
Abstract
Metabotropic glutamate receptors (mGluRs) are important modulators of excitatory transmission, and have been implicated in anxiety and stress-related behaviors. Previously, we showed that group III mGluR agonists could depress excitatory synaptic transmission in the bed nucleus of the stria terminalis (BNST), an integral component of the anxiety circuitry. Here, we provide converging evidence indicating that this effect is mediated primarily by mGluR8, is exerted presynaptically, and is modulated by noradrenergic signaling and stress. The effects of the group III mGluR agonist L-AP4 on excitatory transmission are not potentiated by the mGluR4-selective allosteric potentiator PHCCC, but are mimicked by the mGluR8-selective agonist DCPG. Consistent with these results, mGluR8-like immunoreactivity is seen in the BNST, and the actions of L-AP4 on excitatory transmission are absent in slices from mGluR8 knockout (KO) mice. Application of DCPG is associated with an increase in paired-pulse evoked glutamate synaptic currents, and a decrease in spontaneous glutamate synaptic current frequency, consistent with a primarily presynaptic action. mGluR8-mediated suppression of excitatory transmission is disrupted ex vivo by activation of α1 adrenergic receptors (α1 ARs). BNST mGluR8 function is also disrupted by both acute and chronic in vivo exposure to restraint stress, and in brain slices from α2A AR KO mice. These studies show that mGluR8 is an important regulator of excitatory transmission in the BNST, and suggest that this receptor is selectively disrupted by noradrenergic signaling and by both acute and chronic stress.
Similar content being viewed by others

INTRODUCTION
The bed nucleus of the stria terminalis (BNST), part of the extended amygdala, is a critical relay station for integrating the brain's response to stress and anxiety (Walker and Davis, 2008). Receiving excitatory inputs from other limbic regions such as the basolateral amygdala and medial prefrontal cortex (mPFC), the BNST in turn regulates stress-associated brain regions, including the paraventricular nucleus of the hypothalamus (PVN) (Herman and Cullinan, 1997). The BNST is a key regulator of the PVN (Cecchi et al, 2002), contributing to a basal inhibitory tone preventing hypothalamic–pituitary–adrenal (HPA) axis engagement (Ulrich-Lai and Herman, 2009). Thus, changes in BNST activity directly affect HPA axis activation. Indeed, glutamatergic signaling within this circuitry is engaged during anxiety-related behaviors (Walker and Davis, 1997b; McElligott et al, 2010), and lesioning the BNST prevents recruitment of the PVN by the mPFC (Radley et al, 2009).
Metabotropic glutamate receptors (mGluRs) are G-protein-coupled modulators of neurotransmission, and are involved in a myriad of behaviors and disease states. The eight known receptor subtypes are grouped I–III according to sequence homology, pharmacology, and signal transduction mechanism (Conn and Pin, 1997). Group I (mGluR1 and mGluR5) and group II (mGluR2 and mGluR3) receptors have been implicated in disorders ranging from addiction and fragile X syndrome to schizophrenia and Parkinson's disease (Grueter et al, 2006; Dolen et al, 2007; Bellesi and Conti, 2010; Niswender et al, 2008).
mGluR8 is classified as a group III receptor (mGluRs 4, 6, 7, and 8). These Gi/o-coupled receptors are the least well-studied group of mGluRs because of the relative lack of specific pharmacological tools targeting them. mGluR8 exhibits a diffuse pattern of expression during development that is limited to discrete pockets of expression in the adult mouse, a characteristic unique among presynaptic mGluRs (Duvoisin et al, 1995; Ayala et al, 2008). Group III mGluRs are generally thought to be expressed presynaptically and to serve as autoreceptors (Ayala et al, 2008; Valenti et al, 2005; Abitbol et al, 2008; Schmid and Fendt, 2006). Interestingly, mGluR8 knockout (KO) mice exhibit an anxiety phenotype (Duvoisin et al, 2005, 2010; Linden et al, 2002, but see Fendt et al, 2010), implying involvement of mGluR8 in the anxiety response.
In the BNST, the group III mGluR agonist L-AP4 and the mGluR8-selective agonist DCPG have depressive effects on excitatory transmission, suggesting expression of mGluR8 and potentially other group III mGluRs in this region (Grueter and Winder, 2005). With activity in the BNST being critical during stress exposure, presynaptic mGluRs are well positioned to regulate excitatory inputs coming into this region. The purpose of this study was to determine which group III receptors have measurable effects on excitatory transmission in the BNST, and whether these effects are altered by in vivo exposure to stress.
MATERIALS AND METHODS
mGluR8 Immunohistochemistry
A previously characterized mouse monoclonal antibody specific for mGluR8 was used (Quraishi et al, 2007). Because the secondary rabbit anti-mouse antibody strongly reacts with endogenous IgGs in the vasculature, it was not possible to assess mGluR8 immunoreactivity in mouse brain. Instead, we analyzed the distribution of mGluR8 in rat brain and used a rat secondary antiserum to detect the mouse monoclonal antibody. A rat was killed and its brain was removed and quick-frozen in an isopentane/dry ice bath. Sections, 20 μm thick, were cut on a cryostat and kept frozen. After thawing, sections were fixed for 10 min in 4% paraformaldehyde in 0.1 M phosphate buffer (pH=7.4), washed with PBS, blocked in antibody incubation solution (AIS: 2% normal goat serum, 0.3% Triton X-100, and 0.05% NaN3 in PBS), and incubated with 20 μg/ml purified anti-mGluR8 antibodies in AIS at 4 °C overnight. The next day, sections were washed three times with PBS, incubated with rat anti-mouse IgG antiserum (diluted 1 : 500; Jackson Immunoresearch, West Grove, PA) at room temperature for 1 to 2 h. After three washes with PBS and a 5-min incubation with Hoechst counterstain (Molecular Probes, Eugene, OR), followed by three additional PBS washes, the sections were coverslipped with AquaMount and visualized using a LSM510 Zeiss confocal microscope.
Acute Slice Preparation
Male C57Bl/6J mice (Jackson Laboratories), male α2A AR−/− mice on a C57Bl/6J background, or male mGluR8−/− mice backcrossed into a C57Bl/6J background (Duvoisin et al, 2005), aged 5–10 weeks, were anesthetized with isoflurane and killed according to the IACUC-approved procedure. The brain was rapidly dissected and placed in ice-cold, oxygenated slicing solution (artificial cerebrospinal fluid (ACSF) in which the sodium is replaced with sucrose). A coronal block of tissue containing the BNST was extracted and sliced on a Leica VT1000S vibratome (Wetzlar, Germany) in a bath of ice-cold, oxygenated, slicing solution. For field potential recordings, hemisected sections of 300 μm thickness containing the BNST were transferred to a humidified interface chamber containing warm (28°C), oxygenated ACSF (in mM: 124 NaCl, 4.4 KCl, 1.2 MgSO4•7H2O, 1 NaH2PO4, 2.5 CaCl2•2H2O, 26 NaHCO3, and 10 glucose) perfused continuously at a rate of 2 to 3 ml/min. Slices were allowed to recover for 30 min, at which time picrotoxin (25 μM) was added to block transmission through GABAA receptors. Slices were then recovered for another 30 min before the onset of recording.
Field Potential Recordings
All recordings took place in the presence of picrotoxin (25 μM) to isolate glutamatergic transmission. Local afferents at the dorsal apex of the dorsal BNST (dBNST) were stimulated from 3 to 30 V to determine the range of a response, and a stimulation intensity corresponding to ∼50% of the maximum response amplitude was used. Responses were evoked at a frequency of 0.05 Hz, with a stimulus duration of 0.065 ms. Field potential responses were recorded using borosilicate glass pipettes (resistance 1–3 MΩ) pulled on a Flaming-Brown micropipette puller (Sutter Instrument, Novato, CA). Pipettes were filled with ACSF and positioned ventrally and laterally to the site of afferent stimulation, within the dBNST. All drugs were bath applied. Clampex 8.2 and 9.2 and Clampfit 9.0 were used to record and analyze all experiments (Molecular Devices, Sunnydale, CA).
Whole-Cell Voltage-Clamp Recordings for PPR Experiments
Slices were placed in a submerged chamber (Warner Instruments) and neurons of the dBNST were visualized with an IR-DIC video microscope (Olympus). Recording electrodes (3–5 MΩ) were pulled on a Flaming-Brown Micropipette Puller (Sutter Instruments) using thin-walled borosilicate glass capillaries. For analysis of EPSCs, electrodes were filled with (in mM): K+ gluconate (135), NaCl (5), HEPES (10), EGTA (0.6), ATP (4), GTP (0.4), and biocytin (0.1%). (Biocytin was routinely included in whole-cell recordings, but no imaging data were obtained in this experiment.) Signals were acquired via a Multiclamp amplifier (Axon Instruments), digitized and analyzed via pClamp 9.0 software (Axon Instruments). Input resistance and series resistance were continuously monitored during the experiment. Stimulating electrodes and their placement were the same as for field potential recordings.
Whole-Cell Recordings and Analysis of Spontaneous EPSCs (sEPSCs)
Recording electrodes (4–6 MΩ) were filled with the following for experiments examining spontaneous excitatory transmission (in mM): 117 Cs gluconate, 20 HEPES, 0.4 EGTA, 5 TEA, 2 MgCl, 4 Na2ATP, and 0.3 Na2GTP (pH 7.2–7.4, Osm 270–290). After achieving whole-cell configuration, cells were allowed to equilibrate a minimum of 5 min before sEPSC recordings were started. sEPSC recordings were acquired in 2 min gap-free blocks while voltage-clamped at −70 mV in the presence of picrotoxin (25 μM). Access resistance was monitored between epochs. Cells in which access resistance changed by >20% or had a basal sEPSC frequency of <0.2 Hz were excluded from the analysis. Following a 6-min baseline period, 10 μM DCPG was bath-applied for 10 min followed by a 6-min washout period. Baseline values were calculated as an average of three baseline epochs (6 min baseline period) directly before drug application. Recorded data were analyzed offline via Clampfit 10.2 (Molecular Devices). Individual sEPSC events were identified via the template search option within the Clampfit program, and each event was visually inspected to ensure template accuracy. All correctly identified sEPSC events from a single recording epoch were then saved as a new file for further analysis of frequency and kinetics via Clampfit.
Drugs
All drugs were bath applied. (S)-3,4-Dicarboxyphenylglycine (DCPG), L-(+)-2-Amino-4-phosphonobutyric acid (L-AP4), (E)-1,1a,7,7a-Tetrahydro-7-(hydroxyimino)-N-phencyclopropa[b]chromene-1a-carboxamide (PHCCC), and (1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268) were obtained from Ascent Scientific (Princeton, NJ). DCPG was also obtained from Tocris Bioscience (Ellisville, MO). Methoxamine hydrochloride and picrotoxin were from Sigma-Aldrich (St Louis, MO).
DCPG, L-AP4, methoxamine, and LY379268 were dissolved in water. PHCCC and picrotoxin were dissolved in DMSO at a final concentration of ⩽0.1% DMSO (final concentration, v/v).
Acute (60-Min) Restraint Stress
All restraint experiments involved mice 5–7 weeks old and occurred between 0900 and 1300 hours (2–6 h after lights were on). A mouse was placed inside a 50 ml conical tube modified to have breathing holes. The tube was then placed on a rack inside a sound- and light-attenuating chamber for 60 min. The mouse was anesthetized with isofluorane for decapitation immediately upon removal from the tube, and the brain was processed for electrophysiological recordings, as described above.
Chronic (10-Day) Restraint Stress
The same age and time-of-day restrictions used for the acute restraint were used for chronic restraint. A mouse was placed in a 50 ml conical tube with breathing holes and transferred to a sound- and light-attenuating chamber for 2 h. After restraint, the mouse was placed back inside its home cage. This continued for 10 consecutive days. On the eleventh day, the mouse was killed and the brain was processed for electrophysiological recordings, as described above.
Statistics
For naive single-drug experiments, Student's t-tests were used to assess significance, and are unpaired unless a paired comparison is explicitly stated. In double-drug experiments, because of unequal variances, analyses were performed using Friedman's nonparametric repeated measures ANOVA and Dunn's multiple comparison test. The α2A adrenergic receptor (AR) KO, acute restraint, and chronic restraint experiments were all compared with a shared pool of interleaved naive DCPG control experiments. To assess significant differences in these experiments, analyses were performed using a one-way ANOVA and Dunnett's post hoc test, comparing the pooled interleaved naive controls with each of the three experimental groups. The numbers for each experiment are indicated in the figure legends.
RESULTS
mGluR8 Activation Depresses Excitatory Transmission in the BNST
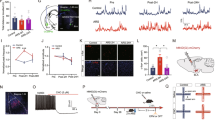
We have previously reported that the group III mGluR agonist L-AP4 depresses excitatory transmission in the BNST (Grueter and Winder, 2005). Here, we used field potential recordings and local afferent stimulation in the anterolateral dBNST to further probe the specific group III mGluR subtypes involved in mediating the effects of L-AP4 in the dBNST (Figure 1a). Consistent with our previous finding, a 20-min bath application of 200 μM L-AP4 produced a small yet significant depression of excitatory transmission in BNST-containing slices (8.8±4.9 peak average percent depression, p<0.05 vs baseline, paired Student's t-test; Figure 1c and f). To investigate the contribution of mGluR4 to the L-AP4 effect on excitatory transmission in the BNST, we utilized the mGluR4-selective allosteric potentiator PHCCC (Maj et al, 2003). PHCCC was applied 15 min before a 20-min co-application of L-AP4 and PHCCC (10.1±2.9 peak average percent depression, Figure 1d and f). PHCCC failed to shift the peak effect of L-AP4, suggesting that mGluR4 does not contribute to the effects of L-AP4 on excitatory transmission in this region (L-AP4: 8.8±4.9 peak average percent depression vs L-AP4+PHCCC: 10.1±2.9 peak average percent depression; Figure 1f).

mGluR8, not mGluR4, mediates the effects of L-AP4 in the BNST. (a) Coronal brain slice showing the dorsal BNST (gray triangle) and the approximate locations of the stimulating and recording electrodes. (b) Confocal image showing a punctate distribution of mGluR8-like immunoreactivity (red) at the dorsal edge of the rat BNST. A Hoechst counterstain (blue) was used to label cell nuclei. (Inset) Anatomical schematic with a small box showing the photographed area. Scale bars in μm. (c) The group III agonist L-AP4 (200 μM) causes a transient yet significant depression in excitatory transmission in BNST (8.8±4.9 peak average percent depression, p<0.05 vs baseline, paired Student's t-test; n=6). (Inset) Representative trace showing the difference in N2 amplitude after L-AP4 (red trace) compared with baseline (black trace). (d) The mGluR4 potentiator PHCCC (30 μM) does not shift the L-AP4 effect in WT mice (n=7). (Inset) Representative trace showing the difference in N2 amplitude after PHCCC+L-AP4 (red trace) compared with baseline (black trace). (e) The effect of PHCCC+L-AP4 is absent in mGluR8 knockout mice (n=6). (Inset) Representative traces showing the lack of difference in N2 amplitude after PHCCC+L-AP4 (red trace) compared with baseline (black trace). (f) No difference in peak effect of average percent depression was observed between PHCCC+L-AP4 and L-AP4 alone, and no effect was present in slices from mGluR8 knockout (KO) mice (L-AP4: 8.8±4.9, L-AP4+PHCCC: 10.1±2.9, mGluR8 KO: 2.6±2.8 peak average percent depression, n=6–7). Scale bars on traces represent 0.2 mV (y axis) and 2 ms (x axis).
We observe mGluR8-like immunoreactivity in the dBNST, suggesting that mGluR8 may be mediating L-AP4 effects in this region (Figure 1b). To test the role of mGluR8 signaling in the actions of L-AP4 in the BNST, we repeated the PHCCC+L-AP4 co-application experiments using brain slices from mGluR8 KO mice (Figure 1e and f). There was no significant effect of L-AP4 or PHCCC in mGluR8 KO mice (WT: 10.1±2.9 peak average percent depression vs mGluR8 KO: 2.6±2.8 peak average percent depression; drug effect in KO compared with baseline fails to reach significance with paired student's t-test; Figure 1f). These data suggest that mGluR8, and not mGluR4, is the primary mediator of the effects of L-AP4 on excitatory transmission in the BNST.
DCPG Depresses Excitatory Transmission in the BNST
To more directly test whether mGluR8 activation has effects on excitatory transmission in the BNST, we utilized the mGluR8-selective agonist DCPG, which has also been previously shown to depress excitatory transmission in this region (Grueter and Winder, 2005). Consistent with this, we found that a 20-min application of DCPG depressed excitatory transmission in the BNST as examined by field potential recordings (Figure 2a). This effect of DCPG was concentration dependent, with 10 and 30 μM DCPG inducing a depression that did not readily reverse (10 μM: 19.9±4.2 peak average percent depression; 30 μM: 16.6±3 peak average percent depression; representative single experiments in Figure 2a). At higher concentrations, DCPG loses selectivity for mGluR8 and can activate mGluR4 as well (Abitbol et al, 2008; Thomas et al, 2001). To determine whether mGluR4 was involved in mediating the effect of DCPG in the BNST, we repeated these experiments with the addition of PHCCC. Adding PHCCC failed to enhance the effect of DCPG, suggesting that mGluR4 is not being recruited by DCPG to regulate excitatory transmission in the BNST (3 μM: 8.9±2.5 peak average percent depression; 3 μM+PHCCC: 8.2±2.8 peak average percent depression; Figure 2c). The finding that PHCCC does not alter the actions of either DCPG or L-AP4 in BNST, coupled with the fact that DCPG has affinity for mGluRs 8 and 4, but not 7, suggests that mGluR8 is the primary receptor mediating the effects of group III agonists on transmission in the BNST.

The mGluR8-selective agonist DCPG depresses excitatory transmission in the dBNST. (a) Representative single experiments demonstrating the effect of 10 μM (closed circles) and 30 μM (open circles) DCPG on excitatory transmission in BNST. (b) DCPG produces a concentration-dependent depression in excitatory transmission (1 μM: 5.4±2.3, 3 μM: 8.9±2.5, 10 μM: 19.9±4.2, and 30 μM: 16.6±3 peak average percent depression, n=6–8). (c) DCPG-induced depression is unaltered by co-application of the mGluR4 potentiator PHCCC (3 μM: 8.9±2.5, 3 μM+30 μM PHCCC: 8.2±2.8 peak average percent depression, n=5–8).
DCPG Modulates Excitatory Transmission Presynaptically in Dorsal BNST
To test the likely site of action of L-AP4 and DCPG, we used paired-pulse ratios (PPRs) of EPCSs. There was an enhancement of PPRs after DCPG application, consistent with group III mGluRs reducing glutamate release probability to depress transmission (1.25±0.14-fold of basal PPRs, data not shown). To further confirm a presynaptic localization of mGluR8, sEPSC frequency and amplitude were examined before and following application of 10 μM DCPG (Figure 3a–d). In agreement with our PPR data, the mean frequency of sEPSCs was significantly decreased following DCPG application (2.4±0.6 Hz control vs 1.5±0.5 Hz post-DCPG, p⩽0.01, paired Student's t-test; Figure 3a, c, and d). The mean amplitude of sEPSCs in the BNST was modestly but significantly decreased by DCPG application (−8.0±0.5 pA control vs −6.5±0.5 pA post-DCPG, p⩽0.005, paired t-test; Figure 3b, c, and d). DCPG had no effect on sEPSC kinetics (Figure 3b). As shown in Figure 3d, however, the effect on frequency was much more pronounced than that on amplitude. Taken together, these data suggest that mGluR8 in the BNST functions as a presynaptic autoreceptor, whose activation reduces glutamate release.

DCPG inhibits glutamatergic transmission in the dBNST. (a) Representative recordings in the dBNST demonstrating the ability of 10 μM DCPG to inhibit the frequency of sEPSC events. (b) Representative sEPSC traces showing the effect of DCPG on sEPSC amplitude. (c) Mean effect of DCPG on sEPSC frequency (Hz; left y axis) and amplitude (pA; right y axis). (Frequency: 2.4±0.6 Hz control vs 1.5±0.5 Hz post-DCPG; *p⩽0.01, paired t-test; amplitude: −8.0±0.5 pA control vs −6.5±0.5 pA post-DCPG; *p⩽0.005, paired t-test; n=8). (d) DCPG reduces sEPSC frequency and amplitude over time, expressed as % control. Note the larger percent inhibition caused by DCPG on sEPSC frequency when compared with sEPSC amplitude.
α1 AR Activation Disrupts DCPG Suppression of Excitatory Transmission
The anxiety phenotype reported in mGluR8 KO mice (Duvoisin et al, 2005, 2010) hints at a potential role for this receptor in counteracting activation of anxiety-promoting circuitry. During a stressor, BNST extracellular levels of norepinephrine (NE) rise dramatically, causing activation of α1 ARs (Cecchi et al, 2002; McElligott et al, 2010). In the PVN, pharmacological or stress-induced activation of α1 ARs leads to a functional inhibition of presynaptic group III mGluRs, and loss of L-AP4 sensitivity (Kuzmiski et al, 2009; Gordon and Bains, 2003). Thus, we hypothesized that stress-induced activation of α1 ARs in the BNST might regulate mGluR8 effects on excitatory transmission.
To determine whether α1 AR activation could alter mGluR8 activity, we first took a pharmacological approach by activating both receptors in series. As previously reported, a 20-min application of 100 μM methoxamine, an α1 AR agonist, produced a robust, persistent depression of excitatory transmission consistent with long-term depression (LTD) (McElligott and Winder, 2008; Figure 5e). After allowing 60 min for the methoxamine effect to be established, DCPG (10 μM) was applied for 20 min (Figure 4b). Consistent with what has been reported for L-AP4 in the PVN (Gordon and Bains, 2003), the effect of DCPG on excitatory transmission was abolished after methoxamine application when compared with the effect of DCPG alone (control DCPG: 19.9±4.2 peak average percent depression vs 1.9±2.1 after methoxamine, p⩽0.005, Friedman's nonparametric repeated measures ANOVA and Dunn's multiple comparison test; Figure 4a and b and inset). To determine whether this lack of an effect of DCPG after methoxamine constituted a shift in potency or efficacy, we repeated the experiment using a high concentration of DCPG (30 μM). The 30 μM concentration of DCPG was able to produce a significant depression after methoxamine, suggesting a shift in the potency of this compound at mGluR8 had occurred (12.5±8.1 peak average percent depression compared with post-methoxamine baseline, p<0.05, paired Student's t-test; Figure 4c and inset). In contrast, when the order of drug application was reversed and slices were pretreated for 20 min with 10 μM DCPG, 100 μM methoxamine still depressed excitatory transmission (control methoxamine: 25.6±6.6 peak average percent depression vs 21.5±4.5 after DCPG, Figure 4d and inset).

Activation of α1 adrenergic receptors disrupts mGluR8 effects on excitatory transmission. (a) The effect of 10 μM DCPG alone (19.9±4.2 peak average percent depression, p⩽0.001 vs baseline, n=8). (b) Methoxamine (100 μM)-induced depression abolishes the effect of 10 μM DCPG (n=6). (Inset) DCPG-induced depression of transmission is lost after methoxamine (control DCPG: 19.9±4.2 peak average percent depression vs 1.9±2.1 after methoxamine, p⩽0.005, Friedman's nonparametric repeated measures ANOVA and Dunn's multiple comparison test, n=6–8). (c) A high concentration of DCPG (30 μM) is able to produce a depression after methoxamine, suggesting a shift in DCPG potency (n=7). 30 μM DCPG does depress transmission significantly after methoxamine (12.5±8.1 peak average percent depression, p<0.05 vs postmethoxamine baseline, paired Student's t-test, n=7). (d) DCPG (10 μM) does not disrupt subsequent methoxamine-induced depression (n=5). (Inset) Methoxamine (100 μM) effect is intact after 10 μM DCPG (control methoxamine: 25.6±6.6 peak average percent depression vs 21.5±4.5 after DCPG, n=5 to 6).
mGluR8 Function Is Selectively Disrupted by Acute In Vivo Stress
Next, we examined whether mGluR8 function is disrupted by acute in vivo stress exposure. Mice underwent one 60-min restraint session and were killed immediately afterward (see Figure 5 schematic). In slices prepared from these mice, the effect of 10 μM DCPG on excitatory transmission was attenuated compared with pooled interleaved naive controls (one-way ANOVA: F(3, 29)=4.802, p=0.0078; stressed: 8.6±1.4 peak average percent depression vs 17.5±2.0 for pooled interleaved naive controls; p⩽0.01, Dunnett's post hoc comparison, Figure 5a and b). However, sensitivity to the group II agonist LY379268 was unaffected after acute stress, demonstrating that the stress-induced disruption is specific to mGluR8 (stressed: 35.7±7.7 peak average percent depression vs 32.5±4.7 for interleaved controls; Figure 5c and d). We have previously reported that chronic restraint stress disrupts α1 AR-mediated LTD (α1-LTD) induction in the BNST (McElligott et al, 2010). Here, we examined the impact of a single restraint stress on this plasticity. Both the peak and late effects of 100 μM methoxamine were modestly but significantly attenuated after a single restraint stress (peak effect: 18.0±1.7 and 24.4±3.0 peak average percent depression for single restraint vs naive controls, respectively; p<0.05, Student's t-test; late effect: 10.2±1.4 and 17.9±2.9 average percent depression for single restraint vs naive controls, respectively; p<0.05, Student's t-test; Figure 5e and f). Taken together with our previous study (McElligott et al, 2010), this suggests that α1 ARs in the BNST were likely activated by the single restraint stress.

A single restraint stress exposure disrupts mGluR8 regulation of transmission in the BNST. (Top) Schematic representing the acute restraint stress timeline. (a) DCPG (10 μM)-induced depression (pooled interleaved naive controls, closed circles) is disrupted in mice that underwent a single restraint stress (open circles; n=8–13). (b) Peak average percent depression of 10 μM DCPG in pooled interleaved naive control mice and acutely restrained mice (stress: 8.6±1.4 peak average percent depression vs 17.5±2.0 for pooled interleaved naive controls; p⩽0.01, Dunnett's post hoc comparison; n=8–13). (c) Group II agonist LY379268-induced depression (1 μM) is intact after restraint (open circles; n=5–7). (d) Peak average percent depression by 1 μM LY379268 in naive and restrained mice (stress: 35.7±7.7 peak average percent depression vs 32.5±4.7 for naive control; n=5–7). (e) Methoxamine-induced depression (100 μM) is altered in acutely restrained mice (open circles; n=4 to 5). (f, left panel) Peak average percent depression by methoxamine is modestly but significantly attenuated in naive vs acutely restrained mice (stress: 18.0±1.7 peak average percent depression vs 24.4±3.0 for naive controls; p<0.05, Student's t-test, n=4 to 5). (f, right panel) Maximal effect of methoxamine is still modestly but significantly attenuated at a late time point (45–52 min after drug removal) (stress: 10.2±1.4 max average percent depression vs 17.9±2.9 for naive controls; p<0.05, Student's t-test, n=4 to 5).
mGluR8 Function Remains Disrupted after Chronic Stress
Finally, we examined whether mGluR8 function is disrupted by a more chronic stressor. Using a protocol shown previously to disrupt α1-LTD in the BNST (McElligott et al, 2010), mice underwent 2 h of restraint stress for 10 consecutive days, and were killed 24 h after their last session, on day 11 (see Figure 6 schematic). As with a single in vivo stress, the effect of 10 μM DCPG was significantly attenuated following chronic restraint stress (chronic restraint: 9.9±3.1 peak average percent depression vs 17.5±2.0 for pooled interleaved naive controls, p<0.05, Dunnett's post hoc comparison; Figure 6a and b). We also assessed mGluR8 function in α2A AR KO mice, which exhibit chronic adrenergic system dysregulation and are considered a chronic stress model (Schramm et al, 2001). The effect of DCPG was also diminished in these mice when compared with pooled interleaved naive controls (α2A AR KO: 9.6±2.6 peak average percent depression vs 17.5±2.0 for pooled interleaved naive controls, p<0.05, Dunnett's post hoc comparison; Figure 6c and d). Taken together, these data reveal that mGluR8 regulation of excitatory transmission can be overridden by adrenergic signaling in the BNST and specifically disrupted by both acute and chronic in vivo stress.

mGluR8 function remains disrupted after chronic stress. (Top) Schematic representing the chronic restraint stress timeline. (a) DCPG (10 μM)-induced depression (pooled interleaved naive controls, closed circles) is disrupted in mice that underwent chronic restraint stress (open circles; n=5–13). (b) Peak average percent depression of 10 μM DCPG in pooled interleaved naive control mice and chronic restraint mice (chronic restraint: 9.9±3.1 peak average percent depression vs 17.5±2.0 for naive controls; p<0.05, Dunnett's post hoc comparison, n=5–13). (c) DCPG-induced depression is also attenuated in α2A AR KO mice, a model of chronic stress. (d) Peak average percent depression of 10 μM DCPG in pooled interleaved naive control mice and α2A AR KO mice (α2A AR KO: 9.6±2.6 peak average percent depression vs 17.5±2.0 for naive controls; p<0.05, Dunnett's post hoc comparison, n=6–13).
DISCUSSION
mGluRs have been shown to alter glutamatergic transmission, playing key roles in several forms of plasticity throughout the brain. In contrast to ionotropic glutamate receptors, the slower nature of mGluR signaling allows for potentially more long-lasting, adaptive changes in synaptic strength. Within the BNST, agonists to group I–III mGluRs depress glutamatergic transmission (Muly et al, 2007; Grueter and Winder, 2005; Grueter et al, 2006). Consistent with previous studies, we report that activation of group III mGluRs with either the general group III agonist L-AP4 or the mGluR8-selective agonist DCPG decreases excitatory transmission in the dBNST. The change in PPRs induced by DCPG, coupled with a significant decrease in sEPSC frequency, suggests that presynaptic mGluR8 is depressing transmission by decreasing glutamate release. As a small effect was also observed on sEPSC amplitude, we cannot currently exclude the possibility that mGluR8 may modulate postsynaptic function as well, although the effects on frequency were much more pronounced. These data are consistent with the autoreceptor function of mGluR8 seen in other brain regions (Ayala et al, 2008; Valenti et al, 2005; Abitbol et al, 2008; Schmid and Fendt, 2006).
We report converging evidence that mGluR8 is the primary mediator of group III ligand effects on excitatory transmission in the BNST. First, the mGluR4 allosteric potentiator PHCCC failed to potentiate the effects of the mGluR8-selective agonist DCPG, suggesting that DCPG is acting solely through mGluR8. Second, PHCCC failed to potentiate the effect of the group III agonist L-AP4, the orthosteric ligand it is commonly paired with. Finally, there was no effect of PHCCC or L-AP4 in slices from mGluR8 KO mice, demonstrating that mGluR8 alone is necessary for the depressive effects of group III ligands on excitatory transmission in this region.
Of the group III receptors expressed in the brain, mGluRs 4 and 8 have a similar affinity for glutamate, and L-AP4 has similar high potency at these two receptors (Schoepp et al, 1999), making mGluR4 the most likely group III receptor other than mGluR8 to be playing a role in regulating excitatory transmission in the BNST. The expression of mGluR6 is restricted to retina, ruling out a contribution in the BNST (Nakajima et al, 1993). However, it is possible that mGluR7 could have effects on transmission in the BNST, as it is widely expressed in the adult brain. mGluR7 is unique among mGluRs in that is has a very low affinity for glutamate, and L-AP4 exhibits very low potency at this receptor (Schoepp et al, 1999). Because of both the low potency of the compound at mGluR7 and also the particularly slow drug on-rate when L-AP4 is used in interface settings, it is unlikely that our concentration of 200 μM L-AP4, bath applied in an interface chamber, yielded any mGluR7 activation. Our own previous studies suggest that even in whole-cell configuration (when slices are submerged, allowing for more efficient drug delivery), near mM concentrations of L-AP4 may be required to activate receptors other than the higher affinity mGluRs 4 and 8 in the BNST (Grueter and Winder 2005). Hence, although it is possible that mGluR7 is present in the BNST and able to regulate transmission, it is highly unlikely to be contributing to the drug effects observed in this study.
We have demonstrated here that mGluR8-expressing, stress-sensitive inputs in the dBNST are regulated by α1 ARs. Previous work has shown that bath application of the α1 AR agonist methoxamine (100 μM) for 20 min induces α1-LTD in the BNST that is modulated by chronic restraint stress (McElligott and Winder, 2008; McElligott et al, 2010). In this study, the same methoxamine application disrupted subsequent mGluR8-induced depression of excitatory transmission in the slice, as did both acute and chronic restraint stress. However, we observed some recovery of mGluR8 function after methoxamine when a high concentration of DCPG (30 μM) was applied. This indicates that the decreased response to 10 μM DCPG seen after methoxamine likely represents a shift in potency. One mechanism that could be underlying this shift is a decrease in mGluR8 signaling caused by desensitization or internalization of the receptor. Importantly, in the reverse experiment, DCPG is unable to disrupt α1-LTD, suggesting that either α1 AR activation is upstream of mGluR8 activation or α1 ARs regulate transmission at mGluR8-expressing and mGluR8-nonexpressing inputs.
Currently, the source of the stress-sensitive, mGluR8-expressing inputs into the dBNST is unknown. The BNST receives excitatory inputs from several limbic brain regions and projects to the PVN, a circuitry thought to be activated during anxiety-related behavior (Walker and Davis, 1997b; McElligott et al, 2010). Both the medial infralimbic cortex (mPFC) and the basolateral amygdala express mGluR8 mRNA, suggesting that these regions could be sending stress-sensitive processes into the BNST (Allen Mouse Brain Atlas, 2009). Indeed, the BNST is a necessary intermediary in the recruitment of the PVN by the mPFC (Radley et al, 2009). Local afferent stimulation as performed in the present study likely indiscriminately activates excitatory inputs from many regions; thus, an interesting possibility is that the modest maximal effect of group III agonists reported here reflects mGluR8 modulation of only a sub-population of afferents.
It should be noted that only mice on a C57Bl/6J background were used in this study. Previous work has shown that anxiety/stress phenotypes vary widely across mouse strains (Mozhui et al, 2010). C57Bl/6J is the line in which all of our previous work examining α1 AR and mGluR effects on excitatory transmission in the BNST was done, and both of the KO mouse strains we used are on a C57Bl/6J background. The impact of knocking out mGluR8 on the adrenergic system is also unknown. We therefore recognize the possibility that mouse strains with an anxiety profile unlike that of C57Bl/6Js may respond differently to these stressors.
In addition to mGluR8, the function of BNST α1 ARs was also attenuated by a single restraint stress, consistent with the idea that α1 ARs in the BNST are activated by in vivo stress (McElligott et al, 2010). A link between α1 ARs, in vivo stress, and group III mGluRs has previously been made in the PVN (Kuzmiski et al, 2009; Gordon and Bains, 2003). However, several factors differentiate those studies from the one reported here. First, the in vivo stressor used in the PVN experiments was hemorrhage—a profound, systemic physiological stressor. We instead used either a single 60-min session or a 10-day chronic protocol of restraint stress, a paradigm shown to increase NE in the BNST and cause activation of α1 ARs (Cecchi et al, 2002; McElligott et al, 2010). Restraint stress is considered to be a more processive stressor than hemorrhage, engaging brain regions such as the BNST that can oppose or allow further activation of stress circuitry via the HPA axis. Recent work from our lab demonstrated that α1-LTD in the BNST is occluded by chronic restraint stress, suggesting engagement of this form of plasticity during stress (McElligott et al, 2010). We report here that α1-LTD in the ex vivo BNST is reduced after a single restraint stress. By recapitulating the α1 AR-mediated disruption of mGluR8 signaling with acute and chronic restraint stress, as well as in the chronic stress model of α2A AR KO mice (which exhibit adrenergic dysregulation), the current study further supports an occlusion hypothesis, and demonstrates that an acute stressor is sufficient to achieve this occlusion.
The studies carried out in the PVN also outline a mechanism by which presynaptic α1 ARs cause desensitization of presynaptic group III mGluRs, measured by loss of L-AP4 sensitivity. The location of the α1 ARs disrupting mGluR8 function in the BNST is, as yet, unknown. Our lab has shown previously that postsynaptic G-protein signaling is necessary for expression of α1-LTD, suggesting a postsynaptic locus for α1 ARs (McElligott and Winder, 2008). One possibility is that postsynaptic α1 ARs are disrupting presynaptic mGluR8 via a retrograde messenger. However, α1 ARs could exist anywhere at the synapse in BNST, including being present at multiple locations, allowing for other mechanisms of mGluR8 disruption by α1 ARs. Alternatively, in the hippocampus, presynaptic group III mGluRs, including mGluR8 specifically, can be inhibited through phosphorylation by PKA (Cai et al, 2001). Thus, any receptor feeding into the cAMP/PKA pathway could desensitize mGluR8.
Importantly, the regulation of mGluR8 signaling by stress appears to be specific. We did not observe any obvious alteration in sensitivity to the group II agonist LY379268 in acutely stressed animals. Group II receptors are known to be presynaptically expressed in BNST and activate Gi/o signaling, similar to mGluR8 (Muly et al, 2007; Grueter and Winder, 2005). Their insensitivity to disruption by a single in vivo stressor suggests that mGluR2/3 do not regulate the same set of excitatory inputs into the BNST regulated by mGluR8 and α1 ARs. Furthermore, mGluR5-mediated LTD is intact after chronic restraint stress, suggesting that postsynaptic mGluRs in the BNST may not be engaged during stress exposure (McElligott et al, 2010). Although activation of any known mGluRs in the BNST depresses transmission, this selective disruption of mGluR8 signaling by stress highlights the potential importance of this receptor in regulating specific sets of inputs coming into the BNST.
We have previously reported that the depressive effects of NE on excitatory transmission in the BNST are time dependent (McElligott and Winder, 2008). A 10-min application of NE transiently depresses excitatory transmission in the BNST, whereas a 20-min application induces α1-LTD, which in turn disinhibits the PVN and allows for HPA axis activation (Cecchi et al, 2002). We propose that under weak stress, increased excitability in the BNST leads to mGluR8 activation, which in turn depresses excitatory transmission in the region, presumably toward restoring homeostatic norms. However, during a single, 60-min restraint stress, BNST levels of NE are elevated for prolonged periods, activating α1 ARs and inducing α1-LTD. In prolonging the decrease in glutamatergic transmission, α1-LTD overrides the signaling mechanisms of mGluR8, perhaps through endocannabinoid signaling. This LTD is then able to decrease the basal inhibition of BNST over the PVN, allowing for HPA axis activation in the face of more prolonged stress exposure.
Finally, mGluR8 function is disrupted in α2A AR KO mice, a chronic stress model (Schramm et al, 2001), and remains disrupted after chronic stress, conditions under which α1-LTD is also known to be disrupted in the BNST (McElligott et al, 2010). The lack of difference between acute and chronic stress effects on mGluR8 function suggests several things. First, the failure of these synapses to adapt to α1 AR-mediated loss of mGluR8 function may indicate an important regulatory role for mGluR8 on these inputs. Likewise, it suggests that these mGluR8-expressing, stress-sensitive inputs are bringing information into the BNST that should be overridden in situations of prolonged stress, thus mediating a valuable adaptive response to a stressor. Last, mGluRs throughout the brain play critical roles in gating metaplasticity. α1 AR activation and subsequent diminished mGluR8 function could be necessary in order to permit activation of other receptors and signaling cascades at these synapses, as part of the brain's stress response. In summary, our data indicate a unique role for mGluR8 among group III mGluRs in the modulation of excitatory transmission in the BNST, and identify this regulation as a target of in vivo stress.
References
Abitbol K, Acher F, Daniel H (2008). Depression of excitatory transmission at PF-PC synapse by group III metabotropic glutamate receptors is provided exclusively by mGluR4 in the rodent cerebellar cortex. J Neurochem 105: 2069–2079.
Allen Mouse Brain Atlas (Internet) (2009). Available from http://mouse.brain-map.org.
Ayala JE, Niswender CM, Luo Q, Banko JL, Conn PJ (2008). Group III mGluR regulation of synaptic transmission at the SC-CA1 synapse is developmentally regulated. Neuropharmacology 54: 804–814.
Bellesi M, Conti F (2010). The mGluR2/3 agonist LY379268 blocks the effects of GLT-1 upregulation on prepulse inhibition of the startle reflex in adult rats. Neuropsychopharmacology 35: 1253–1260.
Cai Z, Saugstad JA, Sorensen SD, Ciombor KJ, Shang C, Schaffhauser H et al (2001). Cyclic AMP-dependent protein kinase phosphorylates group III metabotropic glutamate receptors and inhibits their function as presynaptic receptors. J Neurochem 78: 756–766.
Cecchi M, Khoshbouei H, Javors M, Morilak DA (2002). Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience 112: 13–21.
Conn PJ, Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237.
Dolen G, Osterweil E, Shankaranarayana Rao BS, Smith GB, Auerbach BD, Chattarji S et al (2007). Correction of fragile X syndrome in mice. Neuron 56: 955–962.
Duvoisin RM, Villasana L, Pfangkuch T, Raber J (2010). Sex dependency of anxiety and cognitive phenotypes of mice lacking mGluR8. Behav Brain Res 209: 21–26.
Duvoisin RM, Zhang C, Pfankuch TF, O’Connor H, Gayet-Primo J, Quraishi S et al (2005). Increased measures of anxiety and weight gain in mice lacking the group III metabotropic glutamate receptor mGluR8. Eur J Neurosci 22: 425–436.
Duvoisin RM, Zhang C, Ramonell K (1995). A novel metabotropic glutamate receptor expressed in the retina and olfactory bulb. J Neurosci 15: 3075–3083.
Fendt M, Burki H, Imobersteg S, van der Putten H, McAllister K, Leslie JC et al (2010). The effect of mGlu8 deficiency in animal models of psychiatric diseases. Genes Brain Behav 9: 33–44.
Gordon GRJ, Bains JS (2003). Priming of excitatory synapses by alpha1 adrenoceptor-mediated inhibition of group III metabotropic glutamate receptors. J Neurosci 23: 6223–6231.
Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE et al (2006). Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci 26: 10–19.
Grueter BA, Winder DG (2005). Group II and III metabotropic glutamate receptors suppress excitatory synaptic transmission in the dorsolateral bed nucleus of the stria terminalis. Neuropsychopharmacology 30: 1302–1311.
Herman JP, Cullinan WE (1997). Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20: 78–84.
Kuzmiski JB, Pittman QJ, Bains JS (2009). Metaplasticity of hypothalamic synapses following in vivo challenge. Neuron 62: 839–849.
Linden AM, Johnson BG, Peters SC, Shannon HE, Tian M, Wang Y et al (2002). Increase anxiety-related behavior in mice deficient for metabotropic glutamate 8 (mGlu8) receptor. Neuropharmacology 43: 251–259.
Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W et al (2003). (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology 45: 895–906.
McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL et al (2010). Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci USA 107: 2271–2276.
McElligott ZA, Winder DG (2008). 1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology 33: 2313–2323.
Mozhui K, Karlsson RM, Kash TL, Ihne J, Norcross M, Patel S et al (2010). Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J Neurosci 30: 5357–5367.
Muly EC, Mania I, Guo J-D, Rainnie DG (2007). Group II metabotropic glutamate receptors in anxiety circuitry: correspondence of physiological response and subcellular distribution. J Comp Neurol 505: 682–700.
Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N et al (1993). Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J Biol Chem 268: 11868–11873.
Niswender CM, Johnson KA, Weaver D, Jones CK, Xiang Z, Luo Q et al (2008). Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74: 1345–1358.
Quraishi S, Gayet J, Morgans CW, Duvoisin DM (2007). Distribution of group-III metabotropic glutamate receptors in the retina. J Comp Neurol 501: 931–943.
Radley JJ, Gosselink KL, Sawchenko PE (2009). A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci 29: 7330–7340.
Schmid S, Fendt M (2006). Effects of the mGluR8 agonist (S)-3,4-DCPG in the lateral amygdala on acquisition/expression of fear-potentiated startle, synaptic transmission, and plasticity. Neuropharmacology 50: 154–164.
Schoepp DD, Jane DE, Monn JA (1999). Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38: 1431–1476.
Schramm NL, McDonald MP, Limbird LE (2001). The alpha(2a)-adrenergic receptor plays a protective role in mouse behavioral models of depression and anxiety. J Neurosci 21: 4875–4882.
Thomas NK, Wright RA, Howson PA, Kingston AE, Schoepp DD, Jane DE (2001). (S)-3,4-DCPG, a potent and selective mGlu8a receptor agonist, activates metabotropic glutamate receptors on primary afferent terminals in the neonatal rat spinal cord. Neuropharmacology 40: 311–318.
Ulrich-Lai YM, Herman JP (2009). Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10: 397–409.
Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ (2005). Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther 313: 1296–1304.
Walker DL, Davis M (1997b). Double dissociation between the involvement of the bed nucleus of the stria terminalis and the central nucleus of the amygdala in startle increases produced by conditioned versus unconditioned fear. J Neurosci 17: 9375–9383.
Walker DL, Davis M (2008). Role of the extended amygdala in short-duration versus sustained fear: a tribute to Dr Lennart Heimer. Brain Struct Funct 213: 29–42.
Acknowledgements
We thank Dr Zoe McElligott for valuable feedback on earlier versions of this article, Jacqueline Gayet-Primo for help with the immunohistochemistry, and Dr Chris Olsen for help with statistical analyses. This research was supported by MH77647, AA19455, and DA19112.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Gosnell, H., Silberman, Y., Grueter, B. et al. mGluR8 Modulates Excitatory Transmission in the Bed Nucleus of the Stria Terminalis in a Stress-Dependent Manner. Neuropsychopharmacol 36, 1599–1607 (2011). https://doi.org/10.1038/npp.2011.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.40
Keywords
This article is cited by
-
Targeting metabotropic glutamate receptors for novel treatments of schizophrenia
Molecular Brain (2017)
-
The antidepressant roles of Wnt2 and Wnt3 in stress-induced depression-like behaviors
Translational Psychiatry (2016)
-
Pharmacology of the Bed Nucleus of the Stria Terminalis
Current Pharmacology Reports (2016)
-
The Effects of Stress on Glutamatergic Transmission in the Brain
Molecular Neurobiology (2015)
-
Group III Metabotropic Glutamate Receptors: Pharmacology, Physiology and Therapeutic Potential
Neurochemical Research (2014)
