
Abstract
Some psychiatric illnesses involve a learned component. For example, in posttraumatic stress disorder, memories triggered by trauma-associated cues trigger fear and anxiety, and in addiction, drug-associated cues elicit drug craving and withdrawal. Clinical interventions to reduce the impact of conditioned cues in eliciting these maladaptive conditioned responses are likely to be beneficial. Extinction is a method of lessening conditioned responses and involves repeated exposures to a cue in the absence of the event it once predicted. We believe that an improved understanding of the behavioral and neurobiological mechanisms of extinction will allow extinction-like procedures in the clinic to become more effective. Research on the role of glutamate—the major excitatory neurotransmitter in the mammalian brain—in extinction has led to the development of pharmacotherapeutics to enhance the efficacy of extinction-based protocols in clinical populations. In this review, we describe what has been learned about glutamate actions at its three major receptor types (N-methyl--aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and metabotropic glutamate receptors) in the extinction of conditioned fear, drug craving, and withdrawal. We then discuss how these findings have been applied in clinical research.
Similar content being viewed by others

INTRODUCTION
Psychiatric illnesses are caused by an interaction between genetic and environmental factors. For example, post-traumatic stress disorder (PTSD) develops in vulnerable people on the basis of genetics, early life stress, and exposure to a traumatic event, and involves a constellation of symptoms, including intrusive memories and re-experiencing of the trauma (‘flashbacks’) (American Psychiatric Association, 2000). It is believed that these symptoms arise in part through a Pavlovian conditioning process in which cues present at the time of trauma acquire the ability to elicit fear. Over time, generalization from those cues to other cues and situations leads to pervasive, inappropriate fear and anxiety (Rothbaum and Davis, 2003). Addiction also involves a Pavlovian conditioning component in that cues that reliably precede the onset of drug effects, such as drug paraphernalia, acquire the ability to trigger powerful drug craving and withdrawal responses that contribute to the maintenance of and relapse to drug use (Childress et al, 1986).
There has been considerable interest in developing clinical interventions to reduce the impact of conditioned cues in eliciting these maladaptive responses. One method by which Pavlovian-conditioned responses can be lessened is extinction, a protocol involving repeated or prolonged exposure to a cue (known as the conditioned stimulus) in the absence of the event it once predicted (known as the unconditioned stimulus), resulting in a decrease in the magnitude and/or frequency of the conditioned response. Extinction has been applied successfully in the treatment of fear and anxiety disorders; known as exposure therapy, it involves graded exposure to the feared object, situation, or memory in the absence of any aversive event. Attempts to develop an exposure therapy protocol for addicts involving exposure to drug-paired cues without subsequent drug intake have a similar rationale, but thus far have had limited success (Conklin and Tiffany, 2002).
We believe that a better understanding of the behavioral, psychological, and neurobiological mechanisms of extinction will improve the effectiveness of extinction-like protocols in the clinic. There already have been great strides in this regard: research on extinction in animal models has revealed major mechanisms, and insights into the role of glutamate—the major excitatory neurotransmitter in the mammalian brain—have led to the development of pharmacotherapeutics to enhance the efficacy of exposure therapy in clinical populations. In this review, we describe what has been learned about the role of glutamate in the extinction of conditioned fear, conditioned drug craving, and conditioned withdrawal, and discuss how those findings have been applied in clinical research. We begin with essential background information on animal models for the study of extinction, terminology and basic behavioral features of extinction, and the neural circuitry of extinction. We then turn to the literature on glutamate actions in extinction at its three major receptor subtypes: N-methyl-D-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and metabotropic glutamate receptors (mGluRs). Finally, we consider how these findings have been applied clinically and how they might continue to inform clinical research and practice in the future.
EXTINCTION: FUNDAMENTAL CONCEPTS
Pavlovian conditioning is a form of associative learning in which a conditioned stimulus that predicts the occurrence of an unconditioned stimulus comes to elicit a conditioned response. For example, Pavlov (1927) observed that dogs presented with the sound of a metronome (conditioned stimulus) followed by delivery of food (meat powder; unconditioned stimulus) came to exhibit salivation (conditioned response) to the sound of the metronome alone. Since then, innumerable types of conditioned responses, in species ranging from invertebrates to humans, have been observed. These include both conditioned autonomic responses such as salivation and more complex responses, such as fear evoked by cues associated with trauma in PTSD and drug craving in the presence of drug-related cues in addicts (Heather et al, 1991; Rothbaum and Davis, 2003).
Pavlov (1927) observed that conditioned salivation decreased when dogs were exposed repeatedly to the metronome without being given the meat powder. Known as extinction, this decrease in the magnitude and/or frequency of a conditioned response occurs with repeated or prolonged exposure to the conditioned stimulus in the absence of the unconditioned stimulus. All types of conditioned responses are subject to extinction; in the case of emotional conditioned responses such as fear or drug craving, extinction can be conceptualized as reducing the strength of the conditioned response and thereby re-establishing some control.
Modern extinction research makes extensive use of animal models and has uncovered a great deal about the behavioral characteristics, psychological mechanisms, and neurobiology of extinction. A major theme running through the extinction literature is that although extinction is procedurally simple, it is mechanistically complex. For example, among the putative psychological mechanisms underlying extinction are attentional modulation, habituation-like processes, contextual conditioning, modulation of the strength of associations between the conditioned stimulus and the conditioned response, and changes in the activation threshold of the unconditioned stimulus representation (for a review, see Delamater, 2004). Extinction also likely involves an error correction process in which a discrepancy between the actual and predicted unconditioned stimulus results in a proportional decrease in the strength of the association between the conditioned stimulus and the unconditioned stimulus (Rescorla and Wagner, 1972; Wagner and Rescorla, 1972). The error correction view has been particularly well-studied and leads to strong predictions about the conditions under which extinction occurs; for example, it correctly predicts that extinction will not occur when a conditioned stimulus is nonreinforced in compound with a conditioned inhibitor (defined as a cue that has been trained separately to predict the omission of the unconditioned stimulus), because the discrepancy between the actual unconditioned stimulus and the predicted unconditioned stimulus is zero (Rescorla, 2003; Soltysik et al, 1983). The error correction view also is supported by neurobiological evidence, including findings that dopamine signaling originating in the midbrain (Schultz and Dickinson, 2000) and activity of the inferior olive-climbing fiber system in the cerebellum (Robleto et al, 2004) corresponds to an error signal very much like that envisioned by error correction-based mathematical models of Pavlovian conditioning, such as the Rescorla-Wagner model (1972).
Extinction in Animal Models
Both Pavlovian conditioned responses and instrumental responses (defined as behaviors maintained by a response-contingent outcome, such as delivery of food) are subject to extinction. In general, extinction of one or the other type of response is studied using a specialized animal model, although some models include elements of both. In this review, we are interested primarily in the extinction of Pavlovian conditioned responses as observed in animal models appropriate for the study of conditioned fear, drug seeking, and withdrawal. This focus has implications in terms of the types of cues and responses involved in the studies we will describe. With regard to cues, we define Pavlovian conditioned stimuli as elements of a contingency in which conditioned stimulus presentation predicts unconditioned stimulus delivery, independent of any behavior on the part of the subject. We are not concerned with other, similar types of cues that operate through different mechanisms; these include discriminative stimuli and occasion setters, which are cues that signify that a contingency between other events (a response and an outcome, or a conditioned stimulus and an unconditioned stimulus) is in effect. With regard to the response, we are interested in Pavlovian conditioned responses to Pavlovian conditioned stimuli, as we have defined them. We will not consider extinction of other kinds of responses—most notably instrumental responses—even if the two types of extinction occur within the same paradigm. An example is extinction of lever pressing for a drug of abuse in the intravenous (IV) drug self-administration paradigm (see below), which is an instrumental response. We will not discuss the mechanisms of this kind of extinction, but we will discuss extinction of conditioned stimulus-induced drug craving (defined operationally as described below) within this paradigm, which is a Pavlovian conditioned response.
Most extinction studies focus on conditioned fear responses, but the literature on extinction of conditioned drug craving and withdrawal is growing (Myers and Carlezon, 2010b). The most commonly used animal models for studying these types of responses are Pavlovian fear conditioning, IV drug self-administration, and place conditioning. In each, there is an initial phase of training (known as acquisition) in which an animal (often a rat or a mouse) learns an association between a cue (conditioned stimulus) and a salient event (unconditioned stimulus). Extinction occurs in a subsequent phase of training in which the animal is exposed to the conditioned stimulus in the absence of the unconditioned stimulus.
In the Pavlovian fear conditioning paradigm, acquisition occurs when an animal is presented with a cue such as a light or tone, followed by an aversive stimulus, such as foot shock. After one or more pairings of these events, the animal exhibits fear in the presence of the cue. In rodents, fear is defined operationally in any of several ways; the most common measures are freezing (cessation of all bodily movements, except those required for respiration) and fear-potentiated startle (an increase in the amplitude of the acoustic startle response when startle is elicited in the presence of a fear-eliciting cue vs in its absence). Extinction of conditioned fear occurs when the animal is exposed repeatedly to the conditioned stimulus in the absence of the unconditioned stimulus, and consists of a decrease in the magnitude and/or frequency of the behavioral index of fear.
In the IV drug self-administration paradigm, acquisition occurs over the course of several sessions in which an animal learns to emit an operant response (typically a lever press), which is reinforced by an IV infusion of a drug of abuse. A multimodal stimulus consisting of visual (light) and auditory (tone) components occurs simultaneously with drug infusions and serves as the conditioned stimulus. Cues established in this manner are believed to elicit conditioned drug craving, which is defined operationally as sustainment or reinstatement of drug-seeking behavior (lever pressing) when the cue is presented. Extinction of cue-elicited drug craving occurs over the course of one or more test sessions in which the animal is given access to the lever and the cues are presented periodically but drug delivery is discontinued. Behaviorally, extinction consists of a reduction in cue-elicited drug seeking.
The place conditioning paradigm involves a two- or three-chambered apparatus in which the chambers are distinguished by wall color, floor texture, or both. Typically, the apparatus is configured such that a population of subjects shows no inherent preference for one or the other chamber during a pretraining test in which they are permitted to explore freely. Acquisition occurs when an animal is confined in one of the chambers (which serves as the conditioned stimulus) after an injection of a drug of abuse or a drug that precipitates withdrawal in drug-dependent animals. The drug-paired context thus acquires motivational significance, which is shown in a post-training test in which the animal is once again given the opportunity to explore the apparatus freely. Animals showing conditioned place preference spend more time in the drug-paired chamber than in the alternate chamber, whereas animals showing conditioned place avoidance spend less time in the withdrawal-paired chamber than in the alternate chamber. Extinction of this approach or avoidance response can occur in either of two ways: the animal can be given free access to the place conditioning apparatus in repeated test sessions, or the animal can be confined in the formerly drug- or withdrawal-paired context in the absence of drug administration or precipitated withdrawal (ie, after an injection of saline) and subsequently given free access tests to assess extinction. When extinction has occurred, the animal no longer exhibits a preference for or an aversion to the previously drug- or withdrawal-paired chamber; that is, it spends approximately equal amounts of time in each.
Terminology and Behavioral Features of Extinction
The term ‘extinction’ can be used to describe both a behavioral training protocol and the outcome of exposure to that protocol. As a way of disambiguating this term, we will refer to the experimental protocol of presenting the conditioned stimulus in the absence of the unconditioned stimulus as extinction training and the observed decrease in the magnitude and/or frequency of the conditioned response as extinction. As there is ample evidence that they are mediated by different mechanisms, a further distinction is made between within-session extinction, defined as extinction occurring during extinction training, and extinction retention, defined as extinction memory assessed at some point after the completion of extinction training (typically at least 24 h). Extinction, like other forms of learning and memory, involves encoding, consolidation, and retrieval/expression phases, which are mediated by different neural mechanisms (Box 1).
A critical insight that has emerged from behavioral studies of extinction is that extinction is not due to forgetting, ‘unlearning,’ or erasure of the significance of the conditioned stimulus. Pavlovian conditioned responses are long-lasting and resistant to forgetting, such that without exposure to the conditioned stimulus in the absence of the unconditioned stimulus, the conditioned response does not disappear (Myers and Carlezon, 2010a; Stinus et al, 2000; Quirk, 2002). On the other hand, extinction memory is fragile in that extinguished conditioned responses are subject to recovery under several circumstances. For example, exposure to the unconditioned stimulus alone (ie, in the absence of the conditioned stimulus) after the completion of extinction training can be sufficient to restore the conditioned response, a phenomenon known as reinstatement (Rescorla and Heth, 1975). Similarly, exposure to the conditioned stimulus outside the context of extinction training results in a reappearance of the conditioned response, a very robust and reliable effect known as renewal (Bouton and Bolles, 1979; Chaudhri et al, 2008). Finally, extinguished conditioned responses re-emerge over time after extinction training, an effect known as spontaneous recovery (Brooks et al, 2004; Millin and Riccio, 2002; Pavlov, 1927; Quirk, 2002). Reinstatement, renewal, and spontaneous recovery collectively are known as recovery effects and indicate that extinction does not undo original learning, but is itself a form of new learning in which the conditioned response is suppressed in a context- and time-dependent manner (Bouton, 1993). However, because ‘recovery’ is a clinical term used to describe the beneficial outcome of treatments for psychiatric disorders, including treatments involving extinction protocols, we will describe reinstatement, renewal, and spontaneous recovery as measures of relapse rather than recovery.
Neural Circuitry of Extinction
Extensive work in the fear conditioning paradigm, primarily, has begun to reveal critical elements of the neural circuitry underlying extinction (Figure 1). A model has emerged in which interactions among three key components—the amygdala, medial prefrontal cortex (mPFC), and hippocampus—mediate extinction learning and memory and its modulation by context (Quirk and Mueller, 2008). A small but growing literature suggests that a similar scenario is likely to be true in addiction as well (Myers and Carlezon, 2010b; Peters et al, 2009), but our discussion in this section will draw primarily from the much more extensive fear extinction literature.

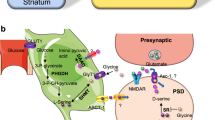
Highly simplified schematic depicting the interactions among the amygdala, infralimbic (IL) region of the medial prefrontal cortex, and hippocampus that are believed to underlie extinction of conditioned fear and, perhaps, extinction of conditioned drug craving and withdrawal as well. The basolateral complex of the amygdala (BLA) is a site of essential plasticity underlying fear memories. BLA receives sensory information about discrete conditioned stimuli, such as lights, tones, and odors, as well as (through the hippocampus) contextual or spatial cues. After acquisition, BLA triggers conditioned fear responses through its projections to the central nucleus of the amygdala (CeA), which in turn innervates hypothalamic and brainstem targets (such as the periaqueductal gray (PAG) and nucleus reticularis pontis caudalis (PnC)) to elicit behavioral indices of fear, including freezing and fear-potentiated startle (FPS). In extinction, the omission of the unconditioned stimulus is detected through a mechanism that is not well understood. CS-related information is relayed to both BLA and IL, and NMDA receptor-dependent synaptic plasticity occurs at both sites. After extinction training is complete, IL contributes to the suppression of fear conditioned responses by inhibiting amygdalar throughput, likely by activating GABAergic interneurons within BLA, GABAergic intercalated cell masses (ICMs) lying between BLA and CeA, or both. A similar scenario may be true for extinction of conditioned drug craving and withdrawal. Drug-related conditioned responses presumably are triggered through different neural intermediaries (such as the ventral tegmental (VTA) or bed nucleus of the stria terminalis (BNST)) than is conditioned fear, but the same basic BLA–mPFC circuitry may be shared, as suggested by findings that NMDA receptor-mediated synaptic plasticity within BLA and mPFC contribute to extinction of cue-induced drug-seeking behavior.
The amygdala is a macrostructure that is the cornerstone of the extinction process. In particular, the basolateral complex (BLA; lateral and basolateral nuclei) and the GABAergic intercalated cell masses (ICMs) that gate impulse traffic from BLA to the central nucleus of the amygdala (CeA) are critical. Individual BLA neurons fire selectively to extinguished cues both as extinction training progresses (Herry et al, 2008) and after extinction training is complete, in a context-dependent manner (Herry et al, 2008; Hobin et al, 2003). Pre-extinction training inactivation of the basal nucleus (Herry et al, 2008) or post-extinction training lesions of the ICMs (Likhtik et al, 2008) impair within-session extinction and extinction retention, respectively (also see Fuchs et al, 2002, 2006). Both encoding and consolidation of extinction memory can be modulated by intra-BLA infusions of a wide variety of drugs, including neurotransmitter receptor agonists and antagonists (Akirav, 2007; Berlau and McGaugh, 2006; Botreau et al, 2006; Falls et al, 1992; Feltenstein and See, 2007; Harris and Westbrook, 1998; Hikind and Maroun, 2008; Kim et al, 2007b; Laurent et al, 2008; Laurent and Westbrook, 2008; Ledgerwood et al, 2003; Lee and Kim, 1998; Lee et al, 2006; Lin et al, 2003b; Mao et al, 2006, 2008; Paolone et al, 2009; Roche et al, 2007; Schroeder and Packard, 2003, 2004; Sotres-Bayon et al, 2007, 2009; Yang et al, 2006; Walker et al, 2002), as well as modulators of downstream second messengers, transcription, and translation (Lin et al, 2003a, 2003b; Lu et al, 2001). Extinction alters the phosphorylation state of second messengers and gene expression patterns within BLA (Cannich et al, 2004; Chhatwal et al, 2005, 2006; Heldt and Ressler, 2007; Herry and Mons, 2004; Lin et al, 2003a, 2003b), and manipulation of gene expression within BLA before extinction training modulates extinction memory consolidation (Chhatwal et al, 2006). Hence, the amygdala is a central locus underlying the encoding, consolidation, and expression of extinction memory.
The mPFC sends dense projections to the amygdala that terminate, in part, on GABAergic interneurons in BLA and on the ICMs (Berretta et al, 2005; Quirk et al, 2003; Rosenkranz and Grace, 2001, 2002; Rosenkranz et al, 2003). Hence, mPFC is in a position to exert inhibitory control over amygdalar throughput, a possible extinction mechanism (Quirk and Mueller, 2008), at least under some circumstances (cf. Garcia et al, 2006; Gewirtz et al, 1997). Electrolytic lesions (Quirk et al, 2000) or localized inactivation (Sierra-Mercado et al, 2006) of the infralimbic (IL) region of mPFC impair extinction retention while having little to no effect on acquisition or within-session extinction, suggesting a role for this region specifically in consolidation and/or expression of extinction memory (also see Hsu and Packard, 2008). Single units within IL fire selectively to presentations of a previously fear conditioned cue during an extinction retention test 24 h after extinction training but not during the extinction training session itself (Milad and Quirk, 2002). Pre-extinction training, intra-mPFC or intra-IL infusions of neurotransmitter receptor agonists or antagonists (Burgos-Robles et al, 2007; Hikind and Maroun, 2008; Laurent and Westbrook, 2008; Mueller et al, 2008; Pfeiffer and Fendt, 2006; Sotres-Bayon et al, 2009; Zushida et al, 2007), as well as modulators of downstream second messengers, transcription, and translation (Hugues et al, 2004, 2006; Mueller et al, 2008; Santini et al, 2004), modulate extinction retention without affecting within-session extinction. Immediate post-extinction training administration of many of these agents has a similar effect. When IL microstimulation is paired with presentations of a previously fear conditioned cue in nonextinguished animals, freezing to those cues is attenuated (Milad and Quirk, 2002; Milad et al, 2004). Collectively, these findings indicate that mPFC has a significant role in many cases in extinction memory consolidation and expression, likely by its interactions with the amygdala.
The hippocampus also sends dense projections to the amygdala (Pitkanen et al, 2000) and substantially innervates mPFC as well (Jay and Witter, 1991). The hippocampus has long been known to mediate spatial and contextual memory (Kim and Fanselow, 1992; Morris et al, 1982), and consistent with this, a role for it in the context dependence of extinction has emerged. When hippocampal function is intact throughout acquisition and extinction training, lesions or temporary inactivation of the hippocampus before an extinction retention test block renewal of extinguished freezing when that test occurs outside of the extinction training context (Corcoran et al, 2005; Corcoran and Maren, 2001, 2004; Hobin et al, 2006; Ji and Maren, 2008; Maren and Hobin, 2007). In other words, contextual modulation of extinction memory expression is diminished in the absence of hippocampal input. This effect is not restricted to behavioral indices of conditioned fear, but is seen with conditioned stimulus-evoked single-unit responses in BLA as well, such that hippocampal inactivation before an extinction retention test blocks the contextual renewal of conditioned stimulus-related single-unit firing (Maren and Hobin, 2007). On the other hand, temporary inactivation of the hippocampus before extinction training enhances the contextual sensitivity of extinction memory, such that renewal is observed in a subsequent extinction retention test even when that test is conducted in the same context as extinction training (Corcoran et al, 2005). One possible interpretation of this finding is that prevention of contextual encoding by the hippocampus during extinction training causes the extinction context not to be recognized as such when hippocampal functionality is restored, leading to renewal. Taken together, these findings indicate that the hippocampus is responsible for encoding contextual information during extinction training and subsequently using that information to promote or impede expression of extinction memory, as appropriate, perhaps by its interactions with the amygdala and mPFC.
NMDA RECEPTORS
The NMDA receptor is an ionotropic receptor that has the unique feature of being doubly gated, requiring both membrane depolarization and ligand binding for activation (Seeburg et al, 1995). NMDA receptors are located throughout the brain and are heterotetramers comprising two obligatory NR1 subunits and two subunits from the NR2 family (denoted NR2A–D) or the more recently discovered NR3 family. They are implicated heavily in learning, memory, and experience-dependent forms of synaptic plasticity, such as long-term potentiation (LTP) (Nicoll and Malenka, 1999), with NR2A- and NR2B-containing NMDA receptors being particularly important in this regard. Subunit composition greatly influences receptor properties; for example, NR2A-containing receptors have lower glutamate affinity, faster kinetics, and greater channel open probability than do NR2B-containing receptors (Cull-Candy and Leszkiewicz, 2004). Functional differences between NR2A- and NR2B-containing receptors have been noted as well (Walker and Davis, 2008), although the nature of these differences is highly dependent on the brain region and developmental stage in question.
The NMDA receptor was the first of the glutamate receptors to be implicated in extinction and continues to be the most thoroughly studied. Owing to the size of the literature, we will consider studies involving NMDA receptor antagonism, positive modulation of NMDA receptor function, and modulation of NMDA subunit expression in separate subsections.
NMDA Receptor Antagonism
Studies involving systemic administration of NMDA receptor antagonists before fear extinction training report dose-dependent impairments of both within-session extinction and extinction retention (Baker and Azorlosa, 1996; Chan and McNally, 2009; Cox and Westbrook, 1994; Dalton et al, 2008; Kelamangalath et al, 2007; Lee et al, 2006; Liu et al, 2009; Santini et al, 2001; Sotres-Bayon et al, 2007, 2009; Storsve et al, 2010; Walker et al, 2002) (Table 1). Systemic NMDA receptor antagonists also impair extinction retention or reinstatement when administered immediately after extinction training (Burgos-Robles et al, 2007; Laurent et al, 2008; Johnson et al, 2000; Laurent and Westbrook, 2008; Liu et al, 2009; Santini et al, 2004; Sotres-Bayon et al, 2009) (Table 1), indicating that NMDA receptors are involved in consolidation as well as encoding of extinction memory.
NMDA receptors within BLA and IL contribute to different aspects or phases of fear extinction. Microinfusions of NMDA receptor antagonists into BLA before fear extinction training impair both within-session extinction and extinction retention (Falls et al, 1992; Feltenstein and See, 2007; Laurent et al, 2008; Laurent and Westbrook, 2008; Lee and Kim, 1998; Lin et al, 2003b; Sotres-Bayon et al, 2007). However, local infusions of NMDA 2A and 2B antagonists into BLA block the expression of several fear-related conditioned responses, including freezing, suggesting that these drugs could artifactually block extinction retention by interfering with synaptic transmission. Arguing against this possibility is the observation that infusions of ifenprodil, an NMDA 2B-preferring antagonist that does not block expression of fear conditioned responses, also blocks extinction retention (Laurent et al, 2008; Laurent and Westbrook, 2008; Sotres-Bayon et al, 2007). Immediate post-extinction training intra-BLA infusions of ifenprodil have no effect on subsequent extinction retention when extinction of fear is measured (Laurent and Westbrook, 2008; Sotres-Bayon et al, 2009), but similar intra-BLA infusions of AP5 do impair extinction retention when cocaine conditioned place preference is measured (Feltenstein and See, 2007). This suggests that NMDA receptor-dependent synaptic plasticity within BLA is involved in encoding fear extinction memory, but that NR2B-containing NMDA receptors within BLA are not required for consolidation, at least for conditioned fear. With regard to IL, pre-extinction training infusions of NMDA receptor antagonists into IL but not the neighboring prelimbic cortex have no effect on within-session extinction but generally impair later extinction retention (Burgos-Robles et al, 2007; Laurent and Westbrook, 2008, 2009a; but see Sotres-Bayon et al, 2009). Immediate post-extinction infusions of NMDA receptor antagonists into IL block extinction retention consistently (Burgos-Robles et al, 2007; Laurent and Westbrook, 2008; Sotres-Bayon et al, 2009), providing strong evidence that NMDA receptor-dependent synaptic plasticity within IL is primarily involved in the consolidation of extinction memory.
Interestingly, the contribution of NMDA receptors to fear extinction shifts somewhat when a cue is extinguished a second time; that is, when the experimental protocol involves fear acquisition and extinction, followed by reacquisition and re-extinction of the same cue. Systemic administration of the NMDA receptor antagonist MK-801 before re-extinction training does not impair subsequent extinction retention, unless re-extinction and the extinction retention test occur in a context different from that of initial acquisition and initial extinction (Chan and McNally, 2009). This may result from a more general phenomenon, namely that NMDA receptor activation is required when extinction events are relatively novel but not when they are relatively familiar (Chan and McNally, 2009). Thus, systemic administration of MK-801 does block second extinction if the re-extinguished conditioned stimulus or the context of re-extinction training is relatively novel by virtue of fewer extinction trials or a shift in context. Conversely, MK-801 fails to block first extinction if the extinguished conditioned stimulus or the context of extinction training is relatively familiar by virtue of pre-exposure to the conditioned stimulus or context. On the other hand, novelty does not seem to matter for fear conditioning itself because AP5 blocks fear acquisition in both a novel and a familiar context (Lee and Kim, 1998; Laurent and Westbrook, 2009b). Effects with localized infusions of NMDA receptor antagonists before second extinction are complex. Intra-BLA infusions of AP5 before re-extinction training have no effect on subsequent extinction retention; however, infusions of ifenprodil impair both within-session extinction and extinction retention (Laurent et al, 2008; Laurent and Westbrook, 2008). Interestingly, intra-BLA infusions of AP5 do block re-extinction if initial extinction was also blocked by intra-BLA AP5 (Laurent et al, 2008), as if blockade of the first extinction restored the sensitivity of the second extinction to NMDA receptor blockade. However, this does not seem to hold for localized infusions into mPFC, where either pre-extinction or immediate post-extinction training infusions of ifenprodil block retention of both first and second extinction (Laurent and Westbrook, 2008).
NMDA receptor antagonism impairs extinction not only of fear conditioned responses but also of conditioned drug craving. Intra-mPFC, pre-extinction training infusion of AP5 impairs extinction of amphetamine conditioned place preference (Hsu and Packard, 2008). In the drug IV self-administration paradigm, systemic pre-extinction training administration of the competitive NMDA receptor antagonist CPP is associated with more robust cocaine-induced reinstatement, perhaps indicative of reduced extinction (Kelamangalath et al, 2007), and intra-BLA infusions of AP5 immediately after each of several test (ie, cue extinction) sessions leads to more persistent cue-maintained drug-seeking behavior, perhaps because extinction of the response-reinstating or response-maintaining value of the cues is impaired (Feltenstein and See, 2007).
Positive Modulation of NMDA Receptor Function
Whereas antagonism of NMDA receptors impairs extinction, enhancement of NMDA receptor function by the NMDA receptor partial agonist D-cycloserine (DCS) facilitates extinction. DCS acts at the glycine modulatory site on the NR1 NMDA receptor subunit to increase calcium influx without causing damage due to neurotoxicity (Sheinin et al, 2001).
In the Pavlovian fear conditioning paradigm, systemic administration of DCS either before (Bouton et al, 2008; Weber et al, 2007; Hefner et al, 2008; Kelley et al, 2007; Langton and Richardson, 2008; Ledgerwood et al, 2003, 2005; Lee et al, 2006; Lin et al, 2009a, 2009b; Mao et al, 2006, 2008; Myers and Carlezon, 2010a; Tomilenko and Dubrovina, 2007; Walker et al, 2002; Weber et al, 2007; Woods and Bouton, 2006; Yamada et al, 2009; Yang and Lu, 2005) or after (Ledgerwood et al, 2003, 2004, 2005; Parnas et al, 2005; Weber et al, 2007; Werner-Seidler and Richardson, 2007) extinction training facilitates extinction (Table 2). Similarly, mutant mice with a single point mutation in the gene encoding D-amino acid oxidase, a catabolic enzyme of D-serine (an endogenous ligand at the NR1 glycine modulatory site), show increased whole-brain D-serine concentrations and facilitated extinction of freezing to contextual, but not auditory, cues (Labrie et al, 2009). Immediate post-extinction injection of spermine, another positive modulator of the NMDA receptor, into the hippocampus facilitates extinction of the inhibitory avoidance response (Gomes et al, 2010). To our knowledge, no published studies have examined the effect of DCS infusions directly into IL, but local infusion of DCS into BLA before (Akirav, 2007; Lee et al, 2006; Mao et al, 2006, 2008; Walker et al, 2002) or after (Ledgerwood et al, 2003) fear extinction training mimics the effects of systemic administration.
DCS may be more effective under some circumstances than others (Table 3). For example, there is a growing body of evidence to suggest that DCS reverses fear extinction deficits that are observed after stress (Akirav, 2007; Matsumoto et al, 2008; Nic Dhonnchadha et al, 2010; Yamamoto et al, 2008), alcohol withdrawal (Bertotto et al, 2006), or REM sleep deprivation (Silvestri and Root, 2008); in genetically modified mice that have a polymorphism in the BDNF gene (val-met substitution) (Yu et al, 2009); and even when muscimol is infused into BLA (Akirav, 2007). Perhaps consistent with these stress-related effects, DCS interacts with stress hormones: DCS blocks the extinction-impairing effect of the corticosteroid synthesis inhibitor metyrapone and enhances the extinction-facilitating effects of the synthetic glucocorticoid dexamethasone (Yang et al, 2007). These observations also may be consistent with findings in clinical studies (described below, in the ‘Targeting glutamate clinically: clinical use of DCS’ section) that DCS facilitates exposure therapy in clinical, but not in subclinical, populations. Finally, DCS facilitates extinction retention only in rats that show some evidence of within-session extinction during extinction training and not in rats that are resistant to extinction (Bouton et al, 2008; Hefner et al, 2008; Weber et al, 2007), and DCS administered before re-extinction training has no effect, similar to the failure of systemic NMDA receptor antagonists to block re-extinction under many circumstances (Langton and Richardson, 2008).
Yang and Lu (2005) examined the molecular mechanisms underlying DCS-induced facilitation of fear extinction. They found that exposure to a suboptimal extinction training protocol modestly increases phospho-MAPK and phospho-Akt (the activated forms of kinases that have important roles in intracellular signaling cascades) in BLA, and that exposure to this same protocol after administration of DCS leads to more robust increases. Consistent with this, intra-BLA infusions of the MAPK inhibitors U0126 or PD98059 or the PI3K inhibitor wortmannin block the behavioral facilitation of extinction by DCS. Intra-BLA infusions of actinomycin D, a transcriptional inhibitor, and anisomycin, a protein synthesis inhibitor, also block DCS-induced facilitation of extinction. In the ‘AMPA receptors’ section, we describe other findings indicating that DCS modulates the expression and trafficking of AMPA receptor subunits in BLA (Lin et al, 2009a, 2009b; Mao et al, 2006; Mao et al, 2008).
Extinction of conditioned drug craving and withdrawal is sensitive to DCS as well. Systemic administration of DCS before or immediately after repeated place preference tests, or localized infusion of DCS into the BLA immediately after repeated tests, facilitates extinction of cocaine conditioned place preference (Botreau et al, 2006; Paolone et al, 2009; Thanos et al, 2009). Pre-extinction training systemic administration of DCS also facilitates extinction of naloxone-induced conditioned place aversion in morphine-dependent rats (Myers and Carlezon, 2010a). DCS does not enhance the rate of extinction of ethanol conditioned place preference but retards subsequent reconditioning, suggesting a facilitation of extinction that may have been obscured by floor or performance effects (Groblewski et al, 2009); similarly, pre-extinction training DCS dose dependently retards subsequent reacquisition of cocaine seeking in both rats and squirrel monkeys (Nic Dhonnchadha et al, 2010; but see Sakurai et al, 2007). Finally, in cocaine (Nic Dhonnchadha et al, 2010) and ethanol (Vengeliene et al, 2008) self-administration paradigms, rats receiving DCS before sessions in which drug delivery was discontinued but response-contingent cues continued to occur stopped responding more rapidly than did rats receiving vehicle, presumably because DCS facilitated extinction of the secondary reinforcing value of the cues. DCS also facilitates extinction of maze running or bar pressing for food (Gabriele and Packard, 2007; Shaw et al, 2009). In some of these studies, DCS-facilitated extinction is especially enduring: DCS reduced cocaine-induced reinstatement (Kelamangalath et al, 2009; Paolone et al, 2009; but see Kelley et al, 2007) and retarded spontaneous recovery (Botreau et al, 2006; Kelley et al, 2007).
Modulation of NMDA Receptor Subunit Expression
Some investigators have noted extinction-related changes in NMDA receptor subunit expression in brain areas including mPFC and hippocampus. In a study examining the effect of previous corticosterone (cort) exposure on subsequent fear learning, cort-exposed rats showed impaired extinction and reduced NR2B expression within vmPFC (Gourley et al, 2009). Rats exhibiting the poorest extinction showed the least NR2B expression, suggesting that the extinction deficit could be a product of changes in vmPFC NMDA receptor expression induced by previous cort exposure. Similarly, rats that had undergone maternal separation as pups showed impaired fear extinction and reduced NR1 expression in IL (Wilber et al, 2009). In a study involving drug-paired cues, rats that received noncontingent (yoked) administrations of cocaine in an operant chamber and which subsequently were exposed to that chamber in the absence of cocaine showed increased expression of the NR1 subunit in mPFC (Crespo et al, 2002). In the hippocampus, contextual fear extinction was found to be associated with decreased expression of NR1, NR2A, and NR2C, and increased expression of NR2B (Yamamoto et al, 2008). Finally, transgenic mice with elevated forebrain NR2B expression show facilitated fear extinction (Tang et al, 1999), suggesting that genetic manipulations that increase NMDA receptor expression can positively impact extinction.
AMPA RECEPTORS
The AMPA receptor is found throughout the brain and is an ionotropic receptor mediating fast synaptic transmission. AMPA receptors are tetramers composed of subunits designated GluR1-4, which assemble as a ‘dimer of dimers.’ In addition to its role as a mediator of basal synaptic transmission, the AMPA receptor also is involved in experience-dependent forms of synaptic plasticity (eg, see Carlezon and Nestler, 2002; Malinow and Malenka, 2002).
The literature on the involvement of AMPA receptors in extinction comes almost entirely from studies of fear extinction (Table 4). An early finding was that infusion of the AMPA receptor antagonist CNQX into BLA before extinction training had no effect on subsequent extinction retention (Falls et al, 1992). The possibility that the small amount of fear seen in the extinction retention test was due to amygdalar neurotoxicity as opposed to intact extinction was ruled out in a separate experiment, in which rats infused with CNQX without undergoing extinction training showed robust fear in a test conducted at the same post-infusion interval. These findings, which have since been replicated by a different group (Lin et al, 2003b), were unexpected and have been somewhat of an enigma in the field, as it is not immediately obvious why blocking the action of glutamate at AMPA receptors in BLA should have no effect, given that BLA activation is considered by most to be critical for fear extinction. However, they do show that extinction can still occur when expression of the conditioned response is blocked, as often occurs with local infusion of AMPA receptor antagonists into BLA (Kim et al, 1993).
A recent finding indicates that the AMPA receptor agonist PEPA dose-dependently facilitates contextual fear extinction when administered systemically to mice before extinction training (Yamada et al, 2009; Zushida et al, 2007). PEPA had a similar facilitatory effect when infused directly into the anterior cingulate/prelimbic region of mPFC and (in a separate experiment) into the basolateral/central amygdala, although the effect size was larger with mPFC infusions (Zushida et al, 2007). Consistent with these behavioral findings, electrophysiological and gene expression (qPCR) analyses indicated that GluR3 and GluR4 subunits and flop AMPA receptor splice variants—which are preferred by PEPA—are enriched in mPFC (Zushida et al, 2007).
A potential mechanism whereby PEPA might exert its behavioral effect is by promotion of AMPA receptor internalization. Conditioned fear acquisition is associated with increased cell-surface expression of GluR1 and GluR2 subunits in BLA (Rumpel et al, 2005); recent reports indicate that fear extinction, under some circumstances, reverses this increase. Kim et al (2007a) found decreased cell-surface expression of GluR1 and GluR2 subunits in BLA in extinguished animals relative to that seen in fear-conditioned controls that did not undergo extinction. Similarly, Mao et al (2006, 2008) reported decreased cell-surface and increased cytoplasmic GluR1 and GluR2 subunit expression in BLA after extinction, when extinction training occurred soon (1 h) after fear acquisition but not at longer intervals (24–48 h), unless extinction training 24–48 h after acquisition occurred under the influence of DCS. Consistent with these biochemical findings, Lin et al (2009a, 2009b) performed whole-cell patch-clamp recordings of BLA neurons in slices obtained from rats that had been fear conditioned and extinguished, and found that increased AMPA/NMDA ratios seen after fear acquisition were reversed after extinction coupled with DCS. Molecular mechanisms of AMPA receptor internalization in extinction include unbinding of GluR subunits from scaffold proteins, including PSD-95 and SAP97 (Mao et al, 2008), activation of the phosphatase calcineurin (Havekes et al, 2008; Lin et al, 2003a), decoupling of protein kinase A from its anchoring protein AKAP (Isiegas et al, 2006; Nijholt et al, 2008), and proteolysis (Mao et al, 2008).
Recently, a novel tool has become available that can be used to manipulate AMPA receptor internalization directly. Known as GluR23Y, it is a synthetic peptide containing a sequence of amino acids found in the carboxy terminus of the GluR2 subunit that is critical to activity-induced AMPA receptor internalization. By competing with endogenous GluR2 subunits, GluR23Y largely blocks AMPA receptor internalization and associated forms of synaptic plasticity, including long-term depression and depotentiation (Brebner et al, 2005). GluR23Y can be dialyzed directly into neurons during whole-cell patch-clamp recordings or conjugated to the HIV Tat protein, which allows it to cross cell membranes, permitting intracerebral or even systemic administration. In amygdala slices, GluR23Y (but not GluR23A, a control construct) blocks the induction of GluR2 internalization (Lin et al, 2009a, 2009b) and synaptic depotentiation in slices obtained from fear-conditioned animals (Kim et al, 2007a). Behaviorally, systemic administration (Dalton et al, 2008) or intra-BLA infusion (Kim et al, 2007a) of Tat-GluR23Y (but not GluR23A) before extinction training impairs within-session extinction and subsequent extinction retention. GluR23Y had no effect when administered in the absence of extinction training (Kim et al, 2007a) or before a test of extinction retention in animals extinguished previously (Dalton et al, 2008). Finally, Lin et al (2009a, 2009b) found that Tat-GluR23Y infused into BLA before extinction training blocks DCS-induced facilitation of extinction and the accompanying decrease in AMPA/NMDA ratios.
AMPA receptor binding is sufficient but not necessary for AMPA receptor internalization. There is evidence for NMDA receptor-dependent AMPA receptor endocytosis in cultured hippocampal (Beattie et al, 2000) and nucleus accumbens neurons (Mangiavacchi and Wolf, 2004) and in the lateral amygdala in vitro (Mao et al, 2008; Yu et al, 2008). In this light, it may be understandable why pre-extinction training intra-BLA infusions of AMPA receptor antagonists have no apparent effect on extinction: if extinction-related AMPA receptor internalization in BLA in vivo were mediated primarily by glutamate binding to NMDA receptors, then AMPA receptor antagonists would not be expected to have an effect. This might provide a mechanism for DCS-induced facilitation of fear extinction: rather than (or in addition to) facilitating extinction-related LTP in BLA, DCS might enhance NMDA receptor-dependent AMPA receptor internalization. Consistent with this, Mao et al (2008) found that DCS facilitated NMDA-induced GluR1 and GluR2 internalization in BLA in vitro.
AMPA receptor internalization is a mechanism of synaptic depotentiation, a form of synaptic plasticity amounting to a reversal of LTP (Huang and Hsu, 2001). Other evidence for a role of BLA depotentiation in extinction includes the finding that application of depotentiation-inducing stimulation to BLA in vivo in previously fear-conditioned animals leads to attenuation of fear memory expression (Lin et al, 2003) and that the induction of BLA depotentiation in vitro is occluded in slices obtained from extinguished animals (Kim et al, 2007a). A puzzle still to be solved in the extinction field is how reversal of BLA LTP—which is believed to underlie fear memory storage (Sigurdsson et al, 2007)—can be reconciled with relapse phenomena indicating that extinction is not ‘unlearning.’ The most likely scenario is that depotentiation is but one of multiple extinction mechanisms. For example, some conditioned stimulus-related single-unit responses in BLA diminish over the course of extinction training, potentially through a depotentiation mechanism, but some persist (Herry et al, 2008; Quirk et al, 1997; Repa et al, 2001) and others develop as extinction training progresses (Herry et al, 2008), suggesting that other forms of synaptic plasticity are involved. In addition, modulation of GABAergic (and not just glutamatergic) neurotransmission has a significant role in extinction in that extinction is impaired by administration of a GABAA receptor inverse agonist (Harris and Westbrook, 1998) or targeted lesions of the ICMs (Likhtik et al, 2008) and is associated with increased frequency and amplitude of IPSCs (Lin et al, 2009a, 2009b), GABA-related gene expression (Chhatwal et al, 2005; Heldt and Ressler, 2007), and GABAA receptor membrane insertion in BLA (Lin et al, 2009a, 2009b). Finally, as we have seen, BLA is one of the several sites of extinction-related plasticity; mPFC and hippocampus are engaged as well and may contribute to the expression of extinction memory by modulating amygdalar activity. A major goal of extinction research is to determine how these varied mechanisms interact to produce context- and time-dependent conditioned response suppression.
The literature on extinction of drug- and withdrawal-paired cues is largely silent on AMPA receptor involvement, with the exception of two studies on Narp, which is a protein that binds to AMPA receptors and contributes to their trafficking and clustering at synapses. Both of these studies were conducted in the place conditioning paradigm; the major findings were that Narp knockout mice show delayed extinction of morphine conditioned place preference (Crombag et al, 2009) and enhanced extinction of morphine withdrawal-induced conditioned place aversion (Reti et al, 2008). It is not immediately obvious how to interpret these findings, both because the knockout was constitutive rather than localized and because so little is known about the neurobiological underpinnings of these two types of extinction, which likely rely on somewhat different substrates. However, at the very least, the data suggest that AMPA receptors have an important role in the extinction of drug- and withdrawal-paired cues, just as they do in fear extinction (also see Sutton et al, 2003). In addition to these studies, there is a recent report that GluR2 expression and AMPA/NMDA current ratios were decreased in mPFC in rats that had been re-exposed to a heroin-associated cue in the IVSA paradigm after extinction of lever pressing (Van den Oever et al, 2008). Although the authors of this study suggest that these changes are associated with cue-induced reinstatement of drug-seeking behavior, an alternative explanation is that the changes are reflective of extinction of the response-reinstating value of the cues over the course of the test session.
METABOTROPIC GLUTAMATE RECEPTORS
Metabotropic glutamate receptors (mGluRs) are G protein-coupled receptors mediating slow synaptic transmission. There are eight subtypes, denoted mGluR1–8, which are classified into three groups based on sequence similarity and signal transduction mechanisms: group I (mGluR1, mGluR5); group II (mGluR2, mGluR3); and group III (mGluR4, mGluR6-8). They are located throughout the brain, with different subtypes having different regional and developmental patterns of expression. Some subtypes (eg, mGluR5) have been implicated in learning and memory and experience-dependent forms of synaptic plasticity (Simonyi et al, 2005).
Studies on the contribution of mGluRs to extinction have appeared relatively recently, and the literature at this time is somewhat limited (Table 4). Thus far, there are data regarding the role of mGluR1, mGluR5, and mGluR7.
Localized infusions of the mGluR1 antagonist CPCCOEt into BLA before extinction training dose-dependently impair within-session extinction and subsequent extinction retention when extinction training occurs 48 h after acquisition, but not 1 h after acquisition (Kim et al, 2007b). The apparent time-dependent nature of mGluR1 involvement is consistent with evidence from other sources that the mechanisms of short- and long-interval extinction may differ (Cain et al, 2005; Mao et al, 2006; Maren and Chang, 2006; Myers et al, 2006). In an electrophysiological study, Kim et al (2007a) found that CPCCOEt blocks ex vivo induction of synaptic depotentiation in BLA in slices obtained from fear-conditioned animals, whereas the group I mGluR agonist DHPG induces depotentiation in these slices (also see Hong et al, 2009). These findings may be consistent with those described in the ‘AMPA receptors’ section suggesting that BLA depotentiation is a mechanism of extinction, at least under some circumstances.
Both mGluR5 knockout mice (Xu et al, 2009) and mGluR7 knockout mice (Callaerts-Vegh et al, 2006; Goddyn et al, 2008) show deficits in fear extinction. Conversely, systemic administration of the newly synthesized mGluR7 agonist AMN082 before extinction training dose-dependently facilitates fear extinction (Fendt et al, 2008). AMN082 has no effect when administered in the absence of extinction training (Fendt et al, 2008).
Extinction of conditioned responses to drug-paired cues is sensitive to manipulations of mGluR function as well, as indicated by the finding that systemic administration of CDPPB, a positive allosteric modulator of mGluR5, before extinction training dose-dependently facilitates extinction of cocaine conditioned place preference (Gass and Olive, 2009). This effect was blocked by coadministration of the mGluR5 antagonist MTEP or the NMDA receptor antagonist MK-801, highlighting the functional interactions between mGluRs and NMDA receptors.
TARGETING GLUTAMATE CLINICALLY: CLINICAL USE OF DCS
The finding that DCS can facilitate fear extinction in animals (Walker et al, 2002) sparked interest in the idea that this drug might also be effective in facilitating exposure therapy for fear and anxiety disorders in people. DCS has been FDA-approved for some time as an antibiotic treatment for tuberculosis at high doses, allowing for clinical studies examining the utility of this compound in humans to be set up relatively quickly and easily.
The first of the DCS clinical studies (Ressler et al, 2004) examined the ability of DCS to enhance exposure therapy for acrophobia, or fear of heights, using virtual reality exposure (VRE) therapy. Previous work had shown improvements in acrophobia outcome measures after 7 weekly VRE therapy sessions (Rothbaum et al, 1995). Volunteer participants who met DSM-IV criteria for acrophobia were assigned randomly to groups receiving placebo or one of two doses of DCS (50 or 500 mg) in conjunction with VRE. Participants underwent a suboptimal amount of exposure therapy for acrophobia (two VRE sessions) and were instructed to take a single dose of study medication 2–4 h before each session. Similar to rats in the preclinical work, participants receiving either dose of DCS exhibited significantly more improvement than did those receiving placebo, with no statistical difference between the two doses. DCS-treated patients exhibited less fear and fewer skin conductance fluctuations in the VR environment, lower overall acrophobia symptoms, increased self-reports of exposure to heights in the ‘real world,’ and higher self-ratings of improvement.
Since then, other groups have found that DCS enhances exposure therapy for other fear and anxiety disorders, including social anxiety disorder (Guastella et al, 2008; Hofmann et al, 2006), obsessive-compulsive disorder (Kushner et al, 2007; Wilhelm et al, 2008; but see Storch et al, 2007), and panic disorder (Otto et al, 2010), indicating that the DCS effect is a relatively general one. The failure in Storch et al (2007) may have resulted from administering DCS 4 h before exposure therapy, which may have been too early. There has been one report of a failure of DCS to facilitate exposure therapy for subclinical spider phobia (Guastella et al, 2007a) and failures of DCS to facilitate extinction of Pavlovian-conditioned fear in a laboratory situation in humans (Guastella et al, 2007b; Grillon, 2009). However, these negative effects may indicate that DCS is useful only in people with clinically significant, maladaptive fear—consistent, perhaps, with the preclinical data suggesting that DCS is particularly effective in stressed animals (described in the ‘Positive modulation of NMDA receptor function’ section).
A very recent study examined the utility of DCS as an adjunct to exposure therapy for nicotine addiction in cigarette smokers (Santa Ana et al, 2009). Smokers underwent two sessions of smoking cue exposure involving handling cigarettes, lighters, and ashtrays, during which skin conductance responses (SCRs) and self-reports of craving were recorded. One hour before each exposure session, participants were administered 50 mg DCS (n=12) or placebo (n=13). In general, exposure therapy for addiction has not been met with great success, for reasons that remain unclear (Conklin and Tiffany, 2002), and consistent with this, participants in the smoking study who received placebo showed little evidence of extinction of SCR or craving during exposure sessions. Intriguingly, however, participants who received DCS showed facilitated within-session extinction of both measures.
Another recent study examined the effect of DCS on extinction of cocaine cue-induced craving in addicts, using a design similar to that of the smoking cue study in that it involved two sessions in which participants handled paraphernalia and simulated cocaine (Price et al, 2009). Two hours before each session, participants received 50 mg DCS (n=5) or placebo (n=5). One week later, a follow-up session was conducted without previous administration of DCS or placebo. Although not statistically significant, there was a trend in follow-up toward decreased self-reported craving in the DCS group relative to that in the placebo group. Owing to the small group sizes, these results are preliminary, but they are suggestive of a facilitation of extinction of cocaine cue-induced craving by DCS. However, DCS also significantly enhanced drug craving during exposure sessions, leading the authors of this study to question the clinical utility of DCS in the treatment of cocaine addiction.
There are two other potential problems with the clinical use of DCS that have emerged in the preclinical (animal) literature, and which need to be considered alongside the successes. One is the finding (discussed earlier) that DCS facilitates extinction but not re-extinction of a conditioned fear response (Langton and Richardson, 2008). Together with related findings that the mechanisms of re-extinction differ under some circumstances from those of original extinction (Chan and McNally, 2009; Laurent et al, 2008; Laurent and Westbrook, 2008), this finding may imply that DCS will be less effective as a clinical tool in patients who have relapsed after treatment and are seeking treatment again. Relapse is prevalent in both anxiety disorders and addiction; hence, it will be important to determine whether DCS is useful with this population and, if necessary, to develop alternative approaches. A second potential cause for concern is the finding that DCS modulates not only extinction but also reconsolidation (Lee et al, 2006, 2009). Reconsolidation is a form of memory consolidation that occurs after reactivation of a memory formed previously. For example, after training a rat to associate a tone with a foot shock, briefly re-exposing the rat to the tone seems to make the previously consolidated memory sensitive to disruption once again, such that the rat will be amnestic with regard to the tone when tested some time later if it was administered a memory-impairing treatment at the time of memory reactivation (Nader et al, 2000). Perhaps consistent with its function as a cognitive enhancer, DCS has the opposite effect; that is, it potentiates memory reconsolidation when administered in conjunction with a brief re-exposure to a previously conditioned stimulus, such that rats exhibit stronger memories when tested later. This is true for both fear conditioned stimuli (Lee et al, 2006) and drug-related conditioned stimuli (Lee et al, 2009). Reconsolidation and extinction are highly related phenomena, being distinguished procedurally by the duration of CS re-exposure, such that short re-exposures seem to promote reconsolidation and long re-exposures promote extinction (Eisenberg et al, 2003; Pedreira and Maldonado, 2003; Suzuki et al, 2004). This suggests that there may be a danger in using DCS clinically if an exposure therapy session is too brief or otherwise favors memory reconsolidation as opposed to extinction. At present, there are no data by which to assess the impact of these two potential problems in clinical populations, but they will be important considerations if DCS is to advance from being an experimental drug to being accepted and used as a clinical tool.
FUTURE RESEARCH DIRECTIONS
A great deal has been learned about the contribution of glutamate to extinction, but there is still much more work to be done. An unanswered question in the fear extinction literature, in particular, is how the apparently multiple mechanisms underlying extinction interact. Focusing just on BLA, a priority in future research will be identifying the various forms of synaptic plasticity involved, keeping in mind that different ones are likely to occur in different cell types, and that the nature of that plasticity might change depending on the circumstances of extinction training—for example, whether extinction is occurring for the first or second time, or if the interval between acquisition and extinction training is long or short.
The literature on extinction of conditioned drug craving and withdrawal is still in its infancy, but interest in this area is increasing and progress is being made. Immediate, major goals of this work include developing a map of the underlying circuitries and characterizing the interactions among the structures that are involved. It is likely that this type of extinction involves similar cortico-limbic mechanisms as implicated in fear extinction (Myers and Carlezon, 2010b), but more evidence is required to solidify that view. Having done this, teasing apart the contribution of glutamate receptor subtypes to the encoding, consolidation, and expression of drug-related extinction memory will be more feasible.
Clinical Implications
Basic research on glutamate receptor involvement in extinction has had an immediate, direct clinical benefit in the use of DCS to facilitate exposure therapy for fear and anxiety disorders. Particularly in the context of addiction, however, more study is required to determine the generality of the DCS effect. The evidence for clinical efficacy of DCS in exposure therapy for nicotine and cocaine addiction (Price et al, 2009; Santa Ana et al, 2009) is encouraging, and this, together with the growing animal literature on DCS-induced facilitation of extinction of conditioned drug craving and withdrawal (described in the ‘Positive modulation of NMDA receptor function’ section), provides a rationale for further study.
DCS is not the only drug that facilitates extinction. We have described three others that do as well: the AMPA receptor agonist PEPA, and the mGluR agonists AMN082 and CDPPB. Any of these might be an attractive candidate for further development as a potential clinical tool, assuming that they do not have unintended side effects. In this respect, the indirect, modulatory effect of DCS on glutamate function might be the key to its putative safety and efficacy.
Looking further ahead, if our understanding of glutamate actions within specific cell populations or brain regions were increased, technology might evolve to permit targeting treatments to those substrates to facilitate and maximize the generalizability of extinction. For example, gene therapy might become available whereby genes encoding NMDA receptor subunits could be delivered specifically to the amygdala or mPFC, before exposure therapy.
References
Akirav I (2007). NMDA partial agonist reverses blocking of extinction of aversive memory by GABA(A) agonist in the amygdala. Learn Mem 16: 682–686.
American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association: Washington, DC.
Baker JD, Azorlosa JL (1996). The NMDA antagonist MK-801 blocks the extinction of Pavlovian fear conditioning. Behav Neurosci 110: 618–620.
Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M et al (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291–1300.
Behar E, McHugh RK, Packham A, Otto MW (2010). D-cycloserine for the augmentation of an attentional training intervention for trait anxiety. J Anxiety Disord 24: 440–445.
Berlau DJ, McGaugh JL (2006). Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol Learn Mem 86: 123–132.
Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Pare D (2005). Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 132: 943–953.
Bertotto ME, Bustos SG, Molina VA, Martijena ID (2006). Influence of ethanol withdrawal on fear memory: effect of D-cycloserine. Neuroscience 142: 979–990.
Bevilaqua LR, Bonini JS, Rossato JI, Izquierdo LA, Cammarota M, Izquierdo I (2006). The entorhinal cortex plays a role in extinction. Neurobiol Learn Mem 85: 192–197.
Botreau F, Paolone G, Stewart J (2006). D-cycloserine facilitates extinction of a cocaine-induced conditioned place preference. Behav Brain Res 172: 173–178. Evidence that extinction of conditioned drug craving, like extinction of conditioned fear, can be facilitated by D-cycloserine.
Bouton ME (1993). Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull 114: 80–99.
Bouton ME, Bolles RC (1979). Contextual control of the extinction of conditioned fear. Learn Motiv 10: 455–466.
Bouton ME, Vurbic D, Woods AM (2008). D-cycloserine facilitates context-specific fear extinction learning. Neurobiol Learn Mem 90: 504–510.
Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S et al (2005). Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310: 1340–1343.
Britton JC, Gold AL, Feczko EJ, Rauch SL, Williams D, Wright CI (2007). D-cycloserine inhibits amygdala responses during repeated presentations of faces. CNS Spectr 12: 600–605.
Brooks DC, Vaughn JM, Freeman AJ, Woods AM (2004). An extinction cue reduces spontaneous recovery of ataxic ethanol tolerance in rats. Psychopharmacology (Berl) 176: 256–265.
Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ (2007). Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53: 871–880.
Cain CK, Godsil BP, Jami S, Barad M (2005). The L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem 12: 277–284.
Callaerts-Vegh Z, Beckers T, Ball SM, Baeyens F, Callaerts PF, Cryan JF et al (2006). Concomitant deficits in working memory and fear extinction are functionally dissociated from reduced anxiety in metabotropic glutamate receptor 7-deficient mice. J Neurosci 26: 6573–6582.
Cammarota M, Bevilaqua LR, Rossato JI, Ramirez M, Medina JH, Izquierdo I (2005). Relationship between short- and long-term memory and short- and long-term extinction. Neurobiol Learn Mem 84: 25–32.
Cannich A, Wotjak CT, Kamprath K, Hermann H, Lutz B, Marsicano G (2004). CB1 cannabinoid receptors modulate kinase and phosphatase activity during extinction of conditioned fear in mice. Learn Mem 11: 625–632.
Carlezon Jr WA, Nestler EJ (2002). Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci 25: 610–615.
Chan WY, McNally GP (2009). Conditioned stimulus familiarity determines effects of MK-801 on fear extinction. Behav Neurosci 123: 303–314.
Chaudhri N, Sahuque LL, Janak PH (2008). Context-induced relapse of conditioned behavioral responding to ethanol cues in rats. Biol Psychiatry 64: 203–210.
Chhatwal JP, Myers KM, Ressler KJ, Davis M (2005). Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci 25: 502–506.
Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ (2006). Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci 9: 870–872.
Childress AR, McLellan AT, O’Brien CP (1986). Abstinent opiate abusers exhibit conditioned craving, conditioned withdrawal and reductions in both through extinction. Br J Addict 81: 655–660. Early evidence that exposure therapy can reduce conditioned drug craving and withdrawal responses in addicts. Subsequent studies were more equivocal.
Conklin CA, Tiffany ST (2002). Applying extinction research and theory to cue-exposure addiction treatments. Addiction 97: 155–167. A meta-analysis of the drug cue exposure therapy literature through 2001, which found no statistically significant benefit.
Corcoran KA, Desmond TJ, Frey KA, Maren S (2005). Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci 25: 8978–8987.
Corcoran KA, Maren S (2001). Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci 21: 1720–1726. The first of several studies providing evidence that the hippocampus is critically involved in the contextual modulation of extinction memory expression.
Corcoran KA, Maren S (2004). Factors regulating the effects of hippocampal inactivation on renewal of conditional fear after extinction. Learn Mem 11: 598–603.
Cox J, Westbrook RF (1994). The NMDA receptor antagonist MK-801 blocks acquisition and extinction of conditioned hypoalgesic responses in the rat. Q J Exp Psychol B 47: 187–210.
Crespo JA, Oliva JM, Ghasemzadeh MB, Kalivas PW, Ambrosio E (2002). Neuroadaptive changes in NMDAR1 gene expression after extinction of cocaine self-administration. Ann NY Acad Sci 965: 78–91.
Crombag HS, Dickson M, Dinenna M, Johnson AW, Perin MS, Holland PC et al (2009). Narp deletion blocks extinction of morphine place preference conditioning. Neuropsychopharmacology 34: 857–866.
Cull-Candy SG, Leszkiewicz DN (2004). Role of distinct NMDA receptor subtypes at central synapses. Sci STKE 255: re16.
Dalton GL, Wang YT, Floresco SB, Phillips AG (2008). Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology 33: 2416–2426.
Delamater AR (2004). Experimental extinction in Pavlovian conditioning: Behavioural and neuroscience perspectives. Q J Exp Psychol B, Comp Physiol Psychol 57: 97–132.
Eisenberg M, Kobilo T, Berman DE, Dudai Y (2003). Stability of retrieved memory: inverse correlation with trace dominance. Science 301: 1102–1104.
Falls WA, Miserendino MJ, Davis M (1992). Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12: 854–863. The first study to demonstrate that extinction of conditioned fear involves NMDA receptors, but not AMPA receptors, within the basolateral amygdala.
Feltenstein MW, See RE (2007). NMDA receptor blockade in the basolateral amygdala disrupts consolidation of stimulus-reward memory and extinction learning during reinstatement of cocaine-seeking in an animal model of relapse. Neurobiol Learn Mem 88: 435–444. Provides evidence that NMDA receptors within the basolateral amygdala are involved in extinction of conditioned drug craving, similar to extinction of conditioned fear.
Fendt M, Schmid S, Thakker DR, Jacobson LH, Yamamoto R, Mitsukawa K et al (2008). mGluR7 facilitates extinction of aversive memories and controls amygdala plasticity. Mol Psychiatry 13: 970–979.
Fuchs RA, Feltenstein MW, See RE (2006). The role of the basolateral amygdala in stimulus-reward memory and extinction memory consolidation and in subsequent conditioned cued reinstatement of cocaine seeking. Eur J Neurosci 23: 2809–2813.
Fuchs RA, Weber SM, Rice HJ, Neisewander JL (2002). Effects of excitotoxic lesions of the basolateral amygdala on cocaine-seeking behavior and cocaine conditioned place preference in rats. Brain Research 929: 15–25.
Gabriele A, Packard MG (2007). D-cycloserine enhances memory consolidation of hippocampus-dependent latent extinction. Learn Mem 14: 468–471.
Garcia R, Chang CH, Maren S (2006). Electrolytic lesions of the medial prefrontal cortex do not interfere with long-term memory of extinction of conditioned fear. Learn Mem 13: 14–17.
Gass JT, Olive MF (2009). Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol Psychiatry 65: 717–720.
Gewirtz JC, Falls WA, Davis M (1997). Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medial prefrontal cortex in rats. Behav Neurosci 111: 712–726.
Goddyn H, Callaerts-Vegh Z, Stroobants S, Dirikx T, Vansteenwegen D, Hermans D et al (2008). Deficits in acquisition and extinction of conditioned responses in mGluR7 knockout mice. Neurobiol Learn Mem 90: 103–111.
Gomes GM, Mello CF, Rosa MM, Bochi GV, Ferreira J, Barron S et al (2010). Polyaminergic agents modulate contextual fear extinction in rats. Neurobiol Learn Mem 93: 589–595.
Gourley SL, Kedves AT, Olausson P, Taylor JR (2009). A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology 34: 707–716.
Grillon C (2009). D-cycloserine facilitation of fear extinction and exposure-based therapy might rely on lower-level, automatic mechanisms. Biol Psychiatry 66: 636–641.
Groblewski PA, Lattal KM, Cunningham CL (2009). Effects of D-cycloserine on extinction and reconditioning of ethanol-seeking behavior in mice. Alcohol Clin Exp Res 33: 772–782.
Guastella AJ, Dadds MR, Lovibond PF, Mitchell P, Richardson R (2007a). A randomized controlled trial of the effect of D-cycloserine on exposure therapy for spider fear. J Psychiatr Res 41: 466–471.
Guastella AJ, Lovibond PF, Dadds MR, Mitchell P, Richardson R (2007b). A randomized controlled trial of the effect of D-cycloserine on extinction and fear conditioning in humans. Behav Res Ther 45: 663–672.
Guastella AJ, Richardson R, Lovibond PF, Rapee RM, Gaston JE, Mitchell P et al (2008). A randomized controlled trial of D-cycloserine enhancement of exposure therapy for social anxiety disorder. Biol Psychiatry 63: 544–549.
Harris JA, Westbrook RF (1998). Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology 140: 105–115.
Havekes R, Nijholt IM, Visser AK, Eisel UL, Van der Zee EA (2008). Transgenic inhibition of neuronal calcineurin activity in the forebrain facilitates fear conditioning, but inhibits the extinction of contextual fear memories. Neurobiol Learn Mem 89: 595–598.
Heather N, Stallard A, Tebbutt J (1991). Importance of substance cues in relapse among heroin users: comparison of two methods of investigation. Addict Behav 16: 41–49.
Hefner K, Whittle N, Juhasz J, Norcross M, Karisson RM, Saksida LM et al (2008). Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci 28: 8074–8085.
Heldt SA, Ressler KJ (2007). Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci 26: 3631–3644.
Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A (2008). Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606. Provides evidence that a population of BLA neurons responds to extinguished cues, specifically.
Herry C, Mons N (2004). Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur J Neurosci 20: 781–790.
Hikind M, Maroun M (2008). Microinfusion of the D1 receptor antagonist, SCH23390 into the IL but not the BLA impairs consolidation of extinction of auditory fear conditioning. Neurobiol Learn Mem 90: 217–222.
Hobin JA, Goosens KA, Maren S (2003). Context-dependent neuronal activity in the lateral amygdala represents fear memories after extinction. J Neurosci 23: 8410–8416.
Hobin JA, Ji J, Maren S (2006). Ventral hippocampal muscimol disrupts context-specific fear memory retrieval after extinction in rats. Hippocampus 16: 174–182.
Hofmann SG, Meuret AE, Smits JA, Simon NM, Pollack MH, Eisenmenger K et al (2006). Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry 63: 298–304.
Hong I, Song B, Lee S, Kim J, Kim J, Choi S (2009). Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur J Neurosci 30: 2089–2099.
Hsu E, Packard MG (2008). Medial prefrontal cortex infusions of bupivacaine or AP-5 block extinction of amphetamine conditioned place preference. Neurobiol Learn Mem 89: 504–512. Provides evidence for an involvement of NMDA receptors within medial prefrontal cortex in extinction of conditioned drug craving.
Huang CC, Hsu KS (2001). Progress in understanding the factors regulating reversibility of long-term potentiation. Rev Neurosci 12: 51–68.
Hugues S, Chessel A, Lena I, Marsault R, Garcia R (2006). Prefrontal infusion of PD098059 immediately after fear extinction training blocks extinction-associated prefrontal synaptic plasticity and decreases prefrontal ERK2 phosphorylation. Synapse 60: 280–287.
Hugues S, Deschaux O, Garcia R (2004). Postextinction infusion of a mitogen-activated protein kinase inhibitor into the medial prefrontal cortex impairs memory of the extinction of conditioned fear. Learn Mem 11: 540–543.
Isiegas C, Park A, Kandel ER, Abel T, Lattal KM (2006). Transgenic inhibition of neuronal protein kinase A activity facilitates fear extinction. J Neurosci 26: 12700–12707.
Jay TM, Witter MP (1991). Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol 313: 574–586.
Ji J, Maren S (2008). Differential roles for hippocampal areas CA1 and CA3 in the contextual encoding and retrieval of extinguished fear. Learn Mem 15: 244–251.
Johnson DM, Baker JD, Azorlosa JL (2000). Acquisition, extinction, and reinstatement of Pavlovian fear conditioning: the roles of the NMDA receptor and nitric oxide. Brain Res 857: 66–70.
Kehoe EJ, Macrae M, Hutchinson CL (1996). MK-801 protects conditioned responses from extinction in the rabbit nictitating membrane preparation. Psychobiology 24: 127–135.
Kelamangalath L, Seymour CM, Wagner JJ (2009). D-serine facilitates the effects of extinction to reduce cocaine-primed reinstatement of drug-seeking behavior. Neurobiol Learn Mem 92: 544–551.
Kelamangalath L, Swant J, Stramiello M, Wagner JJ (2007). The effects of extinction training in reducing the reinstatement of drug-seeking behavior: involvement of NMDA receptors. Behav Brain Res 185: 119–128.
Kelley JB, Anderson KL, Itzhak Y (2007). Long-term memory of cocaine-associated context: disruption and reinstatement. Neuroreport 18: 777–780.
Kim J, Lee S, Park K, Hong I, Song B, Son G et al (2007a). Amygdala depotentiation and fear extinction. Proc Natl Acad Sci USA 104: 20955–20960. Detailed study examining extinction-related synaptic depotentiation and associated AMPA receptor internalization within the lateral amygdala.
Kim J, Lee S, Park H, Song B, Hong I, Geum D et al (2007b). Blockade of amygdala metabotropic glutamate receptor subtype 1 impairs fear extinction. Biochem Biophys Res Com 355: 188–193.
Kim JJ, Fanselow MS (1992). Modality-specific retrograde amnesia of fear. Science 256: 675–677.
Kim M, Campeau S, Falls WA, Davis M (1993). Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 59: 5–8.
Kushner MG, Kim SW, Donahue C, Thuras P, Adson D, Kotlyar M et al (2007). D-cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry 62: 835–838.
Labrie V, Duffy S, Wang W, Barger SW, Baker GB, Roder JC (2009). Genetic inactivation of D-amino acid oxidase enhances extinction and reversal learning in mice. Learn Mem 16: 28–37.
Langton JM, Kim JH, Nicholas J, Richardson R (2007). The effect of the NMDA receptor antagonist MK-801 on the acquisition and extinction of learned fear in the developing rat. Learn Mem 14: 665–668.
Langton JM, Richardson R (2008). D-cycloserine facilitates extinction the first time but not the second time: an examination of the role of NMDA across the course of repeated extinction sessions. Neuropsychopharmacology 33: 3096–3102.
Laurent V, Marchand AR, Westbrook RF (2008). The basolateral amygdala is necessary for learning but not relearning extinction of context conditioned fear. Learn Mem 15: 304–314.
Laurent V, Westbrook RF (2008). Distinct contributions of the basolateral amygdala and the medial prefrontal cortex to learning and relearning extinction of context conditioned fear. Learn Mem 15: 657–666. Demonstrates a shift in the contribution of NMDA receptors within the basolateral amygdala and medial prefrontal cortex to initial fear extinction and subsequent re-extinction.
Laurent V, Westbrook RF (2009a). Inactivation of the infralimbic but not the prelimbic cortex impairs consolidation and retrieval of fear extinction. Learn Mem 16: 520–529.
Laurent V, Westbrook RF (2009b). Infusion of the NMDA receptor antagonist, DL-APV, into the basolateral amygdala disrupts learning to fear a novel and a familiar context as well as relearning to fear an extinguished context. Learn Mem 16: 96–105.
Ledgerwood L, Richardson R, Cranney J (2003). Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci 117: 341–349.
Ledgerwood L, Richardson R, Cranney J (2004). D-cycloserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci 118: 505–513.
Ledgerwood L, Richardson R, Cranney J (2005). D-cycloserine facilitates extinction of learned fear: effects on reacquisition and generalized extinction. Biol Psychiatry 57: 841–847.
Lee H, Kim JJ (1998). Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci 18: 8444–8454.
Lee JL, Gardner RJ, Butler VJ, Everitt BJ (2009). D-cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn Mem 16: 82–85.
Lee JL, Milton AL, Everitt BJ (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci 26: 10051–10056.
Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Pare D (2008). Amygdala intercalated neurons are required for expression of fear extinction. Nature 454: 642–645. Makes use of a saporin-conjugated ligand to lesion selectively the ICMs while largely sparing the adjacent BLA. Demonstrates an impairment of extinction retention following ICM lesions.
Lin CH, Lee CC, Gean PW (2003). Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol 63: 44–52.
Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW (2003a). Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23: 1574–1579. Early evidence for an involvement of synaptic depotentiation within the basolateral amygdala in extinction of conditioned fear.
Lin CH, Yeh SH, Lu HY, Gean PW (2003b). The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci 23: 8310–8317.
Lin HC, Mao SC, Gean PW (2009a). Block of gamma-aminobutyric acid-A receptor insertion in the amygdala impairs extinction of conditioned fear. Biol Psychiatry 66: 665–673.
Lin HC, Mao SC, Su CL, Gean PW (2009b). Alterations of excitatory transmission in the lateral amygdala during expression and extinction of fear memory. Int J Neuropsychopharmacol 13: 335–345: 1–11.
Liu JL, Li M, Dang XR, Wang ZH, Rao ZR, Wu SX et al (2009). A NMDA receptor antagonist, MK-801 impairs consolidating extinction of auditory conditioned fear responses in a Pavlovian model. PloS One 4: e7548.
Lu KT, Walker DL, Davis M (2001). Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21: RC162.
Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126.
Mangiavacchi S, Wolf ME (2004). Stimulation of N-methyl-D-aspartate receptors, AMPA receptors or metabotropic glutamate receptors leads to rapid internalization of AMPA receptors in cultured nucleus accumbens neurons. Eur J Neurosci 20: 649–657.
Mao SC, Hsiao YH, Gean PW (2006). Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci 26: 8892–8899. A sophisticated study employing both behavioral and molecular tools to examine the mechanisms by which D-cycloserine facilitates extinction of conditioned fear.
Mao SC, Lin HC, Gean PW (2008). Augmentation of fear extinction by D-cycloserine is blocked by proteasome inhibitors. Neuropsychopharmacology 33: 3085–3095.
Maren S, Chang CH (2006). Recent fear is resistant to extinction. Proc Natl Acad Sci USA 103: 18020–18025.
Maren S, Hobin JA (2007). Hippocampal regulation of context-dependent neuronal activity in the lateral amygdala. Learn Mem 14: 318–324.
Matsumoto M, Togashi H, Konno K, Koseki H, Hirata R, Izumi T et al (2008). Early postnatal stress alters the extinction of context-dependent conditioned fear in adult rats. Pharmacol Biochem Behav 89: 247–252.
Milad MR, Quirk GJ (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74. A seminal study identifying the medial prefrontal cortex as a major contributor to extinction of conditioned fear.
Milad MR, Vidal-Gonzalez I, Quirk GJ (2004). Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci 118: 389–394.
Millin PM, Riccio DC (2002). Spontaneous recovery of tolerance to the analgesic effect of morphine. Physiol Behav 75: 465–471.
Morris RGM, Garrud P, Rawlins JNP, O’Keefe JO (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297: 681–683.
Mueller D, Porter JT, Quirk GJ (2008). Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci 28: 369–375.
Myers KM, Carlezon Jr WA (2010a). D-cycloserine facilitates extinction of naloxone-induced conditioned place aversion in morphine-dependent rats. Biol Psychiatry 67: 85–87.
Myers KM, Carlezon Jr WA (2010b). Extinction of drug- and withdrawal-paired cues in animal models: Relevance to the treatment of addiction. Neurosci Biobehav Rev (e-pub ahead of print).
Myers KM, Ressler KJ, Davis M (2006). Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem 13: 216–223.
Nader K, Schafe GE, LeDoux JE (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722–726.
Nic Dhonnchadha BA, Szalay JJ, Achat-Mendes C, Platt DM, Otto MW, Spealman RD et al (2010). D-cycloserine deters reacquisition of cocaine self-administration by augmenting extinction learning. Neuropsychopharmacology 35: 357–367.
Nicoll RA, Malenka RC (1999). Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann NY Acad Sci 868: 515–525.
Nijholt IM, Ostroveanu A, Scheper WA, Penke B, Luiten PG, Van der Zee EA et al (2008). Inhibition of PKA anchoring to A-kinase anchoring proteins impairs consolidation and facilitates extinction of contextual fear memories. Neurobiol Learn Mem 90: 223–229.
Otto MW, Tolin DF, Simon NM, Pearlson GD, Basden S, Meunier SA et al (2010). Efficacy of D-cycloserine for enhancing response to cognitive-behavior therapy for panic disorder. Biol Psychiatry 67: 365–370.
Paolone G, Botreau F, Stewart J (2009). The facilitative effects of D-cycloserine on extinction of a cocaine-induced conditioned place preference can be long lasting and resistant to reinstatement. Psychopharmacology (Berl) 202: 403–409.
Parnas AS, Weber M, Richardson R (2005). Effects of multiple exposures to D-cycloserine on extinction of conditioned fear in rats. Neurobiol Learn Mem 83: 224–231.
Pavlov IP (1927). Conditioned Reflexes. Oxford University Press: Oxford, UK.
Pedreira ME, Maldonado H (2003). Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron 38: 863–869.
Peters J, Kalivas PW, Quirk GS (2009). Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 16: 279–288.
Pfeiffer UJ, Fendt M (2006). Prefrontal dopamine D4 receptors are involved in encoding fear extinction. Neuroreport 17: 847–850.
Pitkanen A, Pikkarainen M, Nurminen N, Ylinen A (2000). Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. Ann NY Acad Sci 911: 369–391.
Port RL, Seybold KS (1998). Manipulation of NMDA-receptor activity alters extinction of an instrumental response in rats. Physiol Behav 64: 391–393.
Price KL, McRae-Clark AL, Saladin ME, Maria MM, DeSantis SM, Back SE et al (2009). D-cycloserine and cocaine cue reactivity: preliminary findings. Am J Drug Alcohol Abuse 35: 434–438.
Quirk GJ (2002). Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem 9: 402–407.
Quirk GJ, Armony JL, LeDoux JE (1997). Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron 19: 613–624.
Quirk GJ, Likhtik E, Pelletier JG, Pare D (2003). Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800–8807.
Quirk GJ, Mueller D (2008). Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33: 56–72.
Quirk GJ, Russo GK, Barron JL, Lebron K (2000). The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225–6231.
Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE (2001). Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci 4: 724–731.
Rescorla RA (2003). Protection from extinction. Learn Behav 31: 124–132.
Rescorla RA, Heth CD (1975). Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol: Anim Behav Process 1: 88–96.
Rescorla RA, Wagner AR (1972). A Theory of Pavlovian Conditioning: Variations in the Effectiveness of Reinforcement and Nonreinforcement. In: Black AH and Prokasy WF (eds). Classical Conditioning II: Current Research and Theory. Appleton Century Crofts: New York, pp 64–69.
Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Grapp K, Zimand E et al (2004). Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 61: 1136–1144. The first clinical evidence that D-cycloserine facilitates exposure therapy in a clinical population (people with acrophobia, or a fear of heights), similar to its effects on extinction of conditioned fear in rodents.
Reti IM, Crombag HS, Takamiya K, Sutton JM, Guo N, Dinenna ML et al (2008). Narp regulates long-term aversive effects of morphine withdrawal. Behav Neurosci 122: 760–768.
Robleto K, Poulos AM, Thompson RF (2004). Brain mechanisms of extinction of the classically conditioned eyeblink response. Learn Mem 11: 517–524.
Roche M, O’Connor E, Diskin C, Finn DP (2007). The effect of CB(1) receptor antagonism in the right basolateral amygdala on conditioned fear and associated analgesia in rats. Eur J Neurosci 26: 2643–2653.
Rosenkranz JA, Grace AA (2001). Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci 21: 4090–4103.
Rosenkranz JA, Grace AA (2002). Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci 22: 324–337.
Rosenkranz JA, Moore H, Grace AA (2003). The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci 23: 11054–11064. Suggests that mPFC may regulate amygdala activity via interactions with GABAergic interneurons in BLA, a potential extinction mechanism.
Rothbaum BO, Davis M (2003). Applying learning principles to the treatment of post-trauma reactions. Ann NY Acad Sci 1008: 112–121.
Rothbaum BO, Hodges LF, Kooper R, Opdyke D, Williford JS, North M (1995). Effectiveness of computer-generated (virtual reality) graded exposure in the treatment of acrophobia. Am J Psychiatry 152: 626–628.
Rumpel S, LeDoux J, Zador A, Malinow R (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308: 83–88.
Sakurai S, Yu L, Tan SE (2007). Roles of hippocampal N-methyl-D-aspartate receptors and calcium/calmodulin-dependent protein kinase II in amphetamine-produced conditioned place preference in rats. Behav Pharmacol 18: 497–506.
Santa Ana EJ, Rounsaville BJ, Frankforter TL, Nich C, Babuscio T, Poling J et al (2009). D-Cycloserine attenuates reactivity to smoking cues in nicotine dependent smokers: a pilot investigation. Drug Alcohol Depend 104: 220–227. Evidence that D-cycloserine may facilitate exposure therapy for cue-induced drug craving in addicts.
Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ (2004). Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci 24: 5704–5710.
Santini E, Muller RU, Quirk GJ (2001). Consolidation of extinction learning involves transfer from NMDA- independent to NMDA-dependent memory. J Neurosci 21: 9009–9017.
Schroeder JP, Packard MG (2003). Systemic or intra-amygdala injections of glucose facilitate memory consolidation for extinction of drug-induced conditioned reward. Eur J Neurosci 17: 1482–1488.
Schroeder JP, Packard MG (2004). Facilitation of memory for extinction of drug-induced conditioned reward: role of amygdala and acetylcholine. Learn Mem 11: 641–647.
Schultz W, Dickinson A (2000). Neuronal coding of prediction errors. Annu Rev Neurosci 23: 473–500.
Seeburg PH, Burnashev N, Kohr G, Kuner T, Sprengel R, Monyer H (1995). The NMDA receptor channel: molecular design of a coincidence detector. Recent Prog Horm Res 50: 19–34.
Shaw D, Norwood K, Sharp K, Quigley L, McGovern SF, Leslie JC (2009). Facilitation of extinction of operant behaviour in mice by D-cycloserine. Psychopharmacology (Berl) 202: 397–402.
Sheinin A, Shavit S, Benveniste M (2001). Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology 41: 151–158.
Sierra-Mercado Jr D, Corcoran KA, Lebron-Milad K, Quirk GJ (2006). Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci 24: 1751–1758.
Sigurdsson T, Doyere V, Cain CK, LeDoux JE (2007). Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology 52: 215–227.
Silvestri AJ, Root DH (2008). Effects of REM deprivation and an NMDA agonist on the extinction of conditioned fear. Physiol Behav 93: 274–281.
Simonyi A, Schachtman TR, Christoffersen GR (2005). The role of metabotropic glutamate receptor 5 in learning and memory processes. Drug News Perspect 18: 353–361.
Soltysik SS, Wolfe GE, Nicholas T, Wilson WJ, Garcia-Sanchez JL (1983). Blocking of inhibitory conditioning within a serial conditioned stimulus-conditioned inhibitor compound: maintenance of acquired behavior without an unconditioned stimulus. Learn Motivat 14: 1–29.
Sotres-Bayon F, Bush DE, LeDoux JE (2007). Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology 32: 1929–1940.
Sotres-Bayon F, Diaz-Mataix L, Bush DE, Ledoux JE (2009). Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cerebral Cortex 19: 474–482.
Steinglass J, Sysko R, Schebendach J, Broft A, Strober M, Walsh BT (2007). The application of exposure therapy and D-cycloserine to the treatment of anorexia nervose: a preliminary trial. J Psychiatr Pract 13: 238–245.
Stinus L, Caille S, Koob GF (2000). Opiate withdrawal-induced place aversion lasts for up to 16 weeks. Psychopharmacology (Berl) 149: 115–120.
Storch EA, Merlo LJ, Bengtson M, Murphy TK, Lewis MH, Yang MC et al (2007). D-cycloserine does not enhance exposure-response prevention therapy in obsessive-compulsive disorder. Int Clin Psychopharmacol 22: 230–237.
Storsve AB, McNally GP, Richardson R (2010). US habituation, like CS extinction, produces a decrement in conditioned fear responding that is NMDA dependent and subject to renewal and reinstatement. Neurobiol Learn Mem 93: 463–471.
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D et al (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behavior. Nature 421: 70–75.
Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004). Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24: 4787–4795.
Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I (2001). The role of NMDA glutamate receptors, PKA, MAPK, and CaMKII in the hippocampus in extinction of conditioned fear. Hippocampus 13: 53–58.
Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M et al (1999). Genetic enhancement of learning and memory in mice. Nature 401: 63–69.
Thanos PK, Bermeo C, Wang GJ, Volkow ND (2009). D-cycloserine accelerates the extinction of cocaine-induced conditioned place preference in C57bL/c mice. Behav Brain Res 199: 345–349.
Tomilenko RA, Dubrovina NI (2007). Effects of activation and blockade of NMDA receptors on the extinction of a conditioned passive avoidance response in mice with different levels of anxiety. Neurosci Behav Physiol 37: 509–515.
Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffeimeer AN et al (2008). Prefrontal cortex AMPA receptor plasticity is critical for cue-induced relapse to heroin-seeking. Nat Neurosci 11: 1053–1058.
Vengeliene V, Kiefer F, Spanagel R (2008). D-cycloserine facilitates extinction of conditioned alcohol-seeking behaviour in rats. Alcohol Alcohol 43: 626–629.
Wagner AR, Rescorla RA (1972). Inhibition in Pavlovian Conditioning: Application of a Theory. In: Boakes RA and Halliday MS (eds). Inhibition and Learning. Academic Press: London, pp 301–336.
Walker DL, Davis M (2008). Amygdala infusions of an NR2B-selective or an NR2A-preferring NMDA receptor antagonist differentially influence fear conditioning and expression in the fear-potentiated startle test. Learn Mem 15: 67–74.
Walker DL, Ressler KJ, Lu KT, Davis M (2002). Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci 22: 2343–2351. The first study to show that D-cycloserine facilitates extinction of conditioned fear in rodents.
Weber M, Hart J, Richardson R (2007). Effects of D-cycloserine on extinction of learned fear to an olfactory cue. Neurobiol Learn Mem 87: 476–482.
Werner-Seidler A, Richardson R (2007). Effects of D-cycloserine on extinction: consequences of prior exposure to imipramine. Biol Psychiatry 62: 1195–1197.
Wilber AA, Southwood CJ, Wellman CL (2009). Brief neonatal maternal separation alters extinction of conditioned fear and corticolimbic glucocorticoid and NMDA receptor expression in adult rats. Develop Neurobiol 69: 73–87.
Wilhelm S, Buhlmann U, Tolin DF, Meunier SA, Pearlson GD, Reese HE et al (2008). Augmentation of behavior therapy with D-cycloserine for obsessive-compulsive disorder. Am J Psychiatry 165: 335–341.
Woods AM, Bouton ME (2006). D-Cycloserine facilitates extinction but does not eliminate renewal of the conditioned emotional response. Behav Neurosci 120: 1159–1162.
Xu J, Zhu Y, Contractor A, Heinemann SF (2009). mGluR5 has a critical role in inhibitory learning. J Neurosci 29: 3676–3684.
Yamada D, Zushida K, Wada K, Sekiguchi M (2009). Pharmacological discrimination of extinction and reconsolidation of contextual fear memory by a potentiator of AMPA receptors. Neuropsychopharmacology 34: 2574–2584.
Yamamoto S, Morinobu S, Fuchikami M, Kurata A, Kozuru T, Yamawaki S (2008). Effects of single prolonged stress and D-cycloserine on contextual fear extinction and hippocampal NMDA receptor expression in a rat model of PTSD. Neuropsychopharmacology 33: 2108–2116.
Yang YL, Chao PK, Lu KT (2006). Systemic and intra-amygdala administration of glucocorticoid agonist and antagonist modulate extinction of conditioned fear. Neuropsychopharmacology 31: 912–924.
Yang YL, Chao PK, Ro LS, Wo YY, Lu KT (2007). Glutamate NMDA receptors within the amygdala participate in the modulatory effect of glucocorticoids on extinction of conditioned fear in rats. Neuropsychopharmacology 32: 1042–1051.
Yang YL, Lu KT (2005). Facilitation of conditioned fear extinction by D-cycloserine is mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase cascades and requires de novo protein synthesis in basolateral nucleus of amygdala. Neuroscience 134: 247–260. A detailed examination of the molecular mechanisms underlying the facilitation of extinction by D-cycloserine.
Yu H, Wang Y, Pattwell S, Jing D, Liu T, Zhang Y et al (2009). Variant BDNF Val66Met polymorphism affects extinction of conditioned aversive memory. J Neurosci 29: 4056–4064.
Yu SY, Wu DC, Liu L, Ge Y, Wang YT (2008). Role of AMPA receptor trafficking in NMDA receptor-dependent synaptic plasticity in the rat lateral amygdala. J Neurochem 106: 889–899.
Zushida K, Sakurai M, Wada K, Sekiguchi M (2007). Facilitation of extinction learning for contextual fear memory by PEPA: a potentiator of AMPA receptors. J Neurosci 27: 158–166.
Acknowledgements
This work was supported by the National Institute on Drug Abuse (DA012736 to WAC and DA027752 to KMM), the National Institute of Mental Health (MH047840 to MD and MH063266 to WAC), the Science and Technology Center (The Center for Behavioral Neuroscience of the National Science Foundation under Agreement No. IBN-9876754), and the Yerkes Base Grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Karyn M Myers: I declare that, except for income received from my primary employer, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. William A Carlezon, Jr.: I have US patents related to the use of kappa-opioid receptor ligands in the treatment of psychiatric disorders (Assignee: McLean Hospital). In the past 3 years, I have received compensation for professional services from The American College of Neuropsychopharmacology, Huya Bioscience International, Infinity Pharmaceuticals, Lantheus Medical Imaging, The Society for Neuroscience, and Transcept Pharmaceuticals. Michael Davis: The current paper will discuss an off-label use of D-cycloserine as an adjunct to psychotherapy. I have US patents for the use of D-cycloserine as an adjunct to psychotherapy and the use of D-cycloserine to be administered before sleep on the day on which psychotherapy occurs. I am a partner in a company called Therapade and another called Extinction Pharmaceuticals. Extinction Pharmaceuticals will try to license this technology to commercialize the use of D-cycloserine as an adjunct to psychotherapy. Should this occur, I would receive a signing fee and certain milestone payments over the next several years, during which I expect each payment will be more than $10 000. Once the indication is approved by the FDA, I am entitled to royalties which could exceed $10 000 per year. During the past 3 years, I have received research support, an unrestricted gift, and an honorarium from Astra-Zeneca Pharmaceuticals.
Rights and permissions
About this article
Cite this article
Myers, K., Carlezon, W. & Davis, M. Glutamate Receptors in Extinction and Extinction-Based Therapies for Psychiatric Illness. Neuropsychopharmacol 36, 274–293 (2011). https://doi.org/10.1038/npp.2010.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2010.88
Keywords
This article is cited by
-
Inhibition of mGluR5 alters BDNF/TrkB and GLT-1 expression in the prefrontal cortex and hippocampus and ameliorates PTSD-like behavior in rats
Psychopharmacology (2023)
-
TrkA-cholinergic signaling modulates fear encoding and extinction learning in PTSD-like behavior
Translational Psychiatry (2022)
-
N-acetylcysteine facilitates extinction of cued fear memory in rats via reestablishing basolateral amygdala glutathione homeostasis
Acta Pharmacologica Sinica (2022)
-
Environmental enrichment prevents the late effect of acute stress-induced fear extinction deficit: the role of hippocampal AMPA-GluA1 phosphorylation
Translational Psychiatry (2021)
-
Prevention of the return of extinguished fear by disrupting the interaction of neuronal nitric oxide synthase with its carboxy-terminal PDZ ligand
Molecular Psychiatry (2021)
