic patterns that correlate with mnemonic, visuospatial, and mood symptoms in Parkinson's disease. Am J Psychiatry 159: 746–754.
Meyers CA, Albitar M, Estey E (2005). Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 104: 788–793.
Milaneschi Y, Bandinelli S, Penninx BW, Vogelzangs N, Corsi AM, Lauretani F et al (2010). Depressive symptoms and inflammation increase in a prospective study of older adults: a protective effect of a healthy (Mediterranean-style) diet. Mol Psychiatry 16: 589–590.
Miller AH (2008a). Mechanisms of cytokine-induced behavioral changes: psychoneuroimmunology at the translational interface. Brain Behav Immun 23: 149–158.
Miller AH, Maletic V, Raison CL (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65: 732–741.
Miller AH, Vogt GJ, Pearce BD (2002). The phosphodiesterase type 4 inhibitor, rolipram, enhances glucocorticoid receptor function. Neuropsychopharmacology 27: 939–948.
Miller AL (2008b). The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern Med Rev 13: 216–226.
Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R et al (2008). A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 64: 266–272. An elegant demonstration of how microarray data can be subjected to bioinformatics strategies to elaborate relative activities in glucocorticoid and NF- κ B-signaling pathways, which were decreased and increased, respectively, in patients undergoing caregiver stress.
Mitra AK, Gao L, Zucker IH (2010). Angiotensin II-induced upregulation of AT(1) receptor expression: sequential activation of NF-kappaB and Elk-1 in neurons. Am J Physiol Cell Physiol 299: C561–C569.
Molina-Hernandez M, Tellez-Alcantara NP, Perez-Garcia J, Olivera-Lopez JI, Jaramillo-Jaimes MT (2008). Antidepressant-like actions of minocycline combined with several glutamate antagonists. Prog Neuropsychopharmacol Biol Psychiatry 32: 380–386.
Monk JP, Phillips G, Waite R, Kuhn J, Schaaf LJ, Otterson GA et al (2006). Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J Clin Oncol 24: 1852–1859.
Moron JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM et al (2003). Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J Neurosci 23: 8480–8488.
Motivala SJ, Sarfatti A, Olmos L, Irwin MR (2005). Inflammatory markers and sleep disturbance in major depression. Psychosom Med 67: 187–194.
Motivala SJ, Sollers J, Thayer J, Irwin MR (2006). Tai Chi Chih acutely decreases sympathetic nervous system activity in older adults. J Gerontol A Biol Sci Med Sci 61: 1177–1180.
Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B et al (2006). The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 11: 680–684.
Murumkar PR, DasGupta S, Chandani SR, Giridhar R, Yadav MR (2010). Novel TACE inhibitors in drug discovery: a review of patented compounds. Expert Opin Ther Pat 20: 31–57.
Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS et al (2001). Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med 344: 961–966. A pivotal translational study that has changed clinical practice and demonstrated that proactive treatment with antidepressant medication can prevent the development of depression during chronic immune stimulation.
Nadjar A, Bluthe RM, May MJ, Dantzer R, Parnet P (2005). Inactivation of the cerebral NFkappaB pathway inhibits interleukin-1beta-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology 30: 1492–1499. Demonstration that NF- κ B at the blood brain barrier is pivotal for the passage of cytokine signals from the periphery to the brain.
Nance DM, Sanders VM (2007). Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun 21: 736–745.
Nathan C (2008). Epidemic inflammation: pondering obesity. Mol Med 14: 485–492.
Nathan C, Ding A (2010). Nonresolving inflammation. Cell 140: 871–882.
Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M et al (2008). Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab 9: 622–627.
O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J et al (2009). Induction of IDO by bacille Calmette–Guerin is responsible for development of murine depressive-like behavior. J Immunol 182: 3202–3212. The IDO antagonist, 1MT, was shown to inhibit BCG-induced behavioral changes, indicating that IDO is a viable target for neuropharmacologic development.
O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N et al (2008). Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14: 511–522.
O’Mahony C, van der Kleij H, Bienenstock J, Shanahan F, O’Mahony L (2009). Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol 297: R1118–R1126. Compelling finding that the cholinergic anti-inflammatory reflex may involve the induction of anti-inflammatory T cells, suggesting a cellular component to the impact of vagal pathways on inflammatory tone.
Ongur D, Drevets WC, Price JL (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95: 13290–13295.
Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC (2011). CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. J Neuroinflammation 8: 9.
Pace TW, Hu F, Miller AH (2007). Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21: 9–19.
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH et al (2006). Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163: 1630–1633.
Pace TW, Negi LT, Adame DD, Cole SP, Sivilli TI, Brown TD et al (2009). Effect of compassion meditation on neuroendocrine, innate immune and behavioral responses to psychosocial stress. Psychoneuroendocrinology 34: 87–98. The first study showing that meditation practice can reduce the inflammatory response to stress.
Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M (2010). S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: a double-blind, randomized clinical trial. Am J Psychiatry 167: 942–948.
Papakostas GI, Petersen T, Mischoulon D, Green CH, Nierenberg AA, Bottiglieri T et al (2004a). Serum folate, vitamin B12, and homocysteine in major depressive disorder, Part 2: predictors of relapse during the continuation phase of pharmacotherapy. J Clin Psychiatry 65: 1096–1098.
Papakostas GI, Petersen T, Mischoulon D, Ryan JL, Nierenberg AA, Bottiglieri T et al (2004b). Serum folate, vitamin B12, and homocysteine in major depressive disorder, Part 1: predictors of clinical response in fluoxetine-resistant depression. J Clin Psychiatry 65: 1090–1095.
Pariante CM (2004). Glucocorticoid receptor function in vitro in patients with major depression. Stress 7: 209–219.
Pariante CM, Lightman SL (2008). The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31: 464–468.
Pariante CM, Miller AH (2001). Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 49: 391–404.
Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C et al (1999). The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology 140: 4359–4366.
Paulus MP, Feinstein JS, Simmons A, Stein MB (2004). Anterior cingulate activation in high trait anxious subjects is related to altered error processing during decision making. Biol Psychiatry 55: 1179–1187.
Pemberton LA, Kerr SJ, Smythe G, Brew BJ (1997). Quinolinic acid production by macrophages stimulated with IFN-gamma, TNF-alpha, and IFN-alpha. J Interferon Cytokine Res 17: 589–595.
Penuelas I, Encio IJ, Lopez-Moratalla N, Santiago E (1998). cAMP activates transcription of the human glucocorticoid receptor gene promoter. J Steroid Biochem Mol Biol 67: 89–94.
Pitt D, Nagelmeier IE, Wilson HC, Raine CS (2003). Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology 61: 1113–1120.
Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu HQ et al (2010). Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 35: 1734–1742.
Proudfoot AE, Power CA, Schwarz MK (2010). Anti-chemokine small molecule drugs: a promising future? Expert Opin Investig Drugs 19: 345–355.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS et al (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: 453–462.
Quan N, Avitsur R, Stark JL, He L, Shah M, Caligiuri M et al (2001). Social stress increases the susceptibility to endotoxic shock. J Neuroimmunol 115: 36–45.
Quan N, Banks WA (2007). Brain-immune communication pathways. Brain, Behavior, and Immunity 21: 727–735.
Raison CL, Borisov AS, Broadwell SD, Capuron L, Woolwine BJ, Jacobson IM et al (2005a). Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry 66: 41–48.
Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ et al (2009). Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry 65: 296–303. Some of the first data to demonstrate that peripheral cytokine signals can access the brain in humans and induce a CNS inflammatory response involving the release of cytokines and chemokines, which correlate with CNS monoamine metabolites and behavior.
Raison CL, Borisov AS, Woolwine BJ, Massung B, Vogt G, Miller AH (2010a). Interferon-alpha effects on diurnal hypothalamic–pituitary–adrenal axis activity: relationship with proinflammatory cytokines and behavior. Mol Psychiatry 15: 535–547.
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G et al (2010b). CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15: 393–403. Demonstration that cytokine-induced peripheral elaboration of kynurenine can enter the brain and be converted to kynurenic acid and quinolinic acid, which correlate with activation of inflammatory responses and behavior.
Raison CL, Demetrashvili M, Capuron L, Miller AH (2005b). Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs 19: 105–123.
Raison CL, Lowry CA, Rook GA (2010c). Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry 67: 1211–1224.
Raison CL, Woolwine BJ, Demetrashvili MF, Borisov AS, Weinreib R, Staab JP et al (2007). Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment Pharmacol Ther 25: 1163–1174.
Rajkowska G, Miguel-Hidalgo JJ (2007). Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets 6: 219–233.
Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A et al (2001). Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry 58: 445–452.
Rhen T, Cidlowski JA (2005). Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353: 1711–1723.
Ridker PM (2003). Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 107: 363–369.
Rios C, Santamaria A (1991). Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem Res 16: 1139–1143.
Rohrig UF, Awad L, Grosdidier A, Larrieu P, Stroobant V, Colau D et al (2010). Rational design of indoleamine 2,3-dioxygenase inhibitors. J Med Chem 53: 1172–1189.
Saeed SA, Antonacci DJ, Bloch RM (2010). Exercise, yoga, and meditation for depressive and anxiety disorders. Am Fam Physician 81: 981–986.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010). FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10: 490–500.
Sanchez MM, Alagbe O, Felger JC, Zhang J, Graff AE, Grand AP et al (2007). Activated p38 MAPK is associated with decreased CSF 5-HIAA and increased maternal rejection during infancy in rhesus monkeys. Mol Psychiatry 12: 895–897.
Sanchez-Villegas A, Delgado-Rodriguez M, Alonso A, Schlatter J, Lahortiga F, Serra Majem L et al (2009). Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry 66: 1090–1098.
Saxena M, Williams S, Tasken K, Mustelin T (1999). Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol 1: 305–311.
Schwarcz R, Pellicciari R (2002). Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther 303: 1–10. Comprehensive review of the kynurenine pathway and the relevance of kynurenic and quinolinic acid to CNS pathology.
Schwartz M, Shechter R (2010). Protective autoimmunity functions by intracranial immunosurveillance to support the mind: the missing link between health and disease. Mol Psychiatry 15: 342–354.
Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D (2000). Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst 92: 994–1000.
Shelton RC, Hal Manier D, Lewis DA (2009). Protein kinases A and C in postmortem prefrontal cortex from persons with major depression and normal controls. Int J Neuropsychopharmacol 12: 1223–1232.
Shelton RC, Mainer DH, Sulser F (1996). cAMP-dependent protein kinase activity in major depression. Am J Psychiatry 153: 1037–1042.
Shelton RC, Miller AH (2010). Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol 91: 275–299.
Shoelson SE, Herrero L, Naaz A (2007). Obesity, inflammation, and insulin resistance. Gastroenterology 132: 2169–2180.
Si Q, Cosenza M, Kim MO, Zhao ML, Brownlee M, Goldstein H et al (2004). A novel action of minocycline: inhibition of human immunodeficiency virus type 1 infection in microglia. J Neurovirol 10: 284–292.
Simen BB, Duman CH, Simen AA, Duman RS (2006). TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry 59: 775–785.
Slavich GM, Way BM, Eisenberger NI, Taylor SE (2010). Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci USA 107: 14817–14822. Provocative study demonstrating that stress-induced inflammatory cytokines may mediate the impact of stress on cognitive neurocircuits relevant to anxiety, arousal, and alarm.
Sluzewska A, Sobieska M, Rybakowski JK (1997). Changes in acute-phase proteins during lithium potentiation of antidepressants in refractory depression. Neuropsychobiology 35: 123–127.
Smoak KA, Cidlowski JA (2004). Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125: 697–706.
Soczynska JK, Kennedy SH, Goldstein BI, Lachowski A, Woldeyohannes HO, McIntyre RS (2009). The effect of tumor necrosis factor antagonists on mood and mental health-associated quality of life: novel hypothesis-driven treatments for bipolar depression? Neurotoxicology 30: 497–521.
Spetsieris PG, Moeller JR, Dhawan V, Ishikawa T, Eidelberg D (1995). Visualizing the evolution of abnormal metabolic networks in the brain using PET. Comput Med Imaging Graph 19: 295–306.
Spiegel D, Giese-Davis J, Taylor CB, Kraemer H (2006). Stress sensitivity in metastatic breast cancer: analysis of hypothalamic–pituitary–adrenal axis function. Psychoneuroendocrinology 31: 1231–1244.
Stahl SM (2007). Novel therapeutics for depression: L-methylfolate as a trimonoamine modulator and antidepressant-augmenting agent. CNS Spectr 12: 739–744.
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C et al (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42: 151–157.
Stone TW (2001). Kynurenic acid antagonists and kynurenine pathway inhibitors. Expert Opin Investig Drugs 10: 633–645.
Su KP, Huang SY, Peng CY, Lai HC, Huang CL, Chen YC et al (2010). Phospholipase A2 and cyclooxygenase 2 genes influence the risk of interferon-alpha-induced depression by regulating polyunsaturated fatty acids levels. Biol Psychiatry 67: 550–557.
Sutcigil L, Oktenli C, Musabak U, Bozkurt A, Cansever A, Uzun O et al (2007). Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin Dev Immunol 2007: 76396.
Szuster-Ciesielska A, Slotwinska M, Stachura A, Marmurowska-Michalowska H, Dubas-Slemp H, Bojarska-Junak A et al (2008). Accelerated apoptosis of blood leukocytes and oxidative stress in blood of patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry 32: 686–694.
Takahashi N, Tetsuka T, Uranishi H, Okamoto T (2002). Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem 269: 4559–4565.
Tamir A, Granot Y, Isakov N (1996). Inhibition of T lymphocyte activation by cAMP is associated with down-regulation of two parallel mitogen-activated protein kinase pathways, the extracellular signal-related kinase and c-Jun N-terminal kinase. J Immunol 157: 1514–1522.
Tang YY, Ma Y, Fan Y, Feng H, Wang J, Feng S et al (2009). Central and autonomic nervous system interaction is altered by short-term meditation. Proc Natl Acad Sci USA 106: 8865–8870.
Teeling JL, Cunningham C, Newman TA, Perry VH (2010). The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: implications for a role of COX-1. Brain Behav Immun 24: 409–419.
Tesmer LA, Lundy SK, Sarkar S, Fox DA (2008). Th17 cells in human disease. Immunol Rev 223: 87–113.
Thayer JF, Fischer JE (2009). Heart rate variability, overnight urinary norepinephrine and C-reactive protein: evidence for the cholinergic anti-inflammatory pathway in healthy human adults. J Intern Med 265: 439–447.
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001). Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21: 2580–2588.
Tikka TM, Koistinaho JE (2001). Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 166: 7527–7533.
Tilleux S, Hermans E (2007). Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85: 2059–2070.
Tracey KJ (2009). Reflex control of immunity. Nat Rev Immunol 9: 418–428. Most comprehensive review of the anti-inflammatory cholinergic reflex.
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A et al (2006). Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367: 29–35. First double-blind, placebo-controlled trial where a TNF- α antagonist was shown to significantly improve depressive symptoms independent of effects on disease activity in a sample of subjects with psoriasis.
Ursu S, Stenger VA, Shear MK, Jones MR, Carter CS (2003). Overactive action monitoring in obsessive-compulsive disorder: evidence from functional magnetic resonance imaging. Psychol Sci 14: 347–353.
Vogelzangs N, Kritchevsky SB, Beekman AT, Newman AB, Satterfield S, Simonsick EM et al (2008). Depressive symptoms and change in abdominal obesity in older persons. Arch Gen Psychiatry 65: 1386–1393.
Volgraf M, Lumb JP, Brastianos HC, Carr G, Chung MK, Munzel M et al (2008). Biomimetic synthesis of the IDO inhibitors exiguamine A and B. Nat Chem Biol 4: 535–537.
Volterra A, Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6: 626–640.
Wang X, Wu H, Miller AH (2004). Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry 9: 65–75.
Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L et al (1994). Neurocircuitry of illness-induced hyperalgesia. Brain Res 639: 283–299.
Wichmann T, DeLong MR (1999). Oscillations in the basal ganglia. Nature 400: 621–622.
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD et al (2007). Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8: 950–957.
Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A et al (2009). CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182: 3979–3984.
Wong ML, Dong C, Maestre-Mesa J, Licinio J (2008). Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response. Mol Psychiatry 13: 800–812.
Woods JA, Vieira VJ, Keylock KT (2009). Exercise, inflammation, and innate immunity. Immunol Allergy Clin North Am 29: 381–393.
Wu HQ, Rassoulpour A, Schwarcz R (2007). Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain? J Neural Transm 114: 33–41.
Yu YW, Chen TJ, Hong CJ, Chen HM, Tsai SJ (2003). Association study of the interleukin-1 beta (C-511T) genetic polymorphism with major depressive disorder, associated symptomatology, and antidepressant response. Neuropsychopharmacology 28: 1182–1185.
Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA (2009a). Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol 21: 281–285.
Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M et al (2009b). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10: 1000–1007.
Zhu CB, Blakely RD, Hewlett WA (2006). The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 31: 2121–2131. Pharmacologic blockade of the cytokine-signaling pathway, p38 MAPK, was shown to prevent the effects of LPS on SERTs and behavior, indicating the relevance of p38 MAPK as a viable target for neuropharmacologic intervention in immune-based depression.
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD (2005). p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem 280: 15649–15658.
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010). Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35: 2510–2520.
Zielasek J, Hartung HP (1996). Molecular mechanisms of microglial activation. Adv Neuroimmunol 6: 191–122.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N et al (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9: 268–275.
Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A et al (2001). The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 15: 199–226.
Zuany-Amorim C, Sawicka E, Manlius C, Le Moine A, Brunet LR, Kemeny DM et al (2002). Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced allergen-specific regulatory T-cells. Nat Med 8: 625–629.
Acknowledgements
This work was supported in part by grants from the National Institute of Mental Health to EH (award number K23MH091254), CLR (award numbers R01AT004698 and R01MH75102), and AHM (award numbers R01MH087604, R01MH083746, and R01MH075102), as well as PHS Grant UL1 RR025008 from the Clinical and Translational Science Award program and PHS Grant M01 RR0039 from the General Clinical Research Center program, National Institutes of Health, National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All authors declare that there are no conflicts of interest, and all financial disclosures are listed for each author: Charles L Raison serves as a consultant for Pamlab LLC and Biolex Therapeutics; Andrew H Miller has served as a consultant for Abbott Laboratories, AstraZeneca, GlaxoSmithKline, Lundbeck Research USA, F Hoffmann-La Roche, Schering-Plough Research Institute, and Wyeth/Pfizer, and has received research support from Centocor, GlaxoSmithKline, and Schering-Plough Research Institute; Ebrahim Haroon has nothing to declare.
Rights and permissions
About this article
Cite this article
Haroon, E., Raison, C. & Miller, A. Psychoneuroimmunology Meets Neuropsychopharmacology: Translational Implications of the Impact of Inflammation on Behavior. Neuropsychopharmacol 37, 137–162 (2012). https://doi.org/10.1038/npp.2011.205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.205
Keywords
This article is cited by
-
Emotion regulation shows an age- and sex-specific moderating effect on the relationship between chronic stress and cognitive performance
Scientific Reports (2024)
-
Prediction and Validation of Hub Genes Related to Major Depressive Disorder Based on Co-expression Network Analysis
Journal of Molecular Neuroscience (2024)
-
Identifying the mediating role of inflammation on the relationship between socioeconomic status and Alzheimer’s disease: a Mendelian randomization analysis and mediation analysis
Journal of Neurology (2024)
-
Sex-Related and Brain Regional Differences of URB597 Effects on Modulation of MAPK/PI3K Signaling in Chronically Stressed Rats
Molecular Neurobiology (2024)
-
First trimester human umbilical cord perivascular cells (HUCPVC) modulate the kynurenine pathway and glutamate neurotransmission in an LPS-induced mouse model of neuroinflammation
Journal of Inflammation (2023)
